
Abstract
Background
Malaria still poses one of the major threats to human health. Development of effective antimalarial drugs has decreased this threat; however, the emergence of drug-resistant Plasmodium falciparum, a cause of Malaria, is disconcerting. The antimalarial drug chloroquine has been effectively used, but resistant parasites have spread worldwide. Interestingly, the withdrawal of the drug reportedly leads to an increased population of susceptible parasites in some cases. We examined the prevalence of genomic polymorphisms in a malaria parasite P. falciparum, associated with resistance to an antimalarial drug chloroquine, after the withdrawal of the drug from Indonesia.
Results
Blood samples were collected from 95 malaria patients in North Sulawesi, Indonesia, in 2010. Parasite DNA was extracted and analyzed by polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) for pfcrt and pfmdr1. In parallel, multiplex amplicon sequencing for the same genes was carried out with Illumina MiSeq. Of the 59 cases diagnosed as P. falciparum infection by microscopy, PCR–RFLP analysis clearly identified the genotype 76T in pfcrt in 44 cases. Sequencing analysis validated the identified genotypes in the 44 cases and demonstrated that the haplotype in the surrounding genomic region was exclusively SVMNT. Results of pfmdr1 were successfully obtained for 51 samples, where the genotyping results obtained by the two methods were completely consistent. In pfmdr1, the 86Y mutant genotype was observed in 45 cases (88.2%).
Conclusions
Our results suggest that the prevalence of the mutated genotypes remained dominant even 6 years after the withdrawal of chloroquine from this region. Diversified haplotype of the resistance-related locus, potentially involved in fitness costs, unauthorized usage of chloroquine, and/or a short post-withdrawal period may account for the observed high persistence of prevalence.
Similar content being viewed by others


Background
Malaria caused by Plasmodium falciparum remains a major health concern, particularly in tropical and subtropical regions. P. falciparum uses anopheline mosquitoes as a vector and spreads rapidly in the tropics and subtropics, where 3.3 billion people are at risk of contracting the parasite [1]. According to a 2016 World Health Organization report, >212 million cases of malaria were reported annually and approximately 429,000 people died from malaria worldwide [1]. In Southeast Asia, 15 million cases of malaria (7% global cases) and 26,000 deaths (6% global deaths) are reportedly associated with malaria each year [1].
Malaria is curable and preventable; however, the disease has not yet been completely eradicated. P. falciparum develops resistance against available medication, which explains the failed eradication of malaria; medication plays an important role in malaria control programs [2, 3]. Reports suggest that P. falciparum has now developed resistance to most antimalarial drugs, including chloroquine and its derivatives, sulfadoxine–pyrimethamine, mefloquine, and artemisinin [2–6]. Indeed, chloroquine was the standard antimalarial drug; however, chloroquine-resistant P. falciparum emerged in the late 1950s and spread worldwide [2]. It is widely accepted that several polymorphisms play important roles in chloroquine-resistant P. falciparum, particularly a threonine substitution at codon 76 in the P. falciparum chloroquine-resistant transporter (pfcrt) and a tyrosine substitution at codon 86 in P. falciparum multidrug-resistant protein (pfmdr1) [7–11].
Chloroquine resistance is widely distributed; however, reports from some African countries indicate a decline in the resistant parasite population after chloroquine discontinuation [12–16], although this reduction in resistance varied between countries. A study in Malawi demonstrated a marked decrease in the prevalence of the chloroquine-resistant marker from 85 to 15% 13 years after chloroquine discontinuation [16], where its incidence was only 1/685 in 2009 [17]. Similar observations have been reported from other African countries [12, 14]. However, 76T remains highly prevalent in Brazil [18], the Thai–Myanmar border [19], and Pakistan [20].
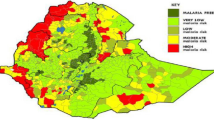
North Sulawesi is one of the malaria-endemic regions of Indonesia. A previous study detected a high prevalence of both 76T and 86Y polymorphisms (94 and 95%, respectively) among isolates from Minahasa, North Sulawesi (Fig. 1) [21]. The Indonesian government had to change its malaria treatment policy because of the high rate of chloroquine resistance, and the administration of artemisinin-based combination therapies began in 2004. Subsequently, dihydroartemisinin piperaquine became the dominant therapy [3]. As mentioned above, chloroquine-sensitive P. falciparum returned to some regions after the discontinuation of chloroquine. If the use of chloroquine can be reconsidered, its benefits would be considerable because chloroquine is a cost-effective drug without known severe side-effects. Most importantly, the reuse of pre-existing drugs would conserve the limited repertoire of antimalarial drugs. To better understand the changes in the resistant genotypes, we determined the prevalence of polymorphisms in pfcrt and pfmdr1 after chloroquine discontinuation in North Sulawesi, Indonesia.

Geographical representation of the sampling sites. Dots represent cities where blood samples were collected. The dashed circle represents the Minahasa region where a previous study was conducted [21]
Methods
Sample collection
Blood samples were collected from 95 malaria patients clinically diagnosed at hospitals in Manado and Bitung, North Sulawesi, Indonesia (Fig. 1), from August to December 2010. The collected blood samples were diagnosed via Giemsa staining and microscopic analysis by medical staff members at each hospital and collected on FTA Elute cards (GE Healthcare Life Sciences, Little Chalfont, UK).
Polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) analysis
Parasite DNA was extracted from dried filter blood spots by boiling them at 95 °C for 15 min. PCR–RFLP analysis of pfcrt and pfmdr1 was conducted as described previously [22]. In brief, PCR was conducted using KAPA 5× buffer (containing 7.5 mM MgCl2), 2.5 mM MgCl2, 0.25 mM each dNTP, 0.5 μM forward and reverse primers (Table 1), 0.625 U Taq, and 5 μL of extracted DNA in a total volume of 20 μL. For the nested PCR analyses, 2 μL of the 100× diluted PCR product from the first amplification step was used as a template in the second step. Restriction enzyme digestion was performed with 7 μL (pfcrt) or 5 μL (pfmdr1) of the PCR products and two units of ApoI for pfcrt or AflIII for pfmdr1. A laboratory clone (3D7) was also amplified and digested along with the samples, which served as a positive control. The digested products were stained with ethidium bromide and separated on a 2% agarose gel.
Multiplex amplicon sequencing with MiSeq and genotyping
The primary PCR reaction was performed as described above. The second nested PCR analyses used 2 μL of the 100× diluted PCR product from the first amplification step as template, with added tab nucleotides (Table 1). For the third PCR, with added dual indices, 2 μL of the 100× diluted PCR product from the second amplification step and TruSeq® DNA HT and RNA HT Sample Prep Kits (Illumina) were used. The amplicons were quantified with an Agilent 2200 TapeStation (Agilent), and then mixed to achieve the same molecular number. The samples were subjected to MiSeq (Illumina), and then sequenced with a MiSeq v3 Reagent Kit v3 (600-cycle). The short read sequences obtained were mapped onto the pfcrt and pfmdr1 coding sequences extracted from P. falciparum genome version 13, which was obtained from plasmoDB [23] by bowtie2 [24]. To identify genetic polymorphisms, the mapped results were analyzed with GATK [25].
Statistical tests
Regional differences in the genotypes were examined with Fisher’s exact test.
Results and discussion
Detection of the frequencies of the mutant genotypes of pfcrt and pfmdr1
In this study, we collected 95 blood samples that had been clinically diagnosed as malaria cases by local doctors. Among these samples, 59 were diagnosed by Giemsa staining as P. falciparum infections or possibly mixed infections of P. falciparum and P. vivax. Thirty cases were infections with P. vivax. No medical evidence was recorded for the remaining 6 cases. Using these samples, we conducted PCR–RFLP assays of the 227th base in pfcrt to identify the drug resistance-related genotypes at amino acid 76 (Fig. 2). We obtained clear results from 45/59 cases, which were initially diagnosed as P. falciparum infections (Fig. 3; Additional file 1: Table S1). In these 45 cases (100%), we identified the genotype “C,” which encodes 76T, and thus, a chloroquine-resistant pfcrt. We conducted similar analyses for the 36 remaining non-P. falciparum cases. We found that the P. falciparum genome was detected in five cases diagnosed as P. vivax infections and two cases without any medical records, possibly due to their ambiguous diagnoses. The identified genotype also encoded a 76T in these cases. In addition, to validate the correct identifications of the genotypes, we subjected the amplified PCR products to multiplex sequencing on the Illumina MiSeq platform (Fig. 4a, b). We examined all 45 cases and confirmed that the genotypes of the P. falciparum infections were 76T (see “Methods” for details of the single-nucleotide polymorphism procedure), except for one where we failed to obtain an amplicon by PCR (Fig. 3; Additional file 1: Table S1). We validated the sequences using the Sanger method for four randomly selected samples, which confirmed that they were identical (Fig. 4c). In addition to validating the sequences, multiplex amplicon sequencing allowed us to identify the exact genotypes in the surrounding region, i.e., the haplotypes. For amino acids 72–76, we found that all cases had the SVMNT haplotype and no case was identified with the CVIET haplotype. Non-synonymous variations at codon 72S were identified as “agt” and “tct” in 41 and 3 specimens, respectively (Additional file 1: Table S1).

Representative images of the PCR–RFLP results. M:100-bp marker, 3D7 Control sample prepared using DNA extracted from the chloroquine-susceptible 3D7 strain

Summary of the diagnosis and genotyping by PCR–RFLP, and the amplicon sequences. a The upper and lower panels show the results for pfcrt and pfmdr1, respectively. As shown by the two bars on the left, 59 samples were diagnosed as P. falciparum or P. falciparum–P. vivax-mixed infections according to Giemsa tests conducted by regional doctors. As shown by the two bars in the middle, 30 were diagnosed as P. vivax infections. As shown by the two bars on the right, six had no medical descriptions. In each set of bars, the left bar represents the genotype determined by PCR–RFLP and the right bar represents that detected by amplicon sequencing. b Cross tables comparing the genotyping results obtained by amplicon sequencing and PCR–RFLP. The tables on the left and right show the results for pfcrt and pfmdr1, respectively. NA not available because of a lack of PCR amplicons, nd no medical descriptions available from the collected sample

Scheme of next generation-based high-throughput multiplex amplicon sequencing and a representative result. a Target regions amplified by nested PCR. In the second PCR, primers with tags for the third PCR were used. In the third PCR, the primers had unique pairs of indices for multiplex sequencing. After sequencing with MiSeq, the reads obtained were separated according to the indices. Red and blue lines represent index sequences with priming targets for Illumina sequencing. b Snapshot of the mapped reads for sample ID A1 with IGV. Arrowheads represent the called variants. The reference is the sequence for the 3D7 strain. c Validation by Sanger sequencing of the corresponding region. Arrowheads represent polymorphisms detected by sequencing
We conducted PCR–RFLP assays of the pfmdr1 gene in parallel (Fig. 3; Additional file 1: Table S1). We obtained clear results from 51/59 samples. We also detected P. falciparum genomes in 10 cases initially diagnosed as P. vivax and 3 cases without any medical records. Among the 51 samples, the 86Y and 86N genotypes were identified in 45 (88.2%) and 6 (11.8%) cases, respectively. We conducted amplicon sequencing of these samples on MiSeq, which confirmed the correct genotype results in all cases. Based on these results, we can make the following conclusions. First, conventional Giemsa staining is occasionally imprecise and further confirmation is needed to precisely detect the infecting parasite species. Second, PCR–RFLP is a convenient and accurate method, although further sequencing analysis can provide extensive information associated with the surrounding bases. Finally, and most importantly, the dominant population of P. falciparum in this area still has a chloroquine-resistant genotype. We also collected and analyzed blood samples from two neighboring regions, i.e., Manado and Bitun, in North Sulawesi, but found no significant differences between the areas in terms of the prevalence of 76T and 86Y mutants (p = 1.000 and 0.218, respectively).
Comparison of the observed prevalence of pfcrt genotype with that in previous studies
Several previous studies have investigated the prevalence of the mutant genotypes of P. falciparum. A study that covered all of Indonesia in 2004 based on PCR–RLFP analyses showed that most of the genotypes in North Sulawesi were 76T [21]. However, compared to previous studies, we found a slight increase in the prevalence of 76T mutation (from 94.4 to 100%), despite the lack of clear statistical significance (p = 0.290).
Another previous study showed that in Lombok, Indonesia, codons 72–76 comprised SVMNT in most cases (87.5%) and only a few isolates possessed the Southeast Asia type of CVIET (10.4%) during 2002 [26]. The high prevalence of 76T was a common feature of all our observations, but we observed no cases of the CVIET haplotype in North Sulawesi. It is known that SVMNT is dominant in the Pacific region, such as Indonesia, Papua New Guinea, and the Philippines in Southeast Asia. By contrast, CVIET is dominant in Thailand, Vietnam, and Myanmar [27, 28], and the CVIET haplotype is assumed to have been spread from there to Africa [29]. Therefore, our finding that SVMNT occurred exclusively in North Sulawesi may reflect the distinct geological origins of the drug-resistant genotypes to some extent.
Intriguingly, the results of our study were very different from those obtained in previous studies conducted in Malawi and other African countries. In Malawi, the prevalence of the pfcrt 76T genotype decreased significantly from 85% in 1992 to 13% in 2000 [16, 30]. Subsequently, it was only present in 1/685 (0.1%) cases in 2009 [17]. In agreement, a field study showed that the susceptibility rate of the parasites to chloroquine increased from 50% in 1993 to 99% in 2005 [30]. Similar reductions in the prevalence of the pfcrt 76T genotype have also been reported from Kenya, Senegal, and other countries that are mainly located in Africa, although their remission rates were less drastic than that in Malawi [12, 14, 31]. Based on these studies, it is suggested that the mutants are fitter than the wild type under the selective pressure of chloroquine. In contrast, the advantage should disappear without the drug. Thus, the corresponding mutation genotypes may have disappeared after a certain time following the drug’s withdrawal.
To rationalize the observed reduction, it should also be hypothesized that the mutants are less fit than the wild type without the drug pressure. It seems to be true in CVIET, which is a major mutated haplotype around the 76T genotype and observed in the regions where the reduction of 76T was reported. In contrast, the SVMNT haplotype, which we found was dominant in North Sulawesi, is likely to be no less fit than the wild type [29]. Indeed, the fixed prevalence of 76T was also reported even after chloroquine withdrawal in Venezuela, where the SVMNT haplotype is similarly dominant [32]. Thus, differences in the biological features of the CVIET and SVMNT haplotypes with respect to their fitness may contribute to the distinct variations in their prevalence. Another even more confusing factor is the administration policy for the use of antimalarial drugs, where incomplete drug withdrawal and unauthorized chloroquine usage may have exposed the parasite to subtherapeutic concentrations, thereby contributing to the fixed mutant genotype. It has been shown that SVMNT is also involved in resistance to amodiaquine, which is used in a combination of artesunate–amodiaquine (AA) [27]. In Indonesia, AA is used despite the current policy recommending dihydroartemisinin piperaquine (DHP). Therefore, suboptimal administration of AA is potentially involved in the sustained SVMNT haplotype, and the inverse effect by strict substitution from AA to DHP may be expected. In addition, misdiagnosis may contribute to the confusion. Hence, further biological studies combined closely with epidemiological and possibly social science studies may yield a more thorough understanding of the prevalence dynamics of the mutant genotypes and their controls.
Prevalence of the genotypes of pfmdr1
It is known that the pfmdr1 86Y genotype is widespread throughout Indonesia (Table 2) [21]. The reported prevalence rates are 15.8 and 26.7% in Flores and East Nusa Tenggara (the eastern part of Indonesia) and in Armopa (West Papua), respectively [21]. By contrast, in western Indonesia, the reported prevalence of 86Y polymorphism is 100% [21]. Sulawesi, located in the middle of these regions, reported the prevalence rates for pfmdr1 86Y polymorphism as 95.0% in Minahasa, North Sulawesi, and 62.5% in Mamuju, South Sulawesi. The prevalence rate detected in the present study was between these two values (88.2%; p = 0.664 and 0.029, respectively). For the Asian countries surrounding Indonesia, the reported prevalence rates are similar to those in western Indonesia or Sulawesi, as well as cases from the most southern part of Thailand (96.3%; p = 0.018, compared with our results). However, the reported prevalence rates in Peninsular Malaysia (5.3%, p = 0.000), Cambodia (2.15%, p = 0.000), western Thailand (3.1%, p = 0.000), and the upper southern part of Thailand (36.4%, p = 0.000) differed from ours [19, 33–36]. Other studies have described almost equal prevalence rates for pfmdr1 86Y in African countries such as Kenya (81.6%, p = 0.545) or even lower prevalence in Benin (57.1%, p = 0.000), Malawi (22.7%, p = 0.000), and Senegal (14.9%, p = 0.000) [13, 14, 30, 37]. Note that one of the lowest prevalence rates for the mutant genotype 86Y was reported from Malawi, where the prevalence of the mutant pfcrt also decreased dramatically [16, 30]. However, as discussed in these studies, the geographical distribution of 86Y polymorphism is complicated and does not always coincide with geographical distribution of pfcrt polymorphism. Furthermore, unlike the mutation at 76T in pfcrt, a clear reduction in the mutant genotype has not yet been reported after the withdrawal of chloroquine. Indeed, it is known that polymorphisms in pfmdr1 are involved in resistance to not only chloroquine but also amodiaquine, mefloquine, and lumefantrine [9, 37–39]. In this study, a small trend of reduction in the six years (95.0 to 88.2%) was observed; however, it was not statistically significant (p = 0.664). Besides, the regions we investigated were not similar enough for the difference to be significant. Therefore, continuous and robust monitoring should be considered to determine the effect of chloroquine withdrawal on the prevalence of 86Y genotype in this region.
Conclusion
In this study, using samples obtained from malaria patients from North Sulawesi, Indonesia, we examined the chloroquine resistance polymorphisms in P. falciparum, i.e., 76T in pfcrt and 86Y in pfmdr1, by PCR–RFLP followed by multiplex amplicon sequencing. We emphasized the power of employing the multiplex amplicon sequencing method as well as confirming the convenience of using the conventional RFLP–PCR method. The cost of sequencing is decreasing rapidly; thus, a more comprehensive overview of the changes in genotypes throughout the world can be obtained by targeted sequencing of a larger number of genes in parasites or even whole genome sequencing. The results obtained in the present study showed that the prevalence rates of the mutant genotypes were 100 and 88.2% for 76T in pfcrt and 86Y in pfmdr1, respectively. We consider that the fixation of 76T mutations can be explained by the incomplete withdrawal of the drug, the short time interval since the withdrawal of chloroquine, and/or the possible equal fitness costs of the susceptible and resistant SMVNT genotypes. This suggests that re-emergence of the use of chloroquine is by no means guaranteed and that national health authorities should consider the results of continuous molecular surveillance when formulating their malaria treatment policies. To obtain definitive conclusion and rationalize these assumptions, the size of our cohort is not comprehensive enough and more detailed epidemiological information might be required. The multiplex amplicon sequencing system we applied is, however, supportive to this end.
Abbreviations
- P. falciparum :
-
Plasmodium falciparum
- P. vivax :
-
Plasmodium vivax
- PCR–RFLP:
-
polymerase chain reaction–restriction fragment length polymorphism
- pfcrt :
-
P. falciparum chloroquine-resistant transporter
- pfmdr1 :
-
P. falciparum multidrug-resistant protein
- AA:
-
artesunate–amodiaquine
- DHP:
-
dihydroartemisinin piperaquine
References
World Health Organization. World Malaria report 2016. World Health Organization; 2016.
Mita T, Tanabe K. Evolution of Plasmodium falciparum drug resistance: implications for the development and containment of artemisinin resistance. Jpn J Infect Dis. 2012;65:465–75.
Kusriastuti R, Surya A. New treatment policy of malaria as a part of malaria control program in Indonesia. Acta Med Indones. 2012;44:265–9.
Wongsrichanalai C, Sibley CH. Fighting drug-resistant Plasmodium falciparum: the challenge of artemisinin resistance. Clin Microbiol Infect. 2013;19:908–16.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–67.
Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–23.
Chaijaroenkul W, Ward SA, Mungthin M, Johnson D, Owen A, Bray PG, et al. Sequence and gene expression of chloroquine resistance transporter (pfcrt) in the association of in vitro drugs resistance of Plasmodium falciparum. Malar J. 2011;10:42.
Sidhu AB, Valderramos SG, Fidock DA. pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–26.
Picot S, Olliaro P, de Monbrison F, Bienvenu AL, Price RN, Ringwald P. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar J. 2009;8:89.
Awasthi G, Satya Prasad GB, Das A. Pfcrt haplotypes and the evolutionary history of chloroquine-resistant Plasmodium falciparum. Mem Inst Oswaldo Cruz. 2012;107:129–34.
Sanchez CP, Mayer S, Nurhasanah A, Stein WD, Lanzer M. Genetic linkage analyses redefine the roles of PfCRT and PfMDR1 in drug accumulation and susceptibility in Plasmodium falciparum. Mol Microbiol. 2011;82:865–78.
Ndiaye M, Faye B, Tine R, Ndiaye JL, Lo A, Abiola A, et al. Assessment of the molecular marker of Plasmodium falciparum chloroquine resistance (Pfcrt) in Senegal after several years of chloroquine withdrawal. Am J Trop Med Hyg. 2012;87:640–5.
Ogouyemi-Hounto A, Ndam NT, Kinde Gazard D, d’Almeida S, Koussihoude L, Ollo E, et al. Prevalence of the molecular marker of Plasmodium falciparum resistance to chloroquine and sulphadoxine/pyrimethamine in Benin seven years after the change of malaria treatment policy. Malar J. 2013;12:147.
Mwai L, Ochong E, Abdirahman A, Kiara SM, Ward S, Kokwaro G, et al. Chloroquine resistance before and after its withdrawal in Kenya. Malar J. 2009;8:106.
Frank M, Lehners N, Mayengue PI, Gabor J, Dal-Bianco M, Kombila DU, et al. A thirteen-year analysis of Plasmodium falciparum populations reveals high conservation of the mutant pfcrt haplotype despite the withdrawal of chloroquine from national treatment guidelines in Gabon. Malar J. 2011;10:304.
Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, et al. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–66.
Frosch AE, Laufer MK, Mathanga DP, Takala-Harrison S, Skarbinski J, Claassen CW, et al. Return of widespread chloroquine-sensitive Plasmodium falciparum to Malawi. J Infect Dis. 2014;210:1110–4.
Inoue J, Lopes D, do Rosario V, Machado M, Hristov AD, Lima GF, et al. Analysis of polymorphisms in Plasmodium falciparum genes related to drug resistance: a survey over four decades under different treatment policies in Brazil. Malar J. 2014;13:372.
Muhamad P, Chaijaroenkul W, Phompradit P, Rueangweerayut R, Tippawangkosol P, Na-Bangchang K. Polymorphic patterns of pfcrt and pfmdr1 in Plasmodium falciparum isolates along the Thai–Myanmar border. Asian Pac J Trop Biomed. 2013;3:931–5.
Khattak AA, Venkatesan M, Jacob CG, Artimovich EM, Nadeem MF, Nighat F, et al. A comprehensive survey of polymorphisms conferring anti-malarial resistance in Plasmodium falciparum across Pakistan. Malar J. 2013;12:300.
Syafruddin D, Asih PB, Casey GJ, Maguire J, Baird JK, Nagesha HS, et al. Molecular epidemiology of Plasmodium falciparum resistance to antimalarial drugs in Indonesia. Am J Trop Med Hyg. 2005;72:174–81.
Warhurst DC. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:299–302.
Aurrecoechea C, Barreto A, Brestelli J, Brunk BP, Cade S, Doherty R, et al. EuPathDB: the eukaryotic pathogen database. Nucleic Acids Res. 2013;41:D684–91.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Huaman MC, Yoshinaga K, Suryanatha A, Suarsana N, Kanbara H. Short report: polymorphisms in the chloroquine resistance transporter gene in Plasmodium falciparum isolates from Lombok, Indonesia. Am J Trop Med Hyg. 2004;71:40–2.
Sa JM, Twu O, Hayton K, Reyes S, Fay MP, Ringwald P, et al. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA. 2009;106:18883–9.
Mita T, Tanabe K, Kita K. Spread and evolution of Plasmodium falciparum drug resistance. Parasitol Int. 2009;58:201–9 (Elsevier Ireland Ltd; [cited 2012 Apr 10]).
Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28:504–14.
Kublin JG, Cortese JF, Njunju EM, Mukadam RA, Wirima JJ, Kazembe PN, et al. Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003;187:1870–5.
Okombo J, Kamau AW, Marsh K, Sutherland CJ, Ochola-Oyier LI. Temporal trends in prevalence of Plasmodium falciparum drug resistance alleles over two decades of changing antimalarial policy in coastal Kenya. Int J Parasitol Drugs Drug Resist. 2014;4:152–63.
Griffing S, Syphard L, Sridaran S, McCollum AM, Mixson-Hayden T, Vinayak S, et al. pfmdr1 amplification and fixation of pfcrt chloroquine resistance alleles in Plasmodium falciparum in Venezuela. Antimicrob Agents Chemother. 2010;54:1572–9.
Atroosh WM, Al-Mekhlafi HM, Mahdy MA, Surin J. The detection of pfcrt and pfmdr1 point mutations as molecular markers of chloroquine drug resistance, Pahang, Malaysia. Malar J. 2012;11:251.
Congpuong K, Na Bangchang K, Mungthin M, Bualombai P, Wernsdorfer WH. Molecular epidemiology of drug resistance markers of Plasmodium falciparum malaria in Thailand. Trop Med Int Heal. 2005;10:717–22.
Mungthin M, Intanakom S, Suwandittakul N, Suida P, Amsakul S, Sitthichot N, et al. Distribution of pfmdr1 polymorphisms in Plasmodium falciparum isolated from Southern Thailand. Malar J. 2014;13:117.
Vinayak S, Alam MT, Sem R, Shah NK, Susanti AI, Lim P, et al. Multiple genetic backgrounds of the amplified Plasmodium falciparum multidrug resistance (pfmdr1) gene and selective sweep of 184F mutation in Cambodia. J Infect Dis. 2010;201:1551–60.
Wurtz N, Fall B, Pascual A, Fall M, Baret E, Camara C, et al. Role of Pfmdr1 in in vitro Plasmodium falciparum susceptibility to chloroquine, quinine, monodesethylamodiaquine, mefloquine, lumefantrine, and dihydroartemisinin. Antimicrob Agents Chemother. 2014;58:7032–40.
Reed MB, Saliba KJ, Caruana SR, Kirk K, Cowman AF. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature. 2000;403:906–9.
Mwai L, Kiara SM, Abdirahman A, Pole L, Rippert A, Diriye A, et al. In vitro activities of piperaquine, lumefantrine, and dihydroartemisinin in Kenyan Plasmodium falciparum isolates and polymorphisms in pfcrt and pfmdr1. Antimicrob Agents Chemother. 2009;53:5069–73.
Authors’ contributions
YS, AEM, SMW, JY, and JT conceived and designed the study. AEM and JT conducted sample collection. PR, VV, IS, IHD, JAK, GEJ, LRR, YE, RM, and JY performed experiments. PR and JY conducted the literature search, performed data extraction and analysis, and interpreted the results. PR drafted and wrote the manuscript. YE, RM, and SMW critically reviewed the manuscript for important intellectual content and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to F. Minamoto and M. Imada for their positive coordination of the Japanese and Indonesian research communities. The authors also gratefully acknowledge support from the Asia-Africa S & T Strategic Cooperation Promotion Program from the Special Coordination Funds for Promoting Science & Technology and a Grant-in-aid for Scientific Research on Innovative Areas “Genome Science” (221S0002) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and JSPS KAKENHI Grant Number 15H05272.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data were included either in the main text or the supplemental materials except MiSeq raw reads as they were summarized in the Additional file 1: Table S1.
Ethics approval and consent to participate
Samples were collected with written informed consent or verbal consent, confirmed by doctors at Sam Ratulangi University, for participants who had difficulties reading the form. Records of these agreements were kept by the doctors. The study protocol was approved by the local ethical committees of Sam Ratulangi University and the University of Tokyo (approved number 10-49).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Reteng, P., Vrisca, V., Sukarno, I. et al. Genetic polymorphisms in Plasmodium falciparum chloroquine resistance genes, pfcrt and pfmdr1, in North Sulawesi, Indonesia. BMC Res Notes 10, 147 (2017). https://doi.org/10.1186/s13104-017-2468-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-017-2468-1