
Abstract
Background
Hosts, parasites, and microbiota interact with each other, forming a complex ecosystem. Alterations to the microbial structure have been observed in various enteric parasitic infections (e.g. parasitic protists and helminths). Interestingly, some parasites are associated with healthy gut microbiota linked to the intestinal eubiosis state. So the changes in bacteria and metabolites induced by parasite infection may offer benefits to the host, including protection from other parasitesand promotion of intestinal health. The only ciliate known to inhabit the hindgut of grass carp, Balantidium ctenopharyngodoni, does not cause obvious damage to the intestinal mucosa. To date, its impact on intestinal microbiota composition remains unknown. In this study, we investigated the microbial composition in the hindgut of grass carp infected with B. ctenopharyngodoni, as well as the changes of metabolites in intestinal contents resulting from infection.
Results
Colonization by B. ctenopharyngodoni was associated with an increase in bacterial diversity, a higher relative abundance of Clostridium, and a lower abundance of Enterobacteriaceae. The family Aeromonadaceae and the genus Citrobacter had significantly lower relative abundance in infected fish. Additionally, grass carp infected with B. ctenopharyngodoni exhibited a significant increase in creatine content in the hindgut. This suggested that the presence of B. ctenopharyngodoni may improve intestinal health through changes in microbiota and metabolites.
Conclusions
We found that grass carp infected with B. ctenopharyngodoni exhibit a healthy microbiota with an increased bacterial diversity. The results suggested that B. ctenopharyngodoni reshaped the composition of hindgut microbiota similarly to other protists with low pathogenicity. The shifts in the microbiota and metabolites during the colonization and proliferation of B. ctenopharyngodoni indicated that it may provide positive effects in the hindgut of grass carp.
Similar content being viewed by others
Background
The intestine is colonized by a huge number of microorganisms, playing an important role in health and disease [1]. These microorganisms have been well adapted to the intestinal mucosa, contributing to the host’s nutrition, metabolism and immune functions [2, 3]. Thus the gut microbiota has been characterized as a microbial organ tightly associated with the host’s health [1, 4].
Grass carp is native to rivers and lakes in eastern Asia, with a wide distribution in China [5]. As an economically important freshwater fish and aquatic plant consumer, it was introduced to many countries for the purpose of human consumption and biological control of aquatic weeds [6, 7]. Microbiota establishes themselves in the intestines of fish larvae, and their diversity increases as the fish grows [8,9,10]. More than ten years ago, it was proposed that the core microbiota in grass carp are composed of Proteobacteria, Firmicutes, and Actinobacteria [11]. However, the microbial composition varies between individuals of the same species, and even within the same individual at different times [8]. Moreover, diets, antibiotics, immune deficiency, environmental factors or infections may lead to variations in bacterial diversity [8, 12]. Shifts in the microbial composition may allow aggravating factors to amplify changes in specific bacterial species, resulting in intestinal dysbiosis, which is a disruption of the symbiotic relationship between microbial communities and hosts [13].
Furthermore, specific intestinal microbial patterns are known to be related to the colonization with parasitic protozoa [14, 15]. Although the mechanisms underlying the association between protozoan infections and microbial variations are still poorly understood [16], the complex interactions among the host, parasite, and host’s intestinal microbiota have important functional implications for the outcomes of parasitic infections and diseases. Long-term colonization of some protists indicates that they may play alternative roles in the intestine, other than directly affecting host health [17, 18]. For example, increased bacterial diversity or higher abundance of probiotics was found in association with the presence of commensal protozoa (e.g. low-pathogenic Entamoeba and Blastocystis).
Balantidium ctenopharyngodoni is an obligate intestinal ciliate inhabiting the hindgut of grass carp, with high prevalence, unique infection features, and asymptomatic carrier status [19, 20]. An in vitro cultivation method of B. ctenopharyngodoni has been explored using a strain of intestinal bacterium [20, 21]. However, the mechanism of interaction between B. ctenopharyngodoni and the intestinal microbiota remains unknown. Additionally, the pathogenicity of B. ctenopharyngodoni is still unclear. In this study, we aim to investigate the associations between B. ctenopharyngodoni and microbiota, the relationships between the infection intensity of B. ctenopharyngodoni and the relative abundance of specific microbial taxa, as well as the changes of metabolites in the hindgut affected by the infection with B. ctenopharyngodoni. Our results indicated that infection with B. ctenopharyngodoni may increase the diversity of microbiota, but decrease the relative abundance of Enterobacteriaceae and Aeromonadaceae. Moreover, it was associated with a high content of creatine in the hindgut. Our findings offer valuable insights into how B. ctenopharyngodoni affects the intestinal health of grass carp. These may further give explanations on why B. ctenopharyngodoni is able to form a stable relationship with its host. Thus, we inferred that the presence of B. ctenopharyngodoni is closely related to a healthy gut microbiota and that it may play a beneficial role in grass carp through modulating intestinal bacteria.
Results
Comparison of bacterial composition and diversity in the hindgut of grass carp
We obtained 2,393,030 high-quality 16 S rRNA sequences from 15 samples of grass carp hindgut contents. A sampling depth of 45,000 reads was resampled according to the rarefaction curves (Fig. S1).
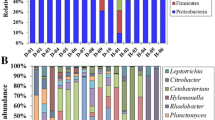
At the class level, 129 classes were shared by all samples, in which the top three classes in terms of relative abundance were Clostridia (BC0H (fish uninfected with B. ctenopharyngodoni): 18.80%±1.68%; BC1H (fish with a low-intensity infection): 19.42%±4.60%; BC2H (fish with a high-intensity infection): 19.78%±2.92%), Gammaproteobacteria (BC0H: 15.74%±5.46%; BC1H: 11.63%±3.60%; BC2H: 9.33%±1.20%) and Bacteroidia (BC0H: 9.39%±1.74%; BC1H: 9.49%±1.83%; BC2H: 9.24%±1.73%) (Fig. 1A). Compared to the BC0H group, Gammaproteobacteria had a low relative abundance in infected groups (BC1H, BC2H), while the relative abundance of Clostridia slightly increased in the infected groups (Fig. 2). At the family level, Enterobacteriaceae (BC0H: 11.45%±5.96%; BC1H: 7.43%±4.13%; BC2H: 4.97%±0.57%) and Ruminococcaceae (BC0H: 7.62%±1.64%; BC1H: 8.14%±4.23%; BC2H: 7.59%±2.21%) were the predominant families in all three groups, and the relative abundance of Enterobacteriaceae was lower in the infected groups (Figs. 1B and 2). Our results also showed that the hindgut of grass carp possesses a core microbiota, and the relative abundance of Firmicutes increased with the intensity of infection of B. ctenopharyngodoni, while the relative abundance of Proteobacteria decreased (Fig. S2).

Bacterial composition in the hindgut of grass carp at the class level (A, top 10 classes) and family level (B, top 20 families). BC0H represents a group not infected with B. ctenopharyngodoni. BC1H represents a group infected with a low abundance of B. ctenopharyngodoni. BC2H represents a group infected with a high abundance of B. ctenopharyngodoni

The relative abundance of specific taxa in the hindgut of three groups of grass carp
The alpha diversity, species richness (ACE and Chao1), and species diversity (Shannon, Simpson) were higher in infected groups than in the BC0H group (Table S1). Moreover, the BC2H group had more unique classes (10, Fig. S3A) and families (44, Fig. S3B). Permutational multivariate analysis of variance (PERMANOVA) showed that there was no significant difference in the composition of bacterial communities among BC0H, BC1H and BC2H groups (BC0H-BC1H, p = 0.800; BC0H-BC2H, p = 0.326; BC1H-BC2H, p = 0.587). Principal coordinates analysis (PCoA) showed that bacterial communities in the three groups were not clearly distinguishable (Fig. S4).
A total of 28 prokaryotic clades were screened out with a linear discriminant analysis (LDA) threshold score of 2.5 (Fig. S5). Microbial taxa with significantly higher abundance in the BC2H group mainly belonged to the families Fusobacteriaceae (genus u114), Gemmatimonadaceae (genus Gemmatimonas), Halomonadaceae, and Sphingobacteriaceae. With regard to the BC0H group, the families Enterobacteriaceae and Aeromonadaceae had significantly higher relative abundance.
Significant changes in predicted bacterial functions
Functional predictions of the results of 16s rRNA sequencing were performed using phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt). Based on the clusters of orthologous group (COG) analysis, the abundance of genes related to “energy production and conversion” and “carbohydrate transport and metabolism” categories exhibited lower abundance in infected groups (BC1H, BC2H); while the abundance of genes related to “nucleotide transport and metabolism” and “translation, ribosomal structure and biogenesis function” were relatively higher in infected groups (Fig. 3A). According to pairwise comparisons of the three groups, the abundance of genes related to “carbohydrate transport and metabolism” significantly decreased along with the intensity of infection (Fig. 3B, C and D).

Functional predictions, clusters and comparison based on the PICRUSt analysis (A. General comparison among groups of fish with differing infection intensities; B, C, D. Pairwise comparison between groups)
Targeted metabolic profiling
Principal component analysis (PCA) revealed similar patterns of metabolites in the 3 groups (Fig. 4A). Since the PCA plot could not differentiate well, Partial least squares-discriminant analysis (PLS-DA) was used to find the differences. The score plot showed that the three groups, BC0H, BC1H and BC2H, were distinguishable (Fig. 4B). A total of 103 metabolites were detected in all three samples. Using the threshold of p-value < 0.05 and |log (fold change)| > 0.5, there were 11 differential metabolites between BC0H and BC2H groups, among which 4 metabolites (gamma-aminobutyric acid, purine, cis-4-hydroxy-D-proline and creatine) were up-regulated and 7 metabolites (L-2-aminoadipic acid, UDP-D-glucose, Glycyl-L-leucine, malic acid, N4-acetylcytidine, argininosuccinic acid, and GDP-L-fucose) were down-regulated in the BC2H group (Fig. 5A and B). Only 7 differential metabolites were identified between BC0H and BC1H groups (Fig. S6). Notably, creatine was the only shared differential metabolite in the two infected groups, and it accounted for a high proportion of all detected metabolites.

Principal component analysis (PCA, A) and partial least squares discrimination analysis (PLS-DA, B) of metabolites in the hindgut of grass carp

Differential metabolites between the hindgut contents of uninfected (BC0H) and high-infected (BC2H) groups identified through the targeted metabolic analysis. (A) Volcano plot for BC0H and BC2H groups. (B) Hierarchical cluster analysis of 11 differential metabolites. (C) Correlation analysis of 11 differential metabolites between BC0H and BC2H groups (p < 0.05). × represents no significant correlation between the two metabolites. (D) Metabolic enrichment analysis of significantly differential metabolites
Furthermore, we examined the correlation among the differential metabolites (BC2H vs. BC0H). The results showed that Glycyl-L-leucine was positively correlated with GDP-L-fucose, malic acid and N4-Acetylcytidine, while it was negatively correlated with creatine, cis-4-Hydroxy-D-proline and purine (Fig. 5C). UDP-D-Glucose was positively correlated with L-2-Aminoadipic acid, and creatine was significantly correlated with cis-4-Hydroxy-D-proline (Fig. 5C). The differential metabolites were mainly involved in ascorbate and aldarate metabolism, arginine and proline metabolism (Fig. 5D).
Co-occurrence of microbial taxa and metabolites
The co-occurrence networks were constructed based on a threshold of both the correlation coefficient of > 0.8 and a p-value of < 0.05. Compared to the uninfected group (BC0H), the infected groups had stronger correlations between microbiota and metabolites. The three groups showed different co-occurrence patterns between microbial taxa and metabolites, and bacteria were predominant in the co-occurrence network (Fig. 6). Specifically, Bacilli, Acidimicrobiia, Clostridia and Fusobacteriia were prominent bacterial responders in the BC0H group (correlation coefficient > 0.9, p < 0.05), in which Bacilli and Acidimicrobiia belonged to a similar pattern correlating negatively with Cytidine, gamma-L-Glutamyl-L-phenylalanine and glycine (Fig. 6A). The correlation coefficients of Bacilli with glycine, cytidine and gamma-L-glutamyl-L-phenylalanine were about 1.0 (p < 0.01, Fig. S7A). In the BC1H group, Clostridia and Epsilonproteobacteria were two core bacterial responders, and many metabolites were associated with them (Fig. 6B). In the BC2H group, Gammaproteobacteria and Saprospirae were two prominent bacteria responders, followed by Betaproteobacteria, Flavobacteriia, Cytophagia, and Anaerolineae. Additionally, UDP-D-Glucuronate and L-Citrulline were the two prominent metabolite responders that were correlated with Clostridia, Betaproteobacteria and Flavobacteria (Fig. 6C, Fig. S7B).

Co-occurrence networks of intestinal bacteria and metabolites in the hindgut of grass carp (A. BC0H group, B. BC1H group, C. BC2H group). Only significant relationships (p < 0.05) are shown. The blue edge indicates a positive relationship between metabolites and bacteria, and the red edge indicated a negative relationship. The width of the edge corresponds to the magnitude of the correlation between the bacteria and metabolites. The node size is proportional to the number of connections
Discussion
The associations between microbiota and parasites have been increasingly investigated in various animal hosts in recent years [22,23,24]. There is evidence emerging that this relationship can influence host health, making the hosts more susceptible, tolerant, or resistant to parasites [17, 25, 26]. The introduction of a parasite can lead to disruptions in microbial composition and diversity [27], moreover, the growth and proliferation of the parasite might also be linked to the associated bacterial microbiome [20, 28]. Thus, the tripartite relationship among the microbiota, parasite and host is complex, with difficult-to-predict outcomes.
Parasite-bacteria interactions may be driven by predation, competition, or other factors. For example, protist predators have an important role in the top-down control of microbial communities [29,30,31]. Balantidium species have one oval cytostome and several food vacuoles containing the intestinal bacteria of hosts [32, 33]. We also found that there is a large proportion of carbohydrate-active enzyme genes encoding carbohydrate-binding module 50 (CBM50) with an affinity for peptidoglycan in the B. ctenopharyngodoni genome (unpublished data). Thus, Balantidium species are also bacterivorous (preying on intestinal bacteria).
Balantidium ctenopharyngodoni alters the diversity and abundance of intestinal microbiota
Disruption of intestinal microbiota has been described in different intestinal inflammatory diseases. Healthier individuals generally harbor greater microbial alpha diversity [18, 34]. Although there were no significant differences in alpha diversity among the three groups, a slight increase was observed in the infected samples.
Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria were the predominant microbiota in all groups. Among these, three are considered to be the core microbiota in the grass carp intestine [11]. Specific gut microbiota patterns have been shown to be linked to the colonization with common parasitic protists [35]. An increase in the relative abundance of Enterobacteriaceae correlates positively with gastrointestinal or systemic inflammation in humans [36, 37]. There was a higher abundance of Gammaproteobacteria (family Enterobacteriaceae) in the patients infected with Blastocystis ST7, which is considered to be a pathogenic subtype of Blastocystis [38, 39]. However, a lower abundance of Enterobacteriaceae and a higher abundance of Clostridia were observed in Blastocystis subtypes with lower pathogenicity [14, 35, 40]. Similar trends in the composition of gut bacteria in response to a Giardia infection may give an explanation of the protective effect of Giardia against moderate to severe diarrhea [41, 42]. Besides, a lower abundance of Clostridiaceae and a higher abundance of Enterobacteriaceae were reported in diarrheal piglets infected with another balantidia species Balantioides coli [43]. In grass carp, infection with B. ctenopharyngodoni also seemed to be associated with changes in specific taxa, including a decrease in the relative abundance of the family Enterobacteriaceae (class Gammaproteobacteria) and an increase in the relative abundance of the genus Clostridium (class Clostridia). From this perspective, B. ctenopharyngodoni can therefore be regarded as a low pathogenic intestinal ciliate.
Balantidium ctenopharyngodoni can reduce the relative abundance of pathogenic bacteria
The intestinal inflammation of grass carp, induced by the pathogen Aeromonas hydrophila, is regarded as one of the most frequently occurring diseases in culture ponds [44]. Citrobacter species are conditionally pathogenic bacteria in fish and the human intestine, and they are responsible for causing enteritis and other infections [45, 46]. In our study, however, the genera Aeromonas and Citrobacter were significantly less abundant in grass carp infected with B. ctenopharyngodoni than in uninfected grass carp. These results indicated that B. ctenopharyngodoni might suppress the abundance of pathogenic bacteria to maintain intestinal homeostasis.
Infection with B. ctenopharyngodoni may also confer positive benefits on the intestine of grass carp
In general, parasite-induced changes to the gut microbiome can modulate the host’s metabolism, so colonization with parasites is associated not only with alterations in the intestinal microbiota but also with changes in host metabolites [35, 47, 48]. In this study, we identified the metabolites present in the hindgut of grass carp, and assessed the interactions between metabolites and microbiota in the infected and uninfected groups. Creatine was enriched in B. ctenopharyngodoni-infected groups (BC1H and BC2H), accounting for quite a large proportion of the detected metabolites (12.7% and 15.2%, respectively). It has been reported that creatine could regulate intestinal epithelial integrity and barrier function, thus producing anti-inflammatory effects [49, 50]. Inflammatory bowel disease (IBD) is a chronic immune-mediated intestinal disease that develops as a result of interactions between environmental, microbial, and immune factors in a host [51]. Since IBD is hard to be cured completely, creatine is a good adjuvant candidate for alleviating the symptoms of IBD [49]. Besides, gut microbiota can express specific enzymes (e.g. creatinase) that mediate creatine breakdown [52]. Thus, the high content of creatine in B. ctenopharyngodoni-infected groups indicated that B. ctenopharyngodoni might further influence the metabolic breakdown of creatine by regulating the composition of intestinal bacteria, and thereby positively affecting the host intestine.
Conclusions
Balantidium ctenopharyngodoni is considered as a commensal organism in the hindgut of grass carp, it does not cause severe intestinal inflammation in most cases, but it may aggravate the symptoms when its host suffers from enteritis [53, 54]. In our study, all the grass carp and their intestines exhibited a normal appearance. The changes of relative abundance in specific bacterial taxa (e.g. decreased relative abundance of Enterobacteriaceae and increased relative abundance of Clostridium) followed similar trends to those in gut microbiota colonized by protists with low pathogenicity. Important pathogens like Aeromonas and Citrobacter showed lower abundance in B. ctenopharyngodoni-infected grass carp. Creatine was significantly enriched in the infected groups, which may have positive effects on the host intestine. Taken together, our results showed that B. ctenopharyngodoni may have some beneficial effects on the intestinal microbiota and metabolites in grass carp.
Methods
Sample collection
Grass carp fed on ryegrass and commercial feed were captured (n = 32) using a cast net from an aquaculture pond in Honghu City, Hubei province, China (30.07°N, 113.75°E). Then, they were quickly transported to the laboratory, where the abdominal cavity was opened after anesthetization using MS-222. The whole intestine was taken out, and the hindgut (from the last bend to the anus) was divided and opened under aseptic conditions. The infection intensity of B. ctenopharyngodoni was observed and enumerated using the direct microscopic counts. The samples were grouped according to the intensity: one group (BC0H) comprised uninfected samples, in which we observed no B. ctenopharyngodoni; another group (BC1H) comprised low-intensity samplesexhibiting 0-103B. ctenopharyngodoni individuals/fish; and the third group (BC2H) comprised high-intensity samples exhibiting more than 103 individuals/fish (Fig. S8). The contents of hindgut were collected into sterile tubes, frozen immediately in liquid nitrogen, and stored at -80 °C for subsequent treatments.
Bacterial DNA extraction, amplification and high-throughput sequencing
Approximately 200 mg of intestinal content was used to extract total genomic DNA via QIAamp® DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. The concentration of extracted DNA was measured with a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, USA). The 16s rRNA V3-V4 region was amplified by PCR using a forward primer with a unique barcode (5’-CCTACGGGNGGCWGCAG-3’) and a reverse primer (5’-GACTACHVGGGTATCTAATCC-3’) [55]. The reactions were performed with an initial denaturation step at 94 °C for 5 min, followed by 25 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 90 s, and then a final extension step at 72 °C for 10 min. The PCR products were separated by 2% agarose gel electrophoresis and purified using a DNA Gel Extraction Kit (Aidlad Biotech, Beijing, China). The concentration of the purified DNA was determined using a NanoDrop 8000 spectrophotometer. Equal amounts of all samples were pooled together for library construction, and sequencing was performed on an Illumina Hiseq 2500 platform.
Sequence data processing
Raw paired-end reads were processed using the QIIME 2 pipeline (version 2021.4.0) [56]. After trimming low-quality bases from de-multiplexed paired-end reads, the processed reads were trimmed, de-noised, and merged using DADA2 plugin incorporated in QIIME 2. Thereafter, chimera sequences were removed, and the generated amplicon sequence variants (ASVs) were summarized, and used to generate a sequence table. The ASVs were classified and taxonomically assigned using the feature classifier in QIIME2 against the Greengenes database (13_8). Then, ASVs derived from mitochondria and chloroplasts were removed. The sequence table was then rarefied to the same sequencing depth (45,000) in all samples according to rarefaction curves. Alpha diversity was evaluated by species richness (ACE and Chao1 indices) and diversity (Shannon and Simpson indices). Permutational multivariate analysis of variance (PERMANOVA) was performed to analyze the statistical significance between two groups based on a weighted UniFrac distance matrix. The principal coordinates analysis (PCoA) plot was also generated based on the distance matrix generated from a weighted UniFrac phylogenetic method. The PICRUSt was used to predict the functional clusters of each group. Linear discriminant analysis effect size (LEfSe) was performed online using the Galaxy workflow framework (http://galaxy.biobakery.org/).
Metabolite extraction and UHPLC-MRM-MS/MS analysis
After 25 mg of each sample was taken and placed into an Eppendorf tube, 500 µl of extraction solution (acetonitrile: methanol: water = 2: 2: 1) was added to each tube. The samples were swirled for 30s, then homogenized at 45 Hz for 4 min, and ultrasound-treated for 5 min with the ice water bath. After the homogenization and ultrasound treatment were repeated three times, the samples were incubated at -40 °C for 1 h to precipitate proteins. The samples were centrifuged at 12,000 rpm for 15 min at 4 °C, and then the supernatants (about 400 µl) were transferred into new Eppendorf tubes and dried in a vacuum concentrator without heating. The dried samples were reconstituted in 100 µl of extraction solution (acetonitrile: water = 1: 1) for 10 min in the ice-water bath. The samples were vortexed for 30 s, sonicated for 10 min, and centrifuged for 15 min at 13,000 rpm, at 4 °C. Finally, 75 µl of supernatant was transferred into a fresh glass vial for LC/MS analysis. The quality control sample was the mixture of an equal aliquot of supernatants from all of the samples.
LC-MS/MS analyses were performed on a UHPLC system (Agilent 1290 Infinity, Agilent Technologies) equipped with a UPLC BEH Amide column (2.1 × 100 mm, 1.7 μm, Waters). The mobile phase consisted of 25 mM ammonium acetate and 25 mM ammonia hydroxide in water (pH = 9.75) (A) and acetonitrile (B) was carried with the elution gradient and flow rate of 0.5 ml/min. The column temperature was set at 25 °C, the sample-plate temperature was 4 °C, and the injected volume was 2 µl. An Agilent 6495 triple quadrupole mass spectrometer (Agilent Technologies) with an electrospray interface and multiple reaction monitoring modes was used for the assay development. The parameters of the typical ion source were as follows, capillary voltage = + 3000/-2500 V, gas (N2) flow = 16 L/min, gas (N2) temperature = 170 °C, sheath gas flow = 12 L/min, sheath gas (N2) temperature = 350 °C, nebulizer = 40 psi. Data were collected and processed with Agilent MassHunter Work Station software (Agilent Technologies). Differential metabolites were analyzed using a log (fold change) of 0.5 and a t-test threshold of 0.05. Principal component analysis (PCA) and partial least squares discrimination analysis (PLS-DA) were performed on the MetaboAnalyst 5.0 platform [57].
Combined analyses of metabolome and microbiome
Co-occurrence analyses of microbiota and metabolites were performed in Rstudio 2022.12.0 + 353 with a correlation coefficient of > 0.8 and a p-value of < 0.05. The Gephi software (Version 0.10.1) was used to analyze and visualize the network graphs under the Fruchterman Reingold algorithm. Correlations between metabolites and bacterial communities were also assessed by Spearman’s correlation analysis, and the graph was generated using the pheatmap package (version 1.0.12, https://cran.r-project.org/web/packages/pheatmap/index.html) in Rstudio.
Data availability
The raw sequencing data used in this study were deposited at the National Center for Biotechnology Information (NCBI) with BioProject ID PRJNA1018602 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1018602). Other data are available from the corresponding author upon reasonable request.
References
Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–20.
Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, et al. Gut microbiome structure and metabolic activity in inflammatory bowel Disease. Nat Microbiol. 2019;4(2):293–305.
Fernández-Veledo S, Vendrell J. Gut microbiota-derived succinate: friend or foe in human metabolic Diseases? Reviews in Endocrine and Metabolic Disorders. 2019;20(4):439–47.
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–93.
Xie C, Li J, Li D, Shen Y, Gao Y, Zhang Z. Grass carp: The fish that feeds half of China. In: Gui J, Tang Q, Li Z, Liu J, Silva SSD, editors. Aquaculture in China: Success stories and modern trends. 2018;93–115.
Sullivan CJ, Weber MJ, Pierce CL, Camacho CA. A comparison of Grass Carp population characteristics upstream and downstream of lock and dam 19 of the Upper Mississippi River. J Fish Wildl Manage. 2020;11(1):99–111.
FAO. The state of world fisheries and aquaculture. Rome; 2022.
Medina-Félix D, Garibay‐Valdez E, Vargas‐Albores F, Martínez‐Porchas M. Fish Disease and intestinal microbiota: a close and indivisible relationship. Reviews in Aquaculture. 2022;15:820–39.
Nayak SK. Role of gastrointestinal microbiota in fish. Aquac Res. 2010;41(11):1553–73.
Egerton S, Culloty S, Whooley J, Stanton C, Ross RP. The gut microbiota of marine fish. Front Microbiol. 2018;9:873.
Wu S, Wang G, Angert ER, Wang W, Li W, Zou H. Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS ONE. 2012;7(2):e30440.
Stecher B, Maier L, Hardt W-D. Blooming’ in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol. 2013;11(4):277–84.
Mukherjee D, Chora ÂF, Mota MM. Microbiota, a third player in the host–plasmodium affair. Trends Parasitol. 2019;36(1):11–8.
Audebert C, Even G, Cian A, Blastocystis Investigation G, Loywick A, Merlin S, et al. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci Rep. 2016;6:25255.
Leon-Coria A, Kumar M, Chadee K. The delicate balance between Entamoeba histolytica, mucus and microbiota. Gut Microbes. 2020;11(1):118–25.
Bar AK, Phukan N, Pinheiro J, Simoes-Barbosa A. The interplay of host microbiota and parasitic protozoans at mucosal interfaces: implications for the outcomes of Infections and Diseases. PLoS Negl Trop Dis. 2015;9(12):e0004176.
Deng L, Tan KSW. Interactions between Blastocystis subtype ST4 and gut microbiota in vitro. Parasites & Vectors. 2022;15(1):80.
Chabe M, Lokmer A, Segurel L. Gut protozoa: friends or foes of the human gut microbiota? Trends Parasitol. 2017;33(12):925–34.
Li M, Li D, Wang J, Zhang J, Gu Z, Gong X. Light and scanning electron microscopic study of Balantidium Ctenopharyngodoni Chen, 1955 (class: Litostomatea) from China. Parasitol Res. 2007;101(1):185–92.
Zhao W, Li M, Hu G, Bu X, Zhou W, Li W, et al. An Infection mechanism of Balantidium Ctenopharyngodoni, based on its prevalence and in vitro cultivation. Aquaculture. 2021;548:737686.
Li M, Bastos Gomes G, Zhao W, Hu G, Huang K, Yoshinaga T, et al. Cultivation of fish ciliate parasites: Progress and prospects. Rev Aquac. 2023;15(1):142–62.
Libertucci J, Young VB. The role of the microbiota in infectious Diseases. Nat Microbiol. 2019;4(1):35–45.
Zhou S, Lu Y, Chen J, Pan Z, Pang L, Wang Y, et al. Parasite reliance on its host gut microbiota for nutrition and survival. ISME J. 2022;16(11):2574–86.
Mayneris-Perxachs J, Arnoriaga-Rodriguez M, Garre-Olmo J, Puig J, Ramos R, Trelis M, et al. Presence of blastocystis in gut microbiota is associated with cognitive traits and decreased executive function. ISME J. 2022;16(9):2181–97.
O’Keeffe KR, Halliday FW, Jones CD, Carbone I, Mitchell CE. Parasites, niche modification and the host microbiome: a field survey of multiple parasites. Mol Ecol. 2021;30(10):2404–16.
Gilchrist CA, Petri SE, Schneider BN, Reichman DJ, Jiang N, Begum S, et al. Role of the gut microbiota of children in diarrhea due to the protozoan parasite Entamoeba histolytica. J Infect Dis. 2015;213(10):1579–85.
Jani AJ, Briggs CJ. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental Infection. Proc Natl Acad Sci. 2014;111(47):E5049–E58.
Jahangiri L, Shinn AP, Pratoomyot J, Bastos Gomes G. Unveiling associations between ciliate parasites and bacterial microbiomes under warm-water fish farm conditions – a review. Rev Aquac. 2020;13(2):1097–118.
Vdacny P, Ersekova E, Soltys K, Budis J, Pecina L, Rurik I. Co-existence of multiple bacterivorous clevelandellid ciliate species in hindgut of wood-feeding cockroaches in light of their prokaryotic consortium. Sci Rep. 2018;8(1):17749.
Jürgens K, Matz C. Predation as a shaping force for the phenotypic and genotypic composition of planktonic bacteria. Antonie Van Leeuwenhoek. 2002;81(1):413–34.
Slaveykova V, Sonntag B, Gutierrez JC. Stress and protists: no life without stress. Eur J Protistol. 2016;55(Pt A):39–49.
Grim JN. Food vacuole contents in the ciliate, Balantidium Jocularum (Balantididae), a symbiont in the intestine of the surgeonfish, Naso tonganus (Acanthuridae). J Eukaryot Microbiol. 2006;53(4):269–74.
Zhao W, Li M, Xiong F, Zhang D, Wu S, Zou H, et al. Identification of intracellular bacteria in the ciliate Balantidium Ctenopharyngodoni (Ciliophora, Litostomatea). J Eukaryot Microbiol. 2020;67(4):417–26.
Peachey LE, Jenkins TP, Cantacessi C. This gut ain’t big enough for both of us. Or is it? Helminth-Microbiota interactions in veterinary species. Trends Parasitol. 2017;33(8):619–32.
Stensvold CR, van der Giezen M. Associations between gut microbiota and common luminal intestinal parasites. Trends Parasitol. 2018;34(5):369–77.
Chung The H, Le S-NH. Dynamic of the human gut microbiome under infectious diarrhea. Curr Opin Microbiol. 2022;66:79–85.
Liu J, Wang M, Chen W, Ma J, Peng Y, Zhang M, et al. Altered gut microbiota taxonomic compositions of patients with sepsis in a pediatric intensive care unit. Front Pead. 2021;9:645060.
Deng L, Lee JWJ, Tan KSW. Infection with pathogenic Blastocystis ST7 is associated with decreased bacterial diversity and altered gut microbiome profiles in diarrheal patients. Parasites & Vectors. 2022;15(1):312.
Wu Z, Mirza H, Tan KSW. Intra-subtype variation in enteroadhesion accounts for differences in epithelial barrier disruption and is associated with metronidazole resistance in Blastocystis subtype-7. PLoS Negl Trop Dis. 2014;8(5):e2885.
Kim M-J, Lee YJ, Kim T-J, Won EJ. Gut microbiome profiles in colonizations with the enteric protozoa Blastocystis in Korean populations. Microorganisms. 2022;10(1):34.
Berry ASF, Johnson K, Martins R, Sullivan MC, Amorim CF, Putre A, et al. Natural Infection with Giardia is associated with altered community structure of the human and canine gut microbiome. mSphere. 2020. https://doi.org/10.1128/mSphere.00670-20.
Bartelt LA, Platts-Mills JA. Giardia: a pathogen or commensal for children in high-prevalence settings? Curr Opin Infect Dis. 2016;29(5):502–7.
He K, Yan W, Sun C, Liu J, Bai R, Wang T, et al. Alterations in the diversity and composition of gut microbiota in weaned piglets infected with Balantioides Coli. Vet Parasitol. 2020;288:109298.
Song X, Zhao J, Bo Y, Liu Z, Wu K, Gong C. Aeromonas hydrophila induces intestinal inflammation in grass carp (Ctenopharyngodon idella): an experimental model. Aquaculture. 2014;434:171–8.
Madurantakam Royam M, Nachimuthu R. Isolation, characterization, and efficacy of bacteriophages isolated against Citrobacter spp. an in vivo approach in a zebrafish model (Danio rerio). Res Microbiol. 2020;171(8):341–50.
Xiong F, Qin L, Hao Y, Zhao D, Li W, Zou H, et al. Gut microbiota modulation and immunity response induced by Citrobacter freundii strain GC01 in grass carp (Ctenopharyngodon Idellus). Aquaculture. 2020;521:735015.
Nieves-Ramírez ME, Partida-Rodríguez O, Laforest-Lapointe I, Reynolds LA, Brown EM, Valdez-Salazar A, et al. Asymptomatic intestinal colonization with protist Blastocystis is strongly associated with distinct microbiome ecological patterns. mSystems. 2018. https://doi.org/10.1128/msystems.00007-18.
Fu P, Xiong F, Feng W, Zou H, Wu S, Li M, et al. Effect of intestinal tapeworms on the gut microbiota of the common carp, Cyprinus carpio. Parasit Vectors. 2019;12(1):252.
Wallimann T, Hall CH, Colgan SP, Glover LE. Creatine supplementation for patients with inflammatory bowel Diseases: a scientific rationale for a clinical trial. Nutrients. 2021;13(5):1429.
Turer E, McAlpine W, Wang K, Lu T, Li X, Tang M, et al. Creatine maintains intestinal homeostasis and protects against Colitis. Proc Natl Acad Sci. 2017;114(7):E1273–E81.
Glassner KL, Abraham BP, Quigley EMM. The microbiome and inflammatory bowel Disease. J Allergy Clin Immunol. 2020;145(1):16–27.
Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80(3):1107–213.
Nie D, Wang J. Biology and Diseases of grass carp. Science Press Beijing; 1999.
Molnár K, Reinhardt M. Intestinal lesions in grasscarp Ctenopharyngodon idella (Valenciennes) infected with Balantidium Ctenopharyngodonis Chen. J Fish Dis. 1978;1(2):151–6.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012;41(1):e1.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, et al. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc. 2022;17(8):1735–61.
Acknowledgements
We are very grateful to Ivan Jakovlić for polishing the English language of the manuscript. Special thanks to Prof. Jie Xiong and Prof. Wei Miao for their valuable assistance with experimental design and data analysis. The research was supported by the Protist 10,000 Genomes (P10K) Project and the Wuhan Branch, Supercomputing Center, Chinese Academy of Sciences, China.
Funding
This work was supported by the China Postdoctoral Science Foundation (No. 2021M703435), the funding for Postdoctoral Innovation Research Post in Hubei Province, the National Natural Science Foundation of China (No. 32303066, 32170437), the National Key Research and Development Program of China (No. 2022YFF1001100), the Second Tibetan Plateau Scientific Expedition and Research Program (STEP) (No. 2019QZKK0304), the earmarked fund for CARS (No. CARS-45), and the National Aquatic Biological Resource Center (NABRC).
Author information
Authors and Affiliations
Contributions
M Li, GT Wang and WS Zhao concepted and designed the study; WS Zhao, XL Bu, WT Zhou, QW Zeng and T Qin executed the experiments; WS Zhao, SG Wu, WX Li and H Zou analyzed data and presented the results; WS Zhao and M Li wrote the original draft. All authors reviewed the manuscript, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental protocols were approved by the Animal Ethical and Welfare Committee of the Institute of Hydrobiology, Chinese Academy of Sciences (No. IHB/LL/2023036). All methods were carried out in accordance with relevant guidelines and regulations. For example, the fish used in this study were gently anesthetized by soaking in MS-222.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, W., Bu, X., Zhou, W. et al. Interactions between Balantidium ctenopharyngodoni and microbiota reveal its low pathogenicity in the hindgut of grass carp. BMC Microbiol 24, 7 (2024). https://doi.org/10.1186/s12866-023-03154-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-03154-8