
Abstract
Background
An assessment of the correlation between anti-malarial treatment outcome and molecular markers would improve the early detection and monitoring of drug resistance by Plasmodium falciparum. The purpose of this systematic review was to determine the risk of treatment failure associated with specific polymorphisms in the parasite genome or gene copy number.
Methods
Clinical studies of non-severe malaria reporting on target genetic markers (SNPs for pfmdr1, pfcrt, dhfr, dhps, gene copy number for pfmdr1) providing complete information on inclusion criteria, outcome, follow up and genotyping, were included. Three investigators independently extracted data from articles. Results were stratified by gene, codon, drug and duration of follow-up. For each study and aggregate data the random effect odds ratio (OR) with 95%CIs was estimated and presented as Forest plots. An OR with a lower 95th confidence interval > 1 was considered consistent with a failure being associated to a given gene mutation.
Results
92 studies were eligible among the selection from computerized search, with information on pfcrt (25/159 studies), pfmdr1 (29/236 studies), dhfr (18/373 studies), dhps (20/195 studies). The risk of therapeutic failure after chloroquine was increased by the presence of pfcrt K76T (Day 28, OR = 7.2 [95%CI: 4.5–11.5]), pfmdr1 N86Y was associated with both chloroquine (Day 28, OR = 1.8 [95%CI: 1.3–2.4]) and amodiaquine failures (OR = 5.4 [95%CI: 2.6–11.3, p < 0.001]). For sulphadoxine-pyrimethamine the dhfr single (S108N) (Day 28, OR = 3.5 [95%CI: 1.9–6.3]) and triple mutants (S108N, N51I, C59R) (Day 28, OR = 3.1 [95%CI: 2.0–4.9]) and dhfr-dhps quintuple mutants (Day 28, OR = 5.2 [95%CI: 3.2–8.8]) also increased the risk of treatment failure. Increased pfmdr1 copy number was correlated with treatment failure following mefloquine (OR = 8.6 [95%CI: 3.3–22.9]).
Conclusion
When applying the selection procedure for comparative analysis, few studies fulfilled all inclusion criteria compared to the large number of papers identified, but heterogeneity was limited. Genetic molecular markers were related to an increased risk of therapeutic failure. Guidelines are discussed and a checklist for further studies is proposed.
Similar content being viewed by others
Background
Early diagnosis and treatment of uncomplicated malaria with anti-malarial drugs remains the mainstay of disease control in endemic areas. The emergence and spread of Plasmodium falciparum resistance to chloroquine (CQ) and sulphadoxine/pyrimethamine (SP) has rendered these two inexpensive, first-line anti-malarials ineffective in most malarious areas of the world, and compromised malaria control programmes [1, 2]. To rationalize alternative anti-malarial drug policy, it is crucial to be able to predict and monitor parasite resistance and yet the challenges are immense [3]. The in vivo test is widely used, but requires substantial logistical and financial support and its interpretation is confounded by factors such as reinfection, immunity, and pharmacokinetics [4]. In vitro tests quantify the anti-malarial activity against parasites isolated from infected individuals, but the correlation between such assays and clinical outcome is mostly unsubstantiated [5]. Identification of the molecular basis of anti-malarial drug resistance and its relationship to therapeutic failure represents a major advance in our ability to monitor anti-malarial drug resistance [6].
Linkage studies with parasite isolates from malaria patients have demonstrated a close association between the pfcrt K76T mutation and the in vitro chloroquine resistant phenotype [7]. Sequence analyses of the multi-drug resistance (Pfmdr) genes, initially thought to confer resistance through gene and P-glycoprotein over-expression, have revealed a series of point mutations that were associated with resistance [8]. More recently, gene copy number has been associated with decreased susceptibility to quinine, mefloquine, artemisinins, lumefantrine and halofantrine [9]. Resistance to antifolates and sulphonamides is conferred by point mutations at specific codons in the genes coding for the dihydrofolate reductase (DHFR) and dihydropteroate synthase (DHPS) enzymes, respectively, resulting in decreased affinity of the enzyme for the drug [10]. The molecular basis of artemisinin susceptibility has not been established yet, although an association with SERCA/ATPase6 has been proposed [11].
Understanding the relationship between putative molecular markers, parasite resistance and treatment failure has become a priority now that artemisin-based combination therapy (ACT) has replaced monotherapies as the first-line treatment of uncomplicated falciparum malaria [12]. In combination, the contribution of the individual components of drug regimen cannot be disentangled from a clinical study. Furthermore, recent studies have highlighted that withdrawal of chloroquine drug pressure may lead to a reversion to chloroquine-susceptible phenotypes [13], and these might have gone undetected if molecular prevalence surveys had not been conducted [14]. Amodiaquine is one of the most widely used artemisinin partner drugs in ACT, but the underlying mechanism of parasite resistance is poorly characterized [15]. Sulphadoxine/pyrimethamine (SP) is the only drug currently studied in detail for intermittent preventive therapy (IPT) in pregnant women and infants, but neither the influence of antifolate resistance on IPT efficacy nor the impact of IPT on the selection of drug resistant parasites has been comprehensively addressed.
Although it is not customary to change treatment policies based on molecular studies alone, molecular studies from Mali and Tanzania have demonstrated that a high prevalence of resistance makers can inform policy change [16, 17]. Hence, it is hoped that the identification of early markers of resistance will facilitate more widespread deployment of rational treatment policies that will retard the emergence of antimalarial drug resistant [18].
A key step in the process of validating experimental findings is to verify the correlation of parasite genetics with clinical response of the host. Collating this information is crucial to our ability to apply specific genetic markers to predict treatment failure. The aim of the current study was to conduct a systematic review and a meta-analysis of clinical trials reporting on putative genetic markers of P. falciparum resistance.
Methods
Study identification
A computerized search was carried out to identify clinical trials of treatments of non-severe malaria recording clinical and parasitological outcomes as well as the presence or absence of genetic polymorphisms or over-expression of genes suspected to be involved in drug resistance. References were screened using a computerized literature search of PubMed (last ten years, ending December 2008) combining the terms [(malaria OR plasmodium)] with different combination according to single nucleotide polymorphisms known to be associated with therapeutic failure: (pfmdr1 OR mdr1 OR mdr OR pfmdr); (pfcrt OR crt); (pfdhfr OR dhfr OR dihydrofolate reductase); (pfdhps OR dhps OR dihydropteroate synthase). Abstracts, case reports, editorials, basic sciences and nonhuman studies were excluded.
Study selection
Three authors (SP, FdM & ALB) independently reviewed abstracts and full text of the references identified to determine suitability for inclusion. Studies were included if they met the criteria allowing a complete extraction of data. Examiners were not blinded to authors, institutions or journal names.
Inclusion criteria
Studies were included in the analysis if it was possible from the publication to obtain all the distinguishing features that follow:
-
1.
Patients presenting non-severe falciparum malaria.
-
2.
Rate of wild/mutated type for any codon position in one or more of the P. falciparum genes known to be involved in drug resistance.
-
3.
Rate of treatment failure/success in the studied population
-
4.
Clinical outcome assessment following WHO criteria (1994 and subsequent versions)
-
5.
Duration of the follow-up
-
6.
Information on the area of the study
-
7.
Drug used, schedule and total dose
Molecular genotyping for recrudescence/reinfection discrimination (whatever the method used and the discriminate gene) was used to distinguish studies: studies using genotyping were specified in the figures.
Data extraction
When possible, relevant information was extracted from published tables or figures. If the data were not provided in tabular form, they were extracted or estimated from the body of the text, mostly by transformation of % to numbers of patients or number of mutations. The number of outcome events (total number of therapeutic failures out of included patients) and denominators (number of mutant type and wild type) were extracted for each resistance gene. Parasitological failures, irrespective of symptoms, were included as treatment failures and, when PCR genotyping was used to distinguish between recrudescence and reinfection, only the data from confirmed failure were used. Studies presenting only final odds ratios, relative risk or genotype failure index, without showing raw data from patients, were not included.
Studies were stratified according to gene; codon; drug; length of the follow up or end-point. Secondary stratification allowed selection of the most accurate study for each gene, according to the drug used, the follow-up duration, and reinfection/recrudescence genotyping.
Analytical strategy and statistical method
The Odds Ratio (OR) was used rather than the Relative Risk (RR) since the OR compares the proportion of therapeutic failures among the mutated parasites to the proportion of therapeutic failures among the wild-type parasites, while the RR compares the incidence of failure between the mutated and the wild-type parasites. Considering the numerous co-factors that could be involved in therapeutic failure, OR seemed more accurate. The same limitation could apply to genotype-failure index (GFI) that was reported by few studies and that failed to take into account the prevalence of the event in general population [12]. For impact assessment, an odds ratio OR > 1 (95%CI) was considered consistent with therapeutic failure attributable to the mutant type of the parasite. A database with the extracted data was created in Comprehensive meta-analysis version 2 (Biostat, Englewood, NJ 07631, USA). For each study, the impact (OR 95%CI), random effects, summary estimates and heterogeneity was calculated according to standard methods [19, 20].
Results are presented as funnel plots where a positive association between a given mutation and failure is depicted by an OR95%CI lying on the right side of the graph ('B side').
Results
Studies selection
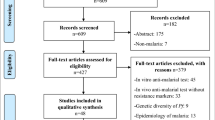
The computerized search identified 963 papers, of which 557 papers describing basic sciences or methodological experiments, and a further 202 papers without clinical follow-up were excluded (figure 1). Efficacy data could not be correlated with parasite genotype in 80 studies. Data were extracted from the remaining 124 studies, although 32 were subsequently excluded due to the lack of clinical failure, the absence of mutation, or the fixation of mutation in the whole parasite population. Hence complete information could be extracted from a total of 92 eligible studies.

Flow chart for the selection of studies published during the last ten years.
Pfmdr1 gene polymorphism
Only the polymorphism at codon 86 (N86Y) was assessed, since few studies addressed the other codons of other known polymorphisms. Of the 38 analysed studies, 12 were excluded mostly because it was impossible to extract data on the relationship between failures and genotypes. 22 studies using chloroquine and six studies using amodiaquine were included. Eleven of the chloroquine studies had a 14-day follow-up [21–31], ten had 28-day follow-up [32–41], and one had a 42-day follow-up [42]. Genotyping for reinfection/recrudescence was available for 3/11, 8/10 and 1/1 of these studies, respectively (Figure 2).

Pfmdr N86Y predictive value of therapeutic failure with chloroquine treatment. (A) Studies with 14 days follow-up (B) Studies with 28 days follow-up. Odds ratios (95% CI) are presented both numerically and graphically. The size of the forest plots is proportional to the relative weight of the study in the meta-analysis. The first row has the name of the first author and the year of publication of the study analysed. Target genes used for distinguishing reinfection to recrudescence are indicated when available. The red plot is the total OR for the listed studies.
The risk of therapeutic failure was greater for patients harbouring the N86Y pfmdr1 polymorphism with an Odds Ratio (OR) of 2.2 (95%CI: 1.6 – 3.1, p < 0.001) for the studies with 14-day follow-up and 1.8 (95%CI: 1.3–2.4, p < 0.001) for those with 28-day follow-up. For the 7 studies with 28-day follow-up in which recurrent infection were genotyped, the OR was 1.9 (95%CI: 1.3–2.7, p < 0.001). To avoid potential publication bias, the number of missing studies that would nullify the observed effect was computed using Classic fail-safe N statistics [20]. The fail-safe number was high (N = 60), indicating the value of this marker will probably not be changed by future studies. All but five studies (two with 14 days follow-up and three with 28 days follow-up) showed an OR > 1, although only eight (36%) had a lower confidence limit > 1.
Six studies using amodiaquine monotherapy were included in the analysis [31, 32, 36, 39, 43, 44], with the N86Y mutation associated with an OR of 5.4 (95%CI: 2.6 – 11.2, p < 0.001) (Figure 3). Genotyping for reinfection/recrudescence was used in five of these studies.

Pfmdr N86Y predictive value of therapeutic failure with amodiaquine treatment. Studies with different follow-up were included. Odds ratios (95% CI), forest plots and studies description are similar to figure 1.
Pfcrt gene polymorphism
Overall 42 studies were identified following chloroquine treatment, of which 13 were excluded due to missing data. Of the 25 studies included (13 with 14 days of follow-up [21–23, 25, 26, 28, 29, 31, 45–49] and 12 with 28 days of follow up [35–40, 50–55]), genotyping was available for 13 (52%), eight of which had a 28-day follow up. The OR for failure associated with the K76T mutation was 2.1 (95%CI: 1.5–3.0, p < 0.001) and 7.2 (95%CI: 4.5 – 11.5, p < 0.001) for the 14-day and the 28-day studies, respectively (Figure 4). All but one study (14-day follow-up) showed an OR > 1 and 11 (44%) had a lower confidence limit > 1. Of the twelve 28-day studies, seven had a lower confidence limit > 1. In the eight studies with genotyping of the recurrent infection the OR for recrudescence by day 28 was 5.1 (95%CI: 3.1–8.45, p < 0.001). The number of missing studies that would nullify the observed effect was 77, meaning that 77 'null' studies would be required in order for the combined 2-tailed p-value to exceed 0.05.

Pfcrt K76T predictive value of therapeutic failure with chloroquine. (A) Studies with 14 days follow-up (B) Studies with 28 days follow-up. Odds ratios (95% CI), forest plots and studies description are similar to previous figures. One study with day 35 end point was included in the day 28 list since the difference seems to be weak in terms of late failure rate.
Pfmdr1 N86Y and Pfcrt K76T associated polymorphisms
In five studies following chloroquine treatment [21–23, 34, 35], data on polymorphisms at both the pfmdr1 N86Y codon and pfcrt K76T codon were available with PCR genotyping, although only two studies had a 28-day follow up [34, 35]. The combined OR was 3.9 (95%CI: 2.6 – 5.8, p < 0.001) with a corresponding fail-safe number of 40 supporting a strong association between these markers and therapeutic failures. All but one study had an OR > 1, and two had a lower confidence limit > 1.
3 studies [31, 43, 44] including 172 patients assessed the relationship of Pfmdr1 N86Y and Pfcrt K76T polymorphisms and amodiaquine efficacy, one with a follow-up of 21 days and the other with 28 days. Failures were distinguished by Msp2 genotyping. The OR was 4.0 (95%CI: 1.1 – 14.6, p < 0.001), although the fail-safe number could not be calculated for two studies.
Pfdhfr gene polymorphism
Polymorphisms at codons 51, 59 and 108 single mutants and the triple mutant (51 + 59 +108) were studied when SP was used to treat patients. Each single mutant was considered irrespective of the presence or absence of other Pfdhfr mutations. It was not possible to derive from published papers if Pfdhfr 108 single mutants were or not linked with double or triple mutations. In total information on Pfdhfr 108 single mutant was available in 18 studies with follow-up to 14 days and 21 days in six [47, 56–60] and two [61, 62] of these studies respectively, leaving ten studies with 28 days follow-up [55, 63–71] (Figure 5). The OR for the 18 studies was 2.1 (95%CI: 1.4–3, p < 0.001) and was 3.5 (95%CI: 1.9–6.3, p < 0.001) for the 10 studies with 28 days of follow-up, with a fail-safe number of 13. Only three studies showed a lower confidence limit > 1, including the study with the highest relative weight and no genotyping. The OR for codon 51 and 59 single mutants were 1.7 (95%CI: 1.0–3.0, p = O.038) and 1.9 (95%CI: 1.4–2.6, p < 0.001), respectively. The same limitation regarding the possible association with other mutations should be taken into account for the interpretation of these results.

Pfdhfr S108N predictive value of therapeutic failure with Sulfadoxine-pyrimethamine. Studies with different follow-up were included. S108N point mutation was considered irrespective of the presence of other mutations at different Pfdhfr codons. Studies were stratified according first to the duration of the follow-up, second to the use of genotyping for recrudescence, third to the date of publication.
Sixteen studies concerning the triple Pfdhfr mutant (51+59+108) were included [55, 56, 61, 63, 65–67, 72–80], of which nine had 28 days of follow up with genotyping available in eight of these. The overall OR was 4.3 (95%CI: 3.0–6.3, p < 0.001) with a fail-safe number of 22 (Figure 6). Four studies with a 28-day follow-up had a lower confidence limit > 1 and a corresponding fail-safe number of 88. In the eight studies with PCR adjusted outcome at day 28 the OR was 3.1 (95%CI: 2.0 – 4.9; p < 0.001).

Pfdhfr N51I+C59R+S108N predictive value of therapeutic failure with Sulfadoxine-pyrimethamine. Studies with different follow-up were included. T he duration of the follow-up of the last study was supposed to be 28 days while not clearly indicated in the method by authors.
For three studies testing one, two and three of the Pfdhfr SNPs, the overall ORs for the triple mutant was 3.9 (95%CI: 2.5 – 6.2, p < 0.001) compared to 2.1 (95%CI: 1.4 – 3.3, p < 0.001) for the single mutant. Only two studies testing on the same patients single (108) and triple (51–59–108) mutants, allowed the comparison of the respective OR: 1.9 (95%CI: 0.4 – 8.9) and 11.1 (95%CI: 2.4 – 51.9). Thus the risk of therapeutic failure increases with the number of mutations in Pfdhfr.
Pfdhps gene polymorphism
Fewer studies addressed the relationship between dihydropteroate synthase (dhps) polymorphisms and therapeutic failures. In 20 studies the A437G or K540E [33, 47, 55–57, 59–61, 63, 64, 67, 69–71, 76, 78–82] single mutations could be correlated with therapeutic failure, with follow up ranging from seven to 28 days and genotyping confirmation available in 50%. The combined OR for A437G was 1.5 (95%CI: 1.0–2.4, p = 0.065) with two of the studies having a lower 95%CI > 1. Pfdhps double mutant (437 + 540) were analysed in a further ten studies with different day follow-up [55, 56, 61, 65, 69, 70, 73, 76, 83, 84]. The overall OR was 3.9 (95%CI: 2.6–5.8, p < 0.001), although recurrent parasites were genotyped in only five of these studies (Figure 7).

Pfdhps 437 – 540 predictive value of therapeutic failure with Sulfadoxine-pyrimethamine. Odds ratios (95% CI), forest plots and studies description are similar to previous figures. Studies were stratified according first to the duration of the follow-up, second to the use of genotyping for recrudescence, third to the date of publication.
Pfdhfr – Pfdhps genes combined polymorphism
Since several treatment regimens were used, the analysis of the combined Pfdhfr and Pfdhps mutants was restricted to SP and the quintuple mutants of Pfdhfr (codons 51–59–108) plus Pfdhps (codons 437 and 540). Three studies with 28-day follow up were included [63, 70, 75] with genotyping of recurrent infections available for two. The OR for these studies was 5.2 (95%CI: 3.2 – 8.8, p < 0.001) with a fail-safe number of 38. All the studies had an OR and a lower confidence limit > 1.
In three studies, the predictive value of the Pfdhfr/Pfdhps quintuple mutants could be compared with that of the Pfdhfr triple mutants. In [63], the OR was 91.6 (95%CI: 11–717, p < 0.001) for triple Pfdhfr mutants and 24.7 (95%CI: 8.3–74.1, p < 0.001) for Pfdhfr/Pfdhps quintuple mutant. In [75], the ORs were 2.2 (95%CI: 1.1–4.7, p < 0.001) and 2.0 (95%CI: 1.0–3.9, p < 0.001), respectively. In [70], the ORs went from 10.3 to 13.4.
Pfmdr1 copy number
Only one study assessing pfmdr1 copy number and the response to the treatment of mefloquine for P. falciparum met the criteria for inclusion [85]. The OR for treatment failure associated with pfmdr1 amplification was 8.6 (95%CI: 3.3–22.9, p < 0.001) for mefloquine monotherapy at 28 days and 2.6 (95%CI: 1.2–5.6) p = 0.01) at 42 days following mefloquine + three days artesunate. In a study from the same site, treatment failure at day 42 following a four day regimen of artemether-lumefantrine was higher in infections with pfmdr1 amplification: OR = 5.1 (95%CI: 1.4–20, p = 0.012).
Publication bias
There was no evidence of major publication bias in any of the analyses from funnel-plot asymmetry. Exclusion of any study did not substantially alter the summary estimates.
Discussion
Despite the large number of studies published on anti-malarial drug efficacy, as reviewed by Myint et al [86], approximately 10% have specifically addressed the in vivo-molecular correlates of resistance with criteria proposed here. In total, 92 met the inclusion criteria, enrolling more than 1,000 patients for each of the major molecular markers of drug resistance. For the drugs presented in this analysis, resistance occurs via two fundamentally different mechanisms. Quinoline resistance is multigenic and epistatic and, at least for chloroquine, affects drug accumulation in the parasite food vacuole [87, 88]. In contrast the underlying molecular mechanism of antifolate resistance involves accumulation of single mutations of the gene encoding for the respective target enzymes [89].
Both pfcrt and pfmdr1 polymorphisms have been associated with chloroquine resistance. The Odds Ratio (OR) of the pfcrt K76T mutation for therapeutic failure after chloroquine exceeded 7.0 at 28 days and 2.0 at day 14. The robustness of this association is confirmed by the high number of null studies (77) required to negate it.
The association between CRT polymorphism and amodiaquine failure has not been adequately addressed. In the analysis presented the pfmdr1 N86Y polymorphism was the most frequently studied mutation and predicted failure to both chloroquine (1.9 (95%CI: 1.3–2.7, p < 0.001)) and amodiaquine (5.4 (95%CI: 2.6 – 11.2, p < 0.001)). However the association of this mutation and clinical response to chloroquine was weak since few null studies would challenge this observation. The predictivity of the combined pfmdr1 + pfcrt was comparable (OR = 3.9 (95%CI: 2.6–5.8)) compared to pfcrt alone and the number needed to nullify this association decreased to 40. However few studies combined both markers.
While the relationship between mutations in the Pfdhfr and Pfdhps genes and parasite resistance to antifolates is well described [90], the relative role of different mutations in either gene in determining treatment outcome is less clear. Although the degree of in vitro resistance and treatment failures to antifolates in this meta-analysis was expected to be proportional to accumulating mutations of Pfdhfr, there was no clear difference in the predictive values of single and triple mutants: OR = 3.5 (95%CI: 1.9–6.3, p < 0.001) and OR = 4.3 (95%CI: 3.0–6.3, p < 0.001) respectively. Most studies failed to analyse the link between mutations at codons 51, 59 and 108. When data were provided on the therapeutic failure rates associated with each of these codons, it was not always possible to carry out a cumulative analysis. The low difference for OR between single and triple mutants suggest that single mutants maybe markers for presence of other point mutations. Due to these limitations, the only predictive value that should be taken into account was the OR for triple mutants. Several other mutant patterns or drug combinations have been studied, but none provided sufficient data to be included in the meta-analysis.
Overall polymorphisms in Pfdhps at positions 437 and 540 were predictive of therapeutic failure (OR = 3.9 (95%CI: 2.6–5.8, p < 0.001), but these data should be considered with caution because of methodological issues with the studies included (different duration of follow-up and different use of genotyping). Pfdhfr + Pfdhps quintuple mutants were analysed from three different studies, providing an OR = 5.2 (95%CI: 3.2–8.8, p < 0.001), with 38 null studies required -suggesting the high predictive value of this composite genotype. It was impossible however to clearly address the question of the predictive role of the increase number of Pfdhfr + Pfdhps mutations since cumulative data from the same patient were rare.
The meta-analysis confirmed and quantified the association of the four genes studied and their underlying associated with the risk of therapeutic failure (Table 1). However there are several caveats. Firstly the resistance of the infecting parasite is only one determinant of treatment outcome. Multiple studies have highlighted the importance of host immunity to the underlying therapeutic efficacy in clinical studies. Such immunity is acquired over time with multiple exposures and thus related to the age of the patient and the transmission intensity [91]. Other contributing factors include the biomass of parasites at the start of treatment, the patient's adherence to treatment, the dose of drug used and its adequate absorption [92]. There were no enough studies in the present analysis for a subgroup or multivariate analysis incorporating age and other confounding factors, which reduces the power of the analysis to detect independent parasite factors associated with treatment failure.
Second, most of the studies included were conducted in Africa over the past 10 years, limiting the relevance of the conclusions in space and time [93]. For instance, during the study period chloroquine resistance was well established, and failure and prevalence of mutations rates were often at saturation, decreasing the power to detect a significant association. In view of the low number of studies meeting inclusion criteria, it was not possible to compare the OR between areas or periods with low mutation rates to areas or periods with mutations close to fixation.
Third, only published studies indexed in PubMed were considered for this meta-analysis, and one cannot exclude a publication bias towards positive studies. However, considering the number of null studies needed to change the data obtained, the effect of unpublished studies is likely to be limited.
Fourth, study methodology varied with respect to inclusion criteria, age of subjects, treatment schedules, PCR methods and reporting, level of transmission at trial site. A frequent reason for excluding a study was insufficient details in the paper to allow coherent data extraction. Moreover, approximately half of the studies included followed patients for only 14 days (Table 1), and as such will not identify late treatment failures, often the earliest manifestation of resistance [94]. Genotyping of recurrent infections to distinguish between re-infection and recrudescence was only available in 53% of studies assessed. When the analyses were restricted to studies where true failures could be determined, the ORs varied significantly and power was lost.
Lastly, the proportion of patients studied for molecular markers represents a fraction of those enrolled or analysed at the end of follow-up. As no explanation is given for patient attrition, a selection bias cannot be excluded.
Conclusion
Recent initiatives, such as the consensus meeting on use of genotyping in clinical trials [95] and the World-Wide Antimalarial Resistance Network [96], will hopefully provide guidelines on how to analyse and report field data on clinical, in vitro, molecular and pharmacokinetic determinants of resistance. As a result of these methodological issues, when inclusion criteria were applied, very few studies were eligible for the meta-analysis compared to the number of studies identified. Despite the limitations listed above, the results of this meta-analysis were reassuringly homogeneous (funnel plots were highly symmetrical) for all markers except pfmdr1 + pfcrt for chloroquine.
While the trials considered studied mostly monotherapies with variable degrees of parasite resistance, these results are still relevant now that combinations have become standard treatment of uncomplicated malaria. Amodiaquine and SP are used combined with artesunate and with each other. SP is currently the drug of choice for intermittent preventive treatment (IPT) in pregnancy and infancy. However, data on amodiaquine are limited and the relevance of the genetic mechanisms of resistance of chloroquine to other quinolines (pyronaridine, piperaquine) used in these combinations remains to be established.
With the extended use of combination therapies including old and newer drugs, genetic markers can discriminate the individual role of each component. Obviously more research is needed into the molecular basis of resistance, which are largely unknown especially for artemisin compounds. Early mapping of known and new resistance genes might be achieved by genome-wide scanning of polymorphisms [97].
Whatever the drug to be tested and the mutation to be surveyed, it is of utmost importance to reach a consensus on the methodology of futures studies, especially if comparison between areas and time is the objective of the network of team involved in molecular surveillance of drug resistance. A checklist is proposed here (Table 2), including a series items which need to be fulfilled before designing a study and before preparing data report. This template could be used by colleagues to increase the portability of molecular studies. It could be amended with the experience of experts in the field.
References
Hastings IM: Malaria control and the evolution of drug resistance: an intriguing link. Trends Parasitol. 2003, 19: 70-73. 10.1016/S1471-4922(02)00017-X.
White NJ: Antimalarial drug resistance. J Clin Invest. 2004, 113: 1084-1092.
Hastings IM, Korenromp EL, Bloland PB: The anatomy of a malaria disaster: drug policy choice and mortality in African children. Lancet Infect Dis. 2007, 7: 739-748. 10.1016/S1473-3099(07)70214-1.
Uhleman AC, Yuthavong Y, Fidock DA: Mechanisms of antimalarial drug action and resistance. Molecular Approaches to Malaria. Edited by: Sherman IW. 2005, Washington: ASM Press, 429-461.
Plowe CV: Monitoring antimalarial drug resistance: making the most of the tools at hand. J Exp Biol. 2003, 206: 3745-3752. 10.1242/jeb.00658.
Woodrow CJ, Krishna S: Antimalarial drugs: recent advances in molecular determinants of resistance and their clinical significance. Cell Mol Life Sci. 2006, 63: 1586-1596. 10.1007/s00018-006-6071-1.
Carlton JM, Fidock DA, Djimdé A, Plowe CV, Wellems TE: Conservation of a novel vacuolar transporter in Plasmodium species and its central role in chloroquine resistance of P. falciparum. Curr Opin Microbiol. 2001, 4: 415-420. 10.1016/S1369-5274(00)00228-9.
Cowman AF: The P-glycoprotein homologues of Plasmodium falciparum : Are they involved in chloroquine resistance?. Parasitol Today. 1991, 7: 70-76. 10.1016/0169-4758(91)90197-V.
Price RN, Cassar C, Brockman A, Duraisingh M, van Vugt M, White NJ, Nosten F, Krishna S: The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob Agents Chemother. 1999, 43: 2943-2949.
Plowe CV, Cortese JF, Djimde A, Nwanyanwu OC, Watkins WM, Winstanley PA, Estrada-Franco JG, Mollinedo RE, Avila JC, Cespedes JL, Carter D, Doumbo OK: Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase and epidemiologic patterns of pyrimethamine-sulfadoxine use and resistance. J Infect Dis. 1997, 176: 1590-1596.
Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O'Neill PM, Bray PG, Ward SA, Krishna S: Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003, 424: 957-961. 10.1038/nature01813.
Laufer MK, Djimdé AA, Plowe CV: Monitoring and deterring drug-resistant malaria in the era of combination therapy. Am J Trop Med Hyg. 2007, 77: 160-169.
Kublin JG, Cortese JF, Njunju EM, Mukadam RA, Wirima JJ, Kazembe PN, Djimdé AA, Kouriba B, Taylor TE, Plowe CV: Reemergence of chloroquine-sensitive Plasmodium falciparum malaria after cessation of chloroquine use in Malawi. J Infect Dis. 2003, 187: 1870-1875. 10.1086/375419.
Plowe CV, Roper C, Barnwell JW, Happi CT, Joshi HH, Mbacham W, Meshnick SR, Mugittu K, Naidoo I, Price RN, Shafer RW, Sibley CH, Sutherland CJ, Zimmerman PA, Rosenthal PJ: World Antimalarial Resistance Network (WARN) III: molecular markers for drug resistant malaria. Malar J. 2007, 6: 121-10.1186/1475-2875-6-121.
Holmgren G, Gil JP, Ferreira PM, Veiga MI, Obonyo CO, Bjorkman A: Amodiaquine resistant Plasmodium falciparum malaria in vivo is associated with selection of pfcrt 76T and pfmdr1 86Y. Infect Genet Evol. 2006, 6: 309-314. 10.1016/j.meegid.2005.09.001.
Djimde A, Doumbo OK, Steketee RW, Plowe CV: Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet. 2001, 358: 890-891. 10.1016/S0140-6736(01)06040-8.
Mugittu K, Ndejembi M, Malisa A, Lemnge M, Premji Z, Mwita A, Nkya W, Kataraihya J, Abdulla S, Beck HP, Mshinda H: Therapeutic efficacy of sulfadoxine-pyrimethamine and prevalence of resistance markers in Tanzania prior to revision of malaria treatment policy: Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase mutations in monitoring in vivo resistance. Am J Trop Med Hyg. 2004, 71: 696-702.
Plowe CV: The evolution of drug-resistant malaria. Trans R Soc Trop Med Hyg. 2009 Apr;103 Suppl 1:S11-4. 2009, 103 (Suppl 1): S11-S14. 10.1016/j.trstmh.2008.11.002.
Lipsey MW, Wilson DB: Practical meta-analysis. 2001, Thousand Oaks: Sage Publications
Hunter JE, Schmidt FL: Methods of meta-analysis: correcting error and bias in research findings. 2004, London: Sage Publications
Mockenhaupt FP, Ehrhardt S, Eggelte A, Agana-Nsiire P, Stollberg K, Mathieu A, Markert M, Otchwemah RN, Bienzle U: Chloroquine-treatment failure in northern Ghana: roles of pfcrt T76 an pfmdr1 Y86. Ann Trop Med Parasitol. 2005, 99: 723-732. 10.1179/136485905X75395.
Huaman MC, Yoshinaga K, Suryanatha A, Suarsana N, Kanbara H: Polymorphisms in the chloroquine resistance transporter gene in Plasmodium falciparum isolates from Lombok, Indonesia. Am J Trop Med Hyg. 2004, 71: 40-42.
Djimde A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, Dicko A, Xin-Zhuan Su, Nomura T, Fidock DA, Wellems TE, Plowe CV: A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001, 344: 257-263. 10.1056/NEJM200101253440403.
Basco LK, Ringwald P: Pfmdr1 Gene mutation and clinical response to chloroquine in Yaoundé, Cameroon. Trans R Soc Trop Med Hyg. 1997, 91: 210-211. 10.1016/S0035-9203(97)90227-3.
Jelinek T, Aida AO, Peyrl-Hoffmann G, Jordan S, Mayor A, Heuschkel C, El Valy AO, Von Sonnenburg F, Christophel EM: Diagnostic value of molecular markers in chloroquine-resistant falciparum malaria in southern Mauritania. Am J Trop Med Hyg. 2002, 67: 449-453.
Tinto H, Ouedraogo JB, Erhart A, Van Overmeir C, Dujardin J-C, Van Marck E, Guiguemde TR, D'Alessandro UD: Relationship between the Pfcrt T76 and the Pfmdr-1 Y86 mutations in Plasmodium falciparum and in vitro/in vivo chloroquine resistance in Burkina Faso, West Africa. Inf Gen Evol. 2003, 3: 287-292. 10.1016/j.meegid.2003.08.002.
Tagelsir N, Ibrahim Z, Medani A, Salih O, Hamad A, Giha H, El-Agib A, Khan B, Saeed N, Ibrahim M: High frequency of Plasmodium falciparum Pfcrt K76T and Pfpgh N86Y in patients clearing infection after chloroquine treatment in the Sudan. Acta Trop. 2006, 97: 19-25. 10.1016/j.actatropica.2005.07.030.
Flüeck TPF, Jelinek T, Kilian AHD, Adagu IS, Kabagambe G, von Sonnenburg F, Warhurst DC: Correlation of in vivo-resistance to chloroquine and allelic polymorphisms in Plasmodium falciparum isolates from Uganda. Trop Med Int Health. 2000, 5: 174-178. 10.1046/j.1365-3156.2000.00543.x.
Khalil IF, Alifrangis M, Tarimo DS, Staalso T, Satti GM, Theander TG, Ronn AM, Bygbjerg IC: The roles of the pfcrt 76 T and pfmdr1 86 Y mutations, immunity and the initial level of parasitaemia, in predicting the outcome of chloroquine treatment in two areas with different transmission intensities. Ann Trop Med Parasitol. 2005, 99: 441-448. 10.1179/136485905X46441.
Dorsey G, Kamya MR, Singh A, Rosenthal PJ: Polymorphisms in the Plasmodium falciparum pfcrt and pfmdr-1 genes and clinical responses to chloroquine in Kampala, Uganda. J Infect Dis. 2001, 183: 1417-1420. 10.1086/319865.
Ochong EO, Broek Van den IVF, Kees Keus, Nzila A: Association between chloroquine and amodiaquine resistance and allelic variation in the Plasmodium falciparum multiple drug resistance 1 gene and the chloroquine resistance transporter gene in isolates from the upper nile in southern Sudan. Am J Trop Med Hyg. 2003, 69: 184-187.
Duraisingh MT, Drakeley CJ, Muller O, Bailey R, Snounou G, Targett GA, Greenwood BM, Warhurst DC: Evidence for selection for the tyrosine-86 allele of the pfmdr1 gene of P lasmodium falciparum by chloroquine and amodiaquine. Parasitology. 1997, 114: 205-211. 10.1017/S0031182096008487.
Nagesha HS, Syafruddin D, Casey GJ, Susanti AI, Fryauff DJ, Reeder JC, Cowman AF: Mutation in the pfmdr1, dhfr and dhps genes of Plasmodium falciparum are associated with in-vivo drug resistance in West Papua, Indonesia. Trans R Soc Trop Med Hyg. 2001, 95: 43-49. 10.1016/S0035-9203(01)90329-3.
Pillai DR, Labbé A-C, Vanisaveth V, Hongvangthong B, Pomphida S, Inkathone S, Zhong K, Kain KC: Plasmodium falciparum malaria in Laos: Chloroquine treatment outcome and predictive value of molecular markers. J Infect Dis. 2001, 183: 789-795. 10.1086/318836.
Happi TC, Thomas SM, Gbotosho GO, Falade CO, Akinboye DO, Gerena L, Hudson T, Sowunmi A, Kyle DE, Milhous W, Wirth DF, Oduola AMJ: Point mutation in the Pfcrt and P fmdr-1 genes of Plasmodium falciparum and clinical response to chloroquine, among malaria patients from Nigeria. Ann Trop Med Parasitol. 2003, 97: 439-451. 10.1179/000349803235002489.
Happi CT, Gbotosho GO, Folarin OA, Bolaji OM, Sowunmi A, Kyle DE, Milhous W, Wirth DF, Oduola AMJ: Association between mutation in Plasmodium falciparum chloroquine resistance transporter and P. falciparum multidrug resistance 1 genes and in vivo amodiaquine resistance in P. falciparum malaria-infected children in Nigeria. Am J Trop Med Hyg. 2006, 75: 155-161.
Sarr O, Myrick A, Daily J, Diop BM, Dieng T, Ndir O, Sow PS, Mboup S, Wirth DF: In vivo and in vitro analysis of chloroquine resistance in Plasmodium falciparum isolates from Senegal. Parasitol Res. 2005, 97: 136-140. 10.1007/s00436-005-1406-7.
Ojurongbe O, Ogungbamigbe TO, Fagbenro-Beyioku AF, Fendel R, Kremsner PG, Kun JF: Rapid detection of Pfcrt and Pfmdr1 mutations in Plasmodium falciparum isolates by FRET and in vivo response to chloroquine among children from Osogbo, Nigeria. Malar J. 2007, 6: 41-10.1186/1475-2875-6-41.
Ursing J, Kofoed PE, Rodrigues A, Rombo L, Gil JP: Plasmodium falciparum genotypes associated with chloroquine and amodiaquine resistance in Guinea-Bissau. Am J Trop Med Hyg. 2007, 76: 844-848.
Sutherland C, Alloueche A, Curtis J, Drakeley CJ, Ord R, Duraisingh M, Greenwood BM, Pinder M, Warhurst D, Targett GAT: Gambian children successfully treated with chloroquine can harbour and transmit Plasmodium falciparum gametocytes carrying resistance genes. Am J Trop Med Hyg. 2002, 67: 578-585.
Mita T, Kaneko A, Koji Lum J, Bwijo B, Takechi M, Zungu IL, Tsukahara T, Tanabe K, Kobayakawa T, Björkman A: Recovery of chloroquine sensitivity and low prevalence of the Plasmodium falciparum chloroquine resistance transporter gene mutation K76T following the discontinuance of chloroquine use in Malawi. Am J Trop Med Hyg. 2003, 68: 413-415.
Bell DJ, Nyirongo SK, Mukaka M, Zijlstra EE, Plowe CV, Molyneux ME, Ward SA, Winstanley PA: Sulfadoxine-pyrimethamine-based combinations for malaria: a randomised blinded trial to compare efficacy, safety and selection of resistance in Malawi. PLoS ONE. 2008, 3: 1578-10.1371/journal.pone.0001578.
Holmgren G, Gil JP, Ferreira PM, Veiga MI, Obonyo CO, Björkman A: Amodiaquine resistant Plasmodium falciparum malaria in vivo is associated with selection of pfcrt 76T and pfmdr1 86Y. Infect Genet Evol. 2006, 6: 309-314. 10.1016/j.meegid.2005.09.001.
Tinto H, Guekoun L, Zongo I, Guiguemdé RT, D'Alessandro U, Ouédraogo JB: Chloroquine-resistance molecular markers (Pfcrt T76 and Pfmdr-1 Y86) and amodiaquine resistance in Burkina Faso. Trop Med Int Health. 2008, 13: 238-40.
Basco LK, Ndounga M, Foumane Ngane V, Soula G: Molecular epidemiology of malaria in Cameroon. XIV. Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene sequences of isolates before and after chloroquine treatment. Am J Trop Med Hyg. 2002, 67: 392-395.
Schneider AG, Premji Z, Felger I, Smith T, Abdulla S, Beck HP, Mshinda H: A point mutation in codon 76 of pfcrt of P. falciparum is positively selected for by Chloroquine treatment in Tanzania. Infect Gen Evol. 2002, 1: 183-189. 10.1016/S1567-1348(01)00021-1.
Berens N, Schwoebel B, Jordan S, Vanisaveth V, Phetsouvanh R, Christophel EM, Phompida S, Jelinek T: Plasmodium falciparum : correlation of in vivo resistance to chloroquine and antifolates with genetic polymorphisms in isolates from the south of Lao PDR. Trop Med Int Health. 2003, 8: 775-782. 10.1046/j.1365-3156.2003.01099.x.
Pati SS, Mishra S, Mohanty S, Mohapatra DN, Sahu PK, Priyadarshi N, Kumar S, Sharma SK, Tyagi PK, Chitnis CE, Das BS: Pfcrt haplotypes and in-vivo chloroquine response in Sundergarh district, Orissa, India. Trans R Soc Trop Med Hyg. 2007, 101: 650-4. 10.1016/j.trstmh.2007.01.008.
Meissner PE, Mandi G, Mockenhaupt FP, Witte S, Coulibaly B, Mansmann U, Frey C, Merkle H, Burhenne J, Walter-Sack I, Müller O: Marked differences in the prevalence of chloroquine resistance between urban and rural communities in Burkina Faso. Acta Trop. 2008, 105: 81-6. 10.1016/j.actatropica.2007.07.014.
Mayor AG, Gomez-Olivé X, Aponte JJ, Casimiro S, Mabunda S, Dgedge M, Barreto A, Alonso PL: Prevalence of the K76T mutation in the putative Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene and its relation to chloroquine resistance in Mozambique. J Infect Dis. 2001, 183: 1413-1416. 10.1086/319856.
Maguire JD, Susanti AI, Krisin , Sismadi P, Fryauff J, Baird JK: The T76 mutation in the pfcrt gene of Plasmodium falciparum and clinical chloroquine resistance phenotypes in Papua, Indonesia. Ann Trop Med Parasitol. 2001, 95: 559-572. 10.1080/00034980120092516.
Nguyen MH, Davis TME, Cox-Singh J, Hewitt S, Quoc Taon T, Bach Kim T, Thi Hanh N, Phuong VN, Nhan DH, Cong LD: Treatment of uncomplicated falciparum Malaria in Southern Vietman: can chloroquine or sulfadoxine-pyrimethamine be reintroducced in combination with Artesunate?. Clin Infect Dis. 2003, 37: 1461-1466. 10.1086/379323.
Nagesha HS, Casey GJ, Rieckmann KH, Fryauff D, Laksana BS, Reeder J, Maguire JD, Baird JK: New haplotypes of the Plasmodium falciparum chloroquine resistance transporter (pfcrt) gene among chloroquine-resistant parasite isolates. Am J Trop Med Hyg. 2003, 68: 398-402.
Ranjit MR, Das A, Chlotray GP, Roth RN, Kar SK: The Pfcrt (K76T) point mutation in Plasmodium falciparum, and its usefulness for monitoring chloroquine resistance. Ann Trop Med Parasitol. 2004, 98: 879-882. 10.1179/000349804X3162.
Mayxay M, Nair S, Sudimack D, Imwong M, Tanomsing N, Pongvongsa T, Phompida S, Phetsouvanh R, White NJ, Anderson TJ, Newton PN: Combined molecular and clinical assessment of Plasmodium falciparum antimalarial drug resistance in the Lao People's Democratic Republic (Laos). Am J Trop Med Hyg. 2007, 77: 36-43.
Basco LK, Tahar R, Keundjian A, Ringwald P: Sequence variations in the genes encoding dihydropteroate synthase and dihydrofolate reductase and clinical response to sulfadoxine-pyrimethamine in patients with acute uncomplicated falciparum malaria. J Infect Dis. 2000, 182: 624-628. 10.1086/315731.
Khalil IF, Ronn AM, Alifrangis M, Gabar HA, Jelinek T, Satti GM, Bygbjerg IC: Response of Plasmodium falciparum to cotrimoxazole therapy: relationship with plasma drug concentrations and dihydrofolate reductase and dihydropteroate synthase genotypes. Am J Trop Med Hyg. 2005, 73: 174-177.
Ibon Rallon N, Osorio LE, Giraldo LE: Lack of an association between the asn-108 mutation in the dihydrofolate reductase gene and in vivo resistance to sulfadoxine/pyrimethamine in Plasmodium falciparum. Am J Trop Med. 1999, 61: 245-248.
Jelinek T, Killan AHD, Curtis J, Duraisingh MT, Kabagambe G, von Sonnenburg F, Warhurst DC: Plasmodium falciparum : selection of serine 108 of dihydrofolate reductase during treatment of uncomplicated malaria with co-trimoxazole in Ugandan children. Am J Trop Med Hyg. 1999, 61: 125-130.
Fryauff DJ, Leksana B, Masbar S, Wiady I, Sismadi P, Susanti AI, Nagesha HS, Syafruddin , Atmosoedjono S, Bangs MJ, Baird JK: The drug sensitivity and transmission dynamics of human malaria on Nias Island, North Sumatra, Indonesia. Ann Trop Med Parasitol. 2002, 96: 447-62. 10.1179/000349802125001249.
Omar SA, Adagu IS, Warhurst DC: Can pre-treatment screening for dhps and dhfr point mutations in Plasmodium falciparum infections be used to predict sulfadoxine-pyrimethamine treatment failure?. Trans R Soc Trop Med Hyg. 2001, 95: 315-319. 10.1016/S0035-9203(01)90250-0.
Curtis J, Duraising MT, Warhurst DC: In vivo selection for a specific genotype of dihydropteroate synthetase of Plasmodium falciparum by pyrimethamine-sulfadoxine but not chlorproguanil-dapsone treatment. J Infect Dis. 1998, 177: 1429-1433. 10.1086/517831.
Aubouy A, Jafari S, Huart V, Migot-Nabias F, Mayombo J, Durand R, Bakary M, Le Bras J, Deloron P: DHFR and DHPS genotypes of Plasmodium falciparum isolates from Gabon correlate with in vitro activity of pyrimethamine and cycloguanil, but not with sulfadoxine-pyrimethamine treatment efficacy. J Antimicrob Chemother. 2003, 52: 43-49. 10.1093/jac/dkg294.
Hamour S, Melaku Y, Keus K, Wambugu J, Atkin S, Montgomery J, Ford N, Hook C, Checchi F: Malaria in the Nuba Mountains of Sudan: baseline genotypic resistance and efficacy of the artesunate plus sulfadoxine-pyrimethamine and artesunate plus amodiaquine combinations. Trans R Soc Trop Med Hyg. 2005, 99: 548-554. 10.1016/j.trstmh.2004.10.003.
Happi CT, Gbotosho GO, Folarin OA, Akinboye DO, Yusuf BO, Ebong OO, Sowumi A, Kyle DE, Milhous W, Wirth DF, Oduola AMJ: Polymorphisms in Plasmodium falciparum dhfr and dhps genes and age related in vivo sulfadoxine-pyrimethamine resistance in malaria-infected patients from Nigeria. Acta Trop. 2005, 95: 183-193. 10.1016/j.actatropica.2005.06.015.
Ndounga M, Tahar R, Basco LK, Casimiro PN, Malonga DA, Ntoumi F: Therapeutic efficacy of sulfadoxine-pyrimethamine and the prevalence of molecular markers of resistance in under 5-year olds in Brazzaville, Congo. Trop Med Int Health. 2007, 12: 1164-71.
Tinto H, Ouedraogo JB, Zongo I, van Overmeir C, van Marck E, Guiguemde TR, D'Alessandro U: Sulfadoxine-pyrimethamine efficacy and selection of Plasmodium falciparum DHFR mutations in Burkina Faso before its introduction as intermittent preventive treatment for pregnant women. Am J Trop Med Hyg. 2007, 76: 608-13.
Khalil I, Alifrangis M, Ronn AM, Gabar HA, Jelinek T, Satti GMH, Bygbjerg IC: Pyrimethamine/sulfadoxine combination in the treatment of uncomplicated falciparum malaria: relation between dihydropteroate synthase/dihydrofolate reductase genotypes, sulfadoxine plasma levels, and treatment outcome. Am J Trop Med Hyg. 2002, 67: 225-229.
Kun JFJ, Lehman LG, Lell B, Schmidt-Ott R, Kremsner PG: Low-dose treatment with sulfadoxine-pyrimethamine combinations selects for drug-resistant Plasmodium falciparum strains. Antimicrobiol Agents Chemother. 1999, 43: 2205-2208.
Alifrangis M, Enosse S, Khalil IF, Tarimo DS, Lemnge MM, Thompson R, Bygbjerg IC, Ronn AM: Prediction of Plasmodium falciparum resistance to sulfadoxine/pyrimethamine in vivo by mutations in the dihydrofolate reductase and dihydropteroate synthetase genes: a comparative study between sites of differing endemicity. Am J Trop Med Hyg. 2003, 69: 601-606.
A-Elbasit IE, Khalil IF, Elbashir MI, Masuadi EM, Bygbjerg IC, Alifrangis M, Giha HA: High frequency of Plasmodium falciparum CICNI/SGEAA and CVIET haplotypes without association with resistance to sulfadoxine/pyrimethamine and chloroquine combination in the Daraweesh area, in Sudan. Eur J Clin Microbiol Infect Dis. 2008, 27: 725-32. 10.1007/s10096-008-0499-1.
Kublin JG, Dzinjalamala FK, Kamwendo DD, Malkin EM, Cortese JF, Martino LM, Mukadam RA, Rogerson SJ, Lescano AG, Molyneux ME, Winstanley PA, Chimpeni P, Taylor TE, Plowe CV: Molecular markers for failure of sulfadoxine-pyrimethamine and chlorproguanil-dapsone treatment of Plasmodium falciparum malaria. J Infect Dis. 2002, 185: 380-388. 10.1086/338566.
Sendagire H, Kyabayinze D, Swedberg G, Kironde F: Plasmodium falciparum : higher incidence of molecular resistance markers for sulfadoxine than for pyrimethamine in Kasangati, Uganda. Trop Med Int Health. 2005, 10: 537-543. 10.1111/j.1365-3156.2005.01414.x.
Djaman JA, Mazabraud A, Basco L: Sulfadoxine-pyrimethamine susceptibilities and analysis of the dihydrofolate reductase and dihydropteroate synthase of Plasmodium falciparum isolates from Côte d'Ivoire. Ann Trop Med Parasitol. 2007, 101: 103-12. 10.1179/136485907X154584.
Eriksen J, Mwankusye S, Mduma S, Kitua A, Sweberg G, Tomson G, Gustafsson LL, Warsame M: Patterns of resistance and DHFR/DHPS genotypes of Plasmodium falciparum in rural Tanzania prior to the adoption of sulfadoxine-pyrimethamine as first-line treatment. Trans R Soc Trop Med Hyg. 2004, 98: 347-353. 10.1016/j.trstmh.2003.10.010.
Checchi F, Durand R, Balkan S, Vonhm BT, Kollie JZ, Biberson P, Baron E, Le Bras J, Guthmann JP: High Plasmodium falciparum resistance to chloroquine and sulfadoxine pyrimethamine in Liberia: results in vivo and analysis of point mutations. Trans R Soc Trop Med Hyg. 2002, 96: 664-669. 10.1016/S0035-9203(02)90346-9.
Kyabayinze D, Cattamanchi A, Kamya MR, Rosenthal PJ, Dorsey G: Validation of a simplified method for using molecular markers to predict sulfadoxine-pyrimethamine treatment failure in African children with falciparum malaria. Am J Trop Med Hyg. 2003, 69: 247-252.
Broek Van den IVF, Wardt Van der S, Talukder L, Chakma S, Brockman A, Nair S, Anderson TC: Drug resistance in Plasmodium falciparum from the Chittagong Hill Tracts, Bangladesh. Trop Med Int Health. 2004, 6: 680-687. 10.1111/j.1365-3156.2004.01249.x.
Mockenhaupt FP, Bousema JT, Eggelte TA, Schreiber J, Ehrhardt S, Wassilew N, Otchwemah R, Sauerwein RW, Bienzle U: Plasmodium falciparum dhfr but not dhps mutations associated with sulphadoxine-pyrimethamine treatment failure and gametocyte carriage in northern Ghana. Trop Med Int Health. 2005, 10: 901-908. 10.1111/j.1365-3156.2005.01471.x.
Basco LK, Tahar R, Ringwald P: Molecular basis of in vivo resistance to sulfadoxine-pyrimethamine in African adult patients infected with Plasmodium falciparum malaria parasites. Antimicrob Agents Chemother. 1998, 42: 1811-1814.
Tahar R, Basco LK: Molecular epidemiology of malaria in Cameroon. XXVII. Clinical and parasitological response to sulfadoxine-pyrimethamine treatment and Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase alleles in Cameroonian children. Acta Trop. 2007, 103 (2): 81-89. 10.1016/j.actatropica.2007.04.008.
Heidari A, Dittrich S, Jelinek T, Kheirandish A, Banihashemi K, Keshavarz H: Genotypes and in vivo resistance of Plasmodium falciparum isolates in an endemic region of Iran. Parasitol Res. 2007, 100: 589-92. 10.1007/s00436-006-0291-z.
Alker AP, Kazadi WM, Kutelemeni AK, Bloland PB, Tshefu AK, Meshnick SR: dhfr and dhps genotype and sulfadoxine-pyrimethamine treatment failure in children with falciparum malaria in the Democratic Republic of Congo. Trop Med Int Health. 2008, 13: 1384-91. 10.1111/j.1365-3156.2008.02150.x.
Kublin JG, Witzig RS, Shankar AH, Zurita JQ, Gilman RH, Guarda JA, Cortese JF, Plowe CV: Molecular assays for surveillance of antifolate-resistant malaria. Lancet. 1998, 351: 1629-1630. 10.1016/S0140-6736(98)24022-0.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S: Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004, 364: 438-47. 10.1016/S0140-6736(04)16767-6.
Myint HY, Tipmanee P, Nosten F, Pukrittayakamee S, Day NPJ, Looareesuwan S, White NJ: A systematic overview of published antimalarial drug trials. Trans R Soc Trop Med Hyg. 2004, 98: 73-81. 10.1016/S0035-9203(03)00014-2.
Foley M, Tilley L: Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther. 1998, 79: 55-87. 10.1016/S0163-7258(98)00012-6.
Hyde JE: Drug-resistant malaria – an insight. FEBS J. 2007, 274: 4688-4698. 10.1111/j.1742-4658.2007.05999.x.
Ekland EH, Fidock DA: Advances in understanding the genetic basis of antimalarial drug resistance. Curr Opin Microbiol. 2007, 10: 363-370. 10.1016/j.mib.2007.07.007.
Watkins WM, Mberu EK, Winstanley PA, Plowe CV: The efficacy of antifolate antimalarial combinations in Africa: a predictive model based on pharmacodynamic and pharmacokinetic analyses. Parasitol Today. 1997, 13: 459-464. 10.1016/S0169-4758(97)01124-1.
Hastings IM, Watkins WM: Intensity of malaria transmission and the evolution of drug resistance. Acta Trop. 2005, 94: 218-229.
Barnes KI, Lindegardh N, Ogundahunsi O, Olliaro P, Plowe CV, Randrianarivelojosia M, Gbotosho GO, Watkins WM, Sibley CH, White NJ: World Antimalarial Resistance Network (WARN) IV: clinical pharmacology. Malar J. 2007, 6: 122-10.1186/1475-2875-6-122.
Mu J, Awadalla P, Duan J, McGee KM, Joy DA, McVean GA, Si XZ: Recombination hotspots and population structure in Plasmodium falciparum. Plos Biology. 2005, e335-10.1371/journal.pbio.0030335.
Stepniewska K, White NJ: Some considerations in the design and interpretation of antimalarial drug trials in uncomplicated falciparum malaria. Malar J. 2006, 5: 127-10.1186/1475-2875-5-127.
World Health Organization: Methods and techniques for clinical trials on anti Malarial drug efficacy: genotyping to identify parasite populations. Geneva. 2007
Sibley CH, Barnes KI, Watkins WM, Plowe CV: A network to monitor antimalarial drug resistance: a plan for moving forward. Trends Parasitol. 2008, 24: 43-48. 10.1016/j.pt.2007.09.008.
Conway DJ: Molecular epidemiology of malaria. Clin Microbiol Rev. 2007, 20: 188-204. 10.1128/CMR.00021-06.
Acknowledgements
We thank Christiane Liou for her assistance in retrieval of the articles surveyed in this study, and Annick Brillant, Elisabeth Inapogui and Françoise Durand-Peyre for editorial assistance. This analysis was supported by a grant of the UNICEF/UNDP/WB/WHO Special Programme for Research and Training in Tropical Diseases (TDR). Financial support was obtained from UNICEF/UNDP/WB/WHO Special Programme for Research and Training in Tropical Diseases (TDR) to acquire the meta-analysis software and to obtain all the published papers.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no conflict of interest. PO and PR are staff members of the World Health Organization. The authors alone are responsible for the views expressed in this publication and they do not necessarily represent the decisions, policy or views of the World Health Organization.
Authors' contributions
SP wrote the protocol, selected the studies, performed the meta-analysis, wrote the first draft, and edited the manuscript. FdM reviewed abstracts. ALB reviewed abstracts and full text and controlled the statistics. All authors contributed to the interpretation of the analysis, read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Picot, S., Olliaro, P., de Monbrison, F. et al. A systematic review and meta-analysis of evidence for correlation between molecular markers of parasite resistance and treatment outcome in falciparum malaria. Malar J 8, 89 (2009). https://doi.org/10.1186/1475-2875-8-89
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-8-89