
Abstract
Background
Parasitic protozoans, helminths, alter the gut microbiota in mammals, yet little is known about the influence of intestinal cestodes on gut microbiota in fish. In the present study, the composition and diversity of the hindgut microbiota were determined in the intestine of common carp (Cyprinus carpio) infected with two tapeworm species, Khawia japonensis and Atractolytocestus tenuicollis.
Results
The intestine contained a core microbiota composed of Proteobacteria, Fusobacteria and Tenericutes. Infection with the two cestode species had no significant effect on the microbial diversity and richness, but it altered the microbial composition at the genus level. PCoA analysis indicated that microbial communities in the infected and uninfected common carp could not be distinguished from each other. However, a Mantel test indicated that the abundance of K. japonensis was significantly correlated with the microbial composition (P = 0.015), while the abundance of A. tenuicollis was not (P = 0.954). According to Pearsonʼs correlation analysis, the abundance of K. japonensis exhibited an extremely significant (P < 0.001) positive correlation with the following gut microbiota taxa: Epulopiscium, U114, Bacteroides, Clostridium and Peptostreptococcaceae (0.8< r < 0.9); and a significant (P < 0.05) correlation with Enterobacteriaceae, Micrococcaceae, Rummeliibacillus, Lysinibacillus boronitolerans, Veillonellaceae, Oxalobacteraceae, Aeromonadaceae (negative), Marinibacillus and Chitinilyticum (0.4< r < 0.7).
Conclusions
These results suggest that the composition of gut microbiota was somewhat affected by the K. japonensis infection. Additionally, increased ratios of pathogenic bacteria (Lawsonia and Plesiomonas) were also associated with the K. japonensis infection, which may therefore increase the likelihood of disease.
Similar content being viewed by others

Background
The gastrointestinal (GI) tract of vertebrates is inhabited by a vast array of organisms, including bacteria, protozoan and helminth parasites [1]. Gut microbiota co-evolve with their hosts and have important functional roles in metabolism, nutrition and immunity [2]. Sharing the same niche with the intestinal microbiota [2], the enteric helminths inevitably interact with the gut microbiota [1], but interactions between gut microbiota and parasites are usually overlooked.
Increasing evidence indicates that the presence of parasitic helminths (including nematodes, cestodes and digeneans) alters the composition and diversity of GI microbiota in mammals [1, 3, 4]. In laboratory experiments, infection with nematodes or cestodes can induce changes in the microbial composition in the intestines of humans and other mammals, sometimes even resulting in a reduced bacterial diversity [5,6,7,8]. However, more often it appears to have a negligible effect on the bacterial diversity [9,10,11,12,13,14,15,16,17]. In field experiments, however, natural infection by nematodes hardly produces any effects on the microbial diversity in mammals [18,19,20], except for one study, which found high microbial diversity in the presence of multiple helminths in wild rodents [21]. In summary, regardless of whether the diversity of microbiota is altered, its composition can be changed by experimental and natural helminth infection both in domestic and wild mammals.
In contrast to mammalian systems, we know very little about interactions between the GI microbiota and helminths in cold-blooded animals such as fish. Although the parasite load of the myxozoan Tetracapsuloides bryosalmonae in the kidney of salmonid fish exhibits a significant positive relationship with the richness of the GI microbiome [22], little else is known about interactions between microbiota and helminths in the digestive tract of fish.
The common carp, Cyprinus carpio, is widely distributed in rivers, lakes, reservoirs and ponds in East Asia (as well as North America, Europe and Africa). The tapeworms Khawia sinensis, K. japonensis and Atractolytocestus sp. are commonly found in the intestine of the common carp [23,24,25,26]. Tapeworms can rob the host of nutrients through the tegument and thereby restrict the host’s growth. However, neither pathological changes nor poor physiological state can be observed in common carp, even when heavily infected by Khawia sp. [24, 27]. It has been hypothesized that GI microbiota associated with the presence of tapeworms may help maintain the homeostasis of the digestive system [28]. Therefore, in this study, we aim to investigate whether the presence of tapeworms influences the composition and diversity of microbiota in the intestine of wild common carp.
Methods
Collection of fish and tapeworms
Live common carp (n = 23) were collected in April 2017 from Liangzi lake (30° 04′–30° 20′ N, 114° 31′–114° 42′ E), Hubei Province, China. Their intestinal tracts were aseptically removed from the abdominal cavity, and then divided into three fragments (foregut, midgut and hindgut). The foregut and midgut were used for the collection of tapeworms, and the hindgut was used for microbiota analysis.
The collected tapeworms were gently rinsed in saline, prefixed in 70% hot alcohol, and then taxonomically identified according to the morphological features of the scolex under a stereomicroscope [26]. Owing to the similar scolex morphology of Atractolytocestus spp., A. tenuicollis was identified using the number of testes [23]. Abundance and maturity of tapeworms were also recorded.
Sample preparation and total bacterial DNA extraction
The intestinal content of each fish was gently squeezed into a sterile tube and thoroughly mixed. The samples were frozen immediately in a freezer and stored at − 80 °C. The total bacterial DNA was extracted from 200 mg of the hindgut content using QIAamp® DNA stool mini kit (Qiagen, New York, USA) according to the manufacturer’s instructions. The purity and concentration of genomic DNA were determined with a spectrophotometer (Nanodrop 8000; Thermo Fisher Scientific, Wilmington, USA). DNA was stored at − 20 °C for later use.
16S rDNA amplification and Illumina high-throughput sequencing
The universal primer pair 515F (5′-GTG YCA GCM GCC GCG GTA-3′), with a unique 12-nt barcode at the 5′-end, and 909R (5′-CCC GYC AAT TCM TTT RAG T-3′) [29], were used to amplify the V4-V5 hypervariable region of the bacterial 16S rDNA gene. The PCR reaction system (25 μl) contained 50 ng of DNA template, 1 μM of each primer, and 12.5 μl of 2× Go Taq Green Master Mix polymerase (Promega, Madison, USA). The PCR procedure was as follows: 5 min at 94 °C as an initial step, followed by 23 cycles of 30 s at 94 °C, 30 s at 55 °C and 90 s at 72 °C, with a final step of 10 min at 72 °C. Replicated PCR products of the same sample were assembled into a PCR tube and subjected to electrophoresis using a 2% agarose gel. The correct band (about 400 bp) was recovered by AidQuick Gel Extraction Kit (Aidlad Biotech, Beijing, China). A spectrophotometer (Nanodrop 8000) was used to determine the DNA concentration and purity. All samples were pooled, with an equal molar amount from each sample, and sent to Novogene Bioinformatics Technology Co. for PCR-free library construction. Sequencing was performed using the PE250 strategy on an Illumina Hiseq 2500 platform. The sequences are available in the NCBI SRA database under the accession number SRP158810.
Sequence data processing
The raw sequenced data were processed using QIIME Pipeline-Version 1.8.0 (http://qiime.org/) [30]. Overlapping paired-end reads were merged using FLASH-1.2.8 software [31]. Only the merged sequences with high-quality reads (length > 250 bp, without ambiguous bases BN, and average base quality score > 30) were used for further analysis. All sequences were trimmed and assigned to each sample on the basis of their barcodes (barcode mismatches = 0). Chimeras were removed using the UCHIME algorithm [32]. Non-chimera sequences were subsampled to the same sequence depth (14,581 reads per sample) using daisychopper.pl [33]. This subset of sequences was clustered into OTUs at a 97% identity threshold using CD-HIT [34]. Singletons were filtered out. Operational taxonomic unit (OTU) identities were assigned in Greengenes database (release 13.8) [35] using UCLUST [36]. Sequences classified as unassigned and C_Chloroplast were removed.
The following alpha diversity indices were calculated: Chao1, ACE, Simpson and Shannon index. Linear discriminant analysis coupled with effect size (Lefse) (http://huttenhower.sph.harvard.edu/galaxy) was used to study the significance of species differences at the genus level [37]. Principal coordinates analysis plots (PCoA) were used to visualize similarities between groups with weighted_unifrac distance [38]. Permutational multivariate analyses of variance (PERMANOVA) were performed to test the significance of differences between groups applying the Vegan package in R. The Pearsonʼs correlation coefficient was used to investigate the degree of linear correlation between the abundance of tapeworms and the abundance of bacteria using PAST 2.16 [39]. Association between the abundance of tapeworms and intestinal microbial community was analysed by a Mantel test using PASSaGE 2 [40]. A Venn diagram of shared and unique OTUs was used to describe the similarities and differences between infected and uninfected common carp (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Statistical analyses
Statistical analyses were performed using SPSS20 (IBM Corporation, Armonk, NY, USA). Pairwise comparisons between infected and uninfected groups were assessed using Student’s t-test at the 0.05 significance threshold.
Results
Tapeworm infection prevalence and mean intensity
Two species of the Caryophyllidea, Khawia japonensis and Atractolytocestus tenuicollis (Additional file 1: Figure S1; Additional file 2: Figure S2), were found in the intestine of common carp (Additional file 3: Table S1). The prevalence and mean abundance (± standard deviation, SD) were respectively 60.9% (14/23) and 3.9 ± 6.9 (0–31) for K. japonensis, and 73.9% (17/23) and 7.3 ± 9.5 (0–31) for A. tenuicollis (Table 1).
Microbial composition in infected and uninfected common carp
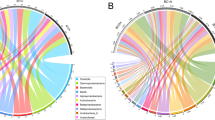
On the basis of a 97% similarity threshold, 664 OTUs were identified at the phylum level, and 49.6% (329) of OTUs were assigned to the genus level (Additional file 4: Table S2). At the phylum level, differences in the dominant microbiota between uninfected and infected groups were not significant: Proteobacteria (58.2 ± 35.3% vs 67.7 ± 29.1%; t(23) = − 0.574, P = 0.572), Fusobacteria (19.2 ± 28.3% vs 19.2 ± 21.9%; t(23) = -0.001, P = 0.999), Tenericutes (14.6 ± 23.5% vs 6.7 ± 18.9%; t(23) = 0.733, P = 0.472) (Fig. 1a). At the genus/family level, Aeromonadaceae, Cetobacterium, Enterobacteriaceae and Mycoplasma were the relatively abundant taxa (higher than 1%) both in infected and uninfected common carp. There were no significant differences in the relative abundance of the four genera/families between uninfected and infected groups (Fig. 1b; Aeromonadaceae: t(23) = − 0.075, P = 0.941; Cetobacterium: t(23) = 0.245, P = 0.809; Enterobacteriaceae: t(23) = − 0.463, P = 0.648; Mycoplasma: t(23) = − 0.507, P = 0.617). Compared with the uninfected group, the relative abundance of five genera, Salinivibrio, U114 (Fusobacteria), Clostridium, Lawsonia and Bacteroides, was higher in infected common carp (Fig. 1b). The Lefse analysis found that a total of 22 taxa displayed significant differences in their abundance between uninfected and infected common carp at a stringent cutoff value (absolute LDA score log10 ≥ 2.0) (Fig. 2a). Among the identified OTUs, 409 were shared by the uninfected group samples (61.6% of sequences), and 601 were shared by the infected group samples (90.5%). Among these, 346 OTUs were shared by the two groups (Fig. 3a).

Microbial composition in the intestine of common carp. a the dominant phyla in uninfected and infected groups. b the dominant genera/families in uninfected and infected groups. c the dominant phyla in KJ(−) and KJ(+) groups. d the dominant genera/families in KJ(−) and KJ(+) groups. “Others” includes the sum of different taxa with an abundance less than 1% in the samples. Abbreviations: KJ(+), infected by Khawia japonensis; KJ(−), uninfected by K. japonensis

Bacterial taxa with significant differences (LDA score > 2.0) in the relative abundance identified by Lefse in uninfected and infected groups (a), and KJ(−) and KJ(+) groups (b)

Numbers and sequence proportions of Shared OTUs between uninfected and infected groups (a), and KJ(−) and KJ(+) groups (b)
Alpha diversity of microbiota in infected and uninfected common carp
There were no significant differences in richness (Chao1 and ACE) and diversity (Shannon, Simpson) between the infected and uninfected groups (Chao1: t(23) = − 0.021, P = 0.984; ACE: t(23) = 0.447, P = 0.659; Shannon: t(23) = 0.237, P = 0.815; Simpson: t(23) = − 0.265, P = 0.793) (Fig. 4).

Alpha diversity of microbial communities in the intestines of uninfected and infected common carp
Beta diversity of microbiota in infected and uninfected common carp
PERMANOVA indicated that there was no significant difference in the composition of microbial communities between the infected and uninfected groups (F(1,21) = 0.5262, P = 0.821). PCoA analysis also showed that bacterial communities of the two groups could not be clearly distinguished (Fig. 5a). However, the Mantel test showed that the abundance of K. japonensis had a significant effect on the bacterial composition (P = 0.015), while the abundance of A. tenuicollis had hardly any influence on the composition of gut microbiota (P = 0.954). Therefore, composition and diversity of microbiota were further analyzed between the groups infected KJ(+) and uninfected KJ(−) with K. japonensis (Table 1).

Principal coordinates analysis (PCoA) of bacterial community structures. a Triangles and dots represent the samples in infected and uninfected groups, respectively. b Dots and rhombuses represent the samples in KJ(−) and KJ(+) groups, respectively
Microbiota composition in KJ(+) and KJ(−) groups
Ratios of dominant microbial taxa at the phylum level differed between KJ(−) and KJ(+) groups, but not significantly: Proteobacteria (74.2 ± 28.6% vs 60.8 ± 30.2%; t(23) = 1.059, P = 0.302), Fusobacteria (13.9 ± 20.4% vs 22.7 ± 23.7%; t(23) = − 0.917, P = 0.370) and Tenericutes (6.6 ± 16.3% vs 9.0 ± 21.7%; t(23) = − 0.284, P = 0.779) (Fig. 1c). Similarly, there were also no significant differences between KJ(−) and KJ(+) groups at the genus/family level (only relatively abundant taxa, higher than 1%, were tested): Aeromonadaceae (t(23) = 0.651, P = 0.522), Enterobacteriaceae (t(23) = − 0.480, P = 0.636), Cetobacterium (t(23) = 0.519, P = 0.609), U114 (t(23) = − 0.747, P = 0.464) and Salinivibrio (t(23) = 0.196, P = 0.864) (Fig. 1d). The relative abundance of the genera Mycoplasma (t(23) = 1.356, P = 0.198), Clostridium (t(23) = 2.228, P = 0.042), Lawsonia (t(23) = 1.196, P = 0.252), Bacteroides (t(23) = 0.723, P = 0.477) and Plesiomonas (t(23) = 1.288, P = 0.212) was higher in the KJ(+) group than in KJ(−) group (Fig. 1d). The Lefse analysis indicated that a total of 21 taxa displayed a significant difference in their abundance between KJ(−) and KJ(+) groups at a stringent cutoff value (absolute LDA score log10 ≥ 2.0) (Fig. 2b). OTUs were determined to investigate shared microbial communities: 484 OTUs were shared by the samples from the KJ(−) group (80.4% of sequences), and 551 OTUs were shared by samples from the KJ(+) group (91.5%). Among these, 433 OTUs were shared between the two groups (Fig. 3b).
Alpha diversity of microbiota in KJ(−) and KJ(+) groups
There were also no significant differences in richness (Chao1 and ACE) and diversity (Shannon, Simpson) between KJ(−) and KJ(+) groups (Chao1: t(23) = 1.635, P = 0.117; ACE: t(23) = 1.728, P = 0.099; Shannon: t(23) = − 0.539, P = 0.596; Simpson: t(23) = − 1.051, P = 0.305) (Fig. 6).

Alpha diversity of microbial communities in KJ(−) and KJ(+) groups
Beta diversity of microbiota in KJ(−) and KJ(+) groups
PERMANOVA indicated that there was also no significant difference in the composition of microbial communities between KJ(−) and KJ(+) groups (F(1,21) = 0.433, P = 0.883). PCoA analysis also showed that bacterial communities of KJ(−) and KJ(+) groups could not be clearly distinguished (Fig. 5b). Pearson correlation coefficient analysis indicated that the abundance of K. japonensis had an extremely significant (P < 0.001) positive correlation with the relative abundance of Epulopiscium, U114, Bacteroides, Clostridium, Peptostreptococcaceae (0.8< r < 0.9); and a significant correlation (P < 0.05) with Enterobacteriaceae, Micrococcaceae, Rummeliibacillus, Lysinibacillus boronitolerans, Veillonellaceae, Oxalobacteraceae, Aeromonadaceae (negative), Marinibacillus and Chitinilyticum (0.4< r < 0.7) (Table 2).
Discussion
Proteobacteria, Fusobacteria and Tenericutes were the dominant microbiota in the hindgut of common carp in our study, which is in disagreement with a previous study of intestinal microbiota of common carp [41]. This may be attributable to differences in diet [42], as the common carp in the previous study was fed a commercial feed, whereas we used a diet composed of crustaceans, snails and detritus in our study.
Increased alpha diversity of the intestinal microbiota is generally associated with a “healthy” gut homeostasis [1]. Although several studies have reported a marked decrease in alpha diversity in animals infected (the acute phase of infection) by parasitic helminths [5,6,7], in most studies conducted to date in a range of animal-helminth systems, the alpha diversity of the gut microbiota remained unchanged following a parasitic infection [9, 11, 18, 21, 43, 44]. In line with this, we did not observe a significant change in alpha diversity in tapeworm-infected common carp in our study. This indicates that parasitic infections are likely to affect only a small fraction of the intestinal microbiota [9, 11]. In addition, it may also be related to the small number of uninfected samples in our trial.
Although two tapeworm species (parasitising the common carp) were discovered in our study, only the abundance of K. japonensis had a significant effect on the composition of microbiota, while the abundance of A. tenuicollis had almost no influence on the composition of microbiota. The relative body surface area of K. japonensis is about two to three times larger than that of A. tenuicollis [23, 45], so the former species can host a much larger number of bacteria. In addition, type 2 immune responses induced by helminth infections can alter the host’s metabolic functions, as well as modify bacterial microbiota populations, but the magnitude of these changes varies among helminth species [3, 21]. We hypothesise that these two factors may explain the much more pronounced effect of K. japonensis on the composition of gut microbiota (compared to A. tenuicollis).
Although none of the observed differences in the composition of intestinal microbiota between infected and uninfected common carp were statistically significant, it may be worth noting that tapeworm infection was associated with an increased relative abundance of two pathogenic bacterial taxa: Lawsonia and Plesiomonas. Lawsonia intracellularis (family Desulfovibrionaceae), the only member of the genus, is an obligate intracellular parasite of intestinal cells, which causes proliferative enteropathy in pigs and some other mammals [46, 47]. Plesiomonas shigelloides, also the sole member of the genus, is ubiquitous in surface water and soil, and can cause gastroenteritis and extraintestinal infections [48, 49]. Thus the increased relative abundance of Lawsonia and Plesiomonas may contribute to infection by the caryophyllidean tapeworms.
Khawia japonensis had a significant positive correlation with U114, Epulopiscium, Bacteroides, Clostridium, Peptostreptococcaceae, Rummeliibacillus, Lysinibacillus boronitolerans, Marinibacillus and Chitinilyticum in our study. U114 might have a role in the regulation of bile acids metabolism [50]; Epulopiscium may be able to break down carbohydrates and complex hemicellulose [51]; Bacteroides has a capacity of breaking down polysaccharides [52]; Clostridium ferments polysaccharides and proteins to produce alcohols and short-chain fatty acids [53]; Peptostreptococcaceae can utilize proteinaceous substrates and carbohydrates [54]; Rummeliibacillus can hydrolyze gelatin and produce acids [55]; Lysinibacillus boronitolerans produces nitrilase, converses iminodiacetonitrile (IDAN) to iminodiacetic acid (IDA) [56], and degrades ioxynil octanoate herbicide [57]; Marinibacillus utilizes cellobiose, trehalose and xylose [58]; and finally, Chitinilyticum degrades chitin [59]. All these microbial taxa are very important for the digestion of polysaccharides and proteins. In addition, due to the lack of guts, cestodes absorb nutrients through the tegument [60]. There is a large amount of microtriche on the surface of teguments of cestodes, and bacteria are abundant around the microtriche [61,62,63,64]. These bacteria can produce enzymes that hydrolyze carbohydrates, disaccharides and proteins [65, 66]. Therefore, the microbiota related to the tapeworm infection is likely to help the cestodes absorb nutrients through the tegument. Whether the microbiota is associated strictly with the tegument of cestodes needs to be further investigated.
Some opportunistic pathogens, such as Enterobacteriaceae, Micrococcaceae, Veillonellaceae, Oxalobacteraceae and Aeromonadaceae, were also significantly correlated with the infection with K. japonensis. Cestodes embed themselves into the intestinal wall via their scolex, leading to blistering, bleeding, and inflammation of the intestinal mucosa, which can be followed by secondary infections by other pathogens [30]. However, previous studies have shown, and our results corroborated, that common carp infected by K. japonensis does not exhibit observable pathological changes [24]. Species of Clostridia (Clostridium especially) are known to tighten the epithelial barrier and decrease the propensity for allergy [67, 68]. The relative abundance of Clostridium was strikingly increased in our study. Therefore, we hypothesize that the increased abundance of Clostridium probably reduced the damage to the intestinal epithelium caused by the tapeworms, which may also be the reason why no obvious pathological changes were observed. However, the ability of Clostridium to reduce allergic reactions in the gut of common carp would first have to be proven by further experiments.
Conclusions
Tapeworm colonization in common carp did not affect the microbial diversity, but it altered the microbial composition at the genus level. Compared with A. tenuicollis, K. japonensis had a greater impact on the composition of intestinal microbiota. Some of the microbial taxa associated with K. japonensis may contribute to nutrient digestion and absorption by the tapeworm. Khawia japonensis infection was associated with increased ratios of some pathogenic bacteria (Lawsonia and Plesiomonas), but it also increased the ratio of Clostridium, which may have reduced the allergic reaction and pathological changes caused by pathogenic bacteria.
Availability of data and materials
The data supporting the findings of this study are included within the article. The sequences were deposited in the NCBI SRA database under the accession number SRP158810.
Abbreviations
- GI:
-
gastrointestinal
- OUT:
-
operational taxonomic unit
- Lefse:
-
linear discriminant analysis coupled with effect size
- PCoA:
-
principal coordinates analysis plots
- PERMANOVA:
-
permutational multivariate analyses of variance
- IDAN:
-
iminodiacetonitrile
- IDA:
-
iminodiacetic acid
References
Peachey LE, Jenkins TP, Cantacessi C. This gut ain’t big enough for both of us. Or is it? Helminth-microbiota interactions in veterinary species. Trends Parasitol. 2017;33:619–32.
Glendinning L, Nausch N, Free A, Taylor DW, Mutapi F. The microbiota and helminths: sharing the same niche in the human host. Parasitology. 2014;141:1255–71.
Brosschot TP, Reynolds LA. The impact of a helminth-modified microbiome on host immunity. Mucosal Immunol. 2018;11:1039–46.
Lee SC, Tang MS, Lim YA, Choy SH, Kurtz ZD, Cox LM, et al. Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis. 2014;8:e2880.
Cattadori IM, Sebastian A, Hao H, Katani R, Albert I, Eilertson KE, et al. Impact of helminth infections and nutritional constraints on the small intestine microbiota. PLoS One. 2016;11:e0159770.
Holm JB, Sorobetea D, Kiilerich P, Ramayo-Caldas Y, Estelle J, Ma T, et al. Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of Lactobacilli. PLoS One. 2015;10:e0125495.
Houlden A, Hayes KS, Bancroft AJ, Worthington JJ, Wang P, Grencis RK, et al. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS One. 2015;10:e0125945.
Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, et al. Helminth infection promotes colonization resistance via type 2 immunity. Science. 2016;352:608–12.
Li RW, Li W, Sun J, Yu P, Baldwin RL, Urban JF. The effect of helminth infection on the microbial composition and structure of the caprine abomasal microbiome. Sci Rep. 2016;6:20606.
Li RW, Wu S, Li W, Navarro K, Couch RD, Hill D, et al. Alterations in the porcine colon microbiota induced by the gastrointestinal nematode Trichuris suis. Infect Immun. 2012;80:2150–7.
McKenney EA, Williamson L, Yoder AD, Rawls JF, Bilbo SD, Parker W. Alteration of the rat cecal microbiome during colonization with the helminth Hymenolepis diminuta. Gut Microbes. 2015;6:182–93.
Rausch S, Held J, Fischer A, Heimesaat MM, Kuhl AA, Bereswill S, et al. Small intestinal nematode infection of mice is associated with increased enterobacterial loads alongside the intestinal tract. PLoS One. 2013;8:e74026.
Reynolds LA, Redpath SA, Yurist-Doutsch S, Gill N, Brown EM, van der Heijden J, et al. Enteric helminths promote Salmonella coinfection by altering the intestinal metabolome. J Infect Dis. 2017;215:1245–54.
Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, et al. Commensal-pathogen interactions in the intestinal tract: Lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes. 2014;5:522–32.
Su C, Su L, Li Y, Long SR, Chang J, Zhang W, Walker WA, et al. Helminth-induced alterations of the gut microbiota exacerbate bacterial colitis. Mucosal Immunol. 2018;11:144–57.
Walk ST, Blum AM, Ewing SA, Weinstock JV, Young VB. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm Bowel Dis. 2010;16:1841–9.
Wu S, Li RW, Li W, Beshah E, Dawson HD, Urban JF, et al. Worm burden-dependent disruption of the porcine colon microbiota by Trichuris suis infection. PLoS One. 2012;7:e35470.
Duarte AM, Jenkins TP, Latrofa MS, Giannelli A, Papadopoulos E, de Carvalho LM, et al. Helminth infections and gut microbiota—a feline perspective. Parasit Vectors. 2016;9:625.
Jenkins TP, Rathnayaka Y, Perera PK, Peachey LE, Nolan MJ, Krause L, et al. Infections by human gastrointestinal helminths are associated with changes in faecal microbiota diversity and composition. PLoS One. 2017;12:e0184719.
Newbold LK, Burthe SJ, Oliver AE, Gweon HS, Barnes CJ, Daunt F, et al. Helminth burden and ecological factors associated with alterations in wild host gastrointestinal microbiota. ISME J. 2017;11:663–75.
Kreisinger J, Bastien G, Hauffe HC, Marchesi J, Perkins SE. Interactions between multiple helminths and the gut microbiota in wild rodents. Phil Trans R Soc B. 2015;370:1675.
Vasemägi A, Visse M, Kisand V, Blader IJ. Effect of environmental factors and an emerging parasitic disease on gut microbiome of wild salmonid fish. mSphere. 2017;2:e00418–517.
Králová-Hromadová I, Štefka J, Bazsalovicsová E, Bokorová S, Oros M. The tapeworm Atractolytocestus tenuicollis (Cestoda: Caryophyllidea)—a sister species or ancestor of an invasive A. huronensis? Parasitol Res. 2013;112:3379–88.
Oros M, Barcak D, Bazsalovicsova E, Hanzelova V. Asian fish tapeworm, Khawia japonensis (Yamaguti, 1934), has expanded its European invasive range. Parasitol Res. 2015;114:2035–9.
Oros M, Hanzelová V, Scholz T. Tapeworm Khawia sinensis: review of the introduction and subsequent decline of a pathogen of carp, Cyprinus carpio. Vet Parasitol. 2009;164:217–22.
Scholz T, Binh TT, Dezfuli BS. Khawia japonensis (Cestoda: Caryophyllidea): another invasive parasite of carp, Cyprinus carpio L., imported to Europe. J Fish Dis. 2011;34:943–9.
Brunanska M, Mackiewicz JS, Mlocicki D, Swiderski Z, Nebesarova J. Early intrauterine embryonic development in Khawia sinensis Hsu, 1935 (Cestoda, Caryophyllidea, Lytocestidae), an invasive tapeworm of carp (Cyprinus carpio): an ultrastructural study. Parasitol Res. 2012;110:1009–17.
Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–73.
Tamaki H, Wright CL, Li X, Lin Q, Hwang C, Wang S, et al. Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next-generation titanium sequencing platform. PLoS One. 2011;6:e25263.
Jasrotia D, Kaur H. Molecular analysis of a novel species, Gangesia punjabensis (Family: Proteocephalidae, Subfamily: Gangesiinae) infecting an Indian freshwater cat fish, Wallago attu evidencing species complex. J Parasit Dis. 2017;41:888–98.
Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–63.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194.
Gibert JA, Field D, Swift P, Newbold L, Oliver A, Smyth T, et al. The seasonal structure of microbial communities in the Western English Channel. Environ Microbiol. 2009;11:3132–9.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658.
Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a Chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb. 2006;72:5069–72.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Navasmolina JA, Peraltasánchez JM, González A, Mcmurdie PJ, Vázquezbaeza Y, Xu Z, et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013;531:371–444.
Hammer O, Harper DTA, Ryan PD. PAST (Paleontological Statistics) version 2.16. Software package for education and data analysis. Paleontol Electrón. 2012;4:1–9.
Rosenberg MS, Anderson CD. PASSaGE: Pattern Analysis, Spatial Statistics and Geographic Exegesis Version 2. Methods Ecol Evol. 2011;2:229–32.
van Kessel MA, Dutilh BE, Neveling K, Kwint MP, Veltman JA, Flik G, et al. Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express. 2011;1:41.
Hao YT, Wu SG, Ivan J, Zou H, Li WX, Wang GT. Impacts of diet on hindgut microbiota and short-chain fatty acids in grass carp (Ctenopharyngodon idellus). Aquac Res. 2017;48:5595–605.
Fricke WF, Song Y, Wang AJ, Smith A, Grinchuk V, Pei C, et al. Type 2 immunity-dependent reduction of segmented filamentous bacteria in mice infected with the helminthic parasite Nippostrongylus brasiliensis. Microbiome. 2015;3:40.
Li RW, Wu S, Li W, Huang Y, Gasbarre LC. Metagenome plasticity of the bovine abomasal microbiota in immune animals in response to Ostertagia ostertagi infection. PLoS One. 2011;6:e24417.
Scholz T, Brabec J, Král’ová-Hromadová I, Oros M, Bazsalovicsova E, Ermolenko A, et al. Revision of Khawia spp. (Cestoda: Caryophyllidea), parasites of cyprinid fish, including a key to their identification and molecular phylogeny. Folia Parasit. 2011;58:197.
Mcorist S, Gebhart CJ, Boid R, Barns SM. Characterization of Lawsonia intracellularis gen. nov., sp. nov., the obligately intracellular bacterium of porcine proliferative enteropathy. Int J Syst Evol Microbiol. 1995;45:820–5.
McOrist S, Gebhart CJ. Genus III. Lawsonia. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s manual of systematic bacteriology. The proteobacteria. Part C, the alpha-, beta-, delta-, and epsilon proteobacteria, vol. 2. 2nd ed. New York: Springer; 2005. p. 940–3.
Brenden RA, Miller MA, Janda JM. Clinical disease spectrum and pathogenic factors associated with Plesiomonas shigelloides infections in humans. Clin Infect Dis. 1988;10:303–16.
Escobar JC, Bhavnani D, Trueba G, Ponce K, Cevallos W, Eisenberg J. Plesiomonas shigelloides infection, Ecuador, 2004–2008. Emerg Infect Dis. 2012;18:322–4.
Zhang J, Xiong F, Wang GT, Li WX, Li M, Zou H, et al. The influence of diet on the grass carp intestinal microbiota and bile acids. Aquac Res. 2017;48:4934–44.
Miyake S, Ngugi DK, Stingl U. Phylogenetic diversity, distribution and cophylogeny of giant bacteria (Epulopiscium) with their surgeonifish hosts in the Red Sea. Front Microbiol. 2015;7:285.
Martens EC, Kelly AG, Tauzin AS, Brumer H. The devil lies in the details: how variations in polysaccharide fine-structure impact the physiology and evolution of gut microbes. J Mol Biol. 2014;426:3851–65.
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–88.
The Slobodkin A, Peptostreptococcaceae Family. In: Rosenberg E, Delong EF, Lory S, Stackebrabdt E, Thompson F, editors. The Prokaryotes. Berlin: Springer; 2014. p. 291–302.
Shivaji S, Srinivas TNR, Reddy GSN. The family Planococcaceae. In: Rosenberg E, Delong EF, Lory S, Stackebrabdt E, Thompson F, editors. The prokaryotes. Berlin: Springer; 2014. p. 303–51.
Muluka H, Sheelu G, Nageshwar YV. Bioconversion of Iminodiacetonitrile to Iminodiacetic acid with whole cells of Lysinibacillus boronitolerans MTCC 107614 (IICT-akl252). Bioprocess Biosyst Eng. 2016;39:413–20.
Oliveira KO, Silva ARM, da Silva BF, Milagre HMS, Milagre CDF. Insights into the microbial degradation pathways of the ioxynil octanoate herbicide. In: Hou CT, Shaw JF, editors. Biocatalysis and agricultural biotechnology. Boca Raton: CRC Press; 2018. p. 258–64.
Ruger HJ. Differentiation of Bacillus globisporus, Bacillus marinus comb. nov., Bacillus aminovorans, and Bacillus insolitus. Int J Syst Evol Micr. 1983;33:157–61.
Chang SC, Chen WM, Wang JT, Wu MC. Chitinilyticum aquatile gen. nov., sp. nov., a chitinolytic bacterium isolated from a freshwater pond used for Pacific white shrimp culture. Int J Syst Evol Microbiol. 2007;57:2854–60.
Poddubnaya LG, Scholz T, Kuchta R, Levron C, Brunanska M. Ultrastructure of the proglottid tegument (neodermis) of the cestode Echinophallus wageneri (Pseudophyllidea: Echinophallidae), a parasite of the bathypelagic fish Centrolophus niger. Parasitol Res. 2007;101:373–83.
Korneva J. Nanobacteria associated with mucous intestines of freshwater fishes and tegument of their parasites (Cestoda). Acta Parasitol. 2008;53:312–4.
Plotnikov AO, Korneva ZV. Morphological and ultrastructural characteristics of symbiotic bacteria colonizing the surface of the helminth Triaenophorus nodulosus and the intestine of pike Esox lucius. Inland Water Biol. 2008;1:25–31.
Poddubnaia LG. Electron microscope investigation of bacteria associated with the tegument of the tapeworm species Eubothrium rugosum, a parasite of the intestine of burbot. Parazitologiya. 2005;39:293.
Zhv K, Plotnikov AO. The symbiotic microflora associated with the tegument of proteocephalidean cestodes and the intestines of their fish hosts. Parazitologiya. 2006;40:313.
Izvekova GI. Activity of carbohydrases of symbiotic microflora and their role in processes of digestion of fish and their parasitizing cestodes (on the example of pike and Triaenophorus nodulosus). J Evol Biochem Phys. 2005;41:406–14.
Izvekova GI. Hydrolytic activity of enzymes produced by symbiotic microflora and its role in digestion processes of bream and its intestinal parasite Caryophyllaeus laticeps (Cestoda, Caryophyllidea). Biol Bull. 2006;33:287–92.
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41.
Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–6.
Acknowledgments
The authors would like to thank Dr I. Jakovlić for his help in English language editing.
Funding
This work was funded by the National Natural Science Foundation of China (31872604, 31572658) and the Earmarked Fund for China Agriculture Research System (CARS-45-15).
Author information
Authors and Affiliations
Contributions
PPF performed the analysis and wrote the manuscript. WXL designed the experiments. FX and WWF performed the laboratory work. All authors contributed to the interpretation of the findings. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Tapeworms were collected from fish in accordance with the recommended guidelines for animal experimentation by the Chinese Association for Laboratory Animal Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Morphological characteristics of Atractolytocestus tenuicollis in the intestine of common carp (Cyprinus carpio) from China.
Additional file 2: Figure S2.
Morphological characteristics of Khawia japonensis in the intestine of common carp (Cyprinus carpio) from China.
Additional file 3: Table S1.
Maturity of the two tapeworms Khawia japonensis and Atractolytocestus tenuicollis in the intestine of common carp (Cyprinus carpio) from China.
Additional file 4: Table S2.
Complete OTU table of gut microbiota at the genus level in the intestine of common carp (Cyprinus carpio).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fu, P.P., Xiong, F., Feng, W.W. et al. Effect of intestinal tapeworms on the gut microbiota of the common carp, Cyprinus carpio. Parasites Vectors 12, 252 (2019). https://doi.org/10.1186/s13071-019-3510-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-019-3510-z