
Abstract
The microbes in the gastrointestinal (GI) tract are of high importance for the health of the host. In this study, Roche 454 pyrosequencing was applied to a pooled set of different 16S rRNA gene amplicons obtained from GI content of common carp (Cyprinus carpio) to make an inventory of the diversity of the microbiota in the GI tract. Compared to other studies, our culture-independent investigation reveals an impressive diversity of the microbial flora of the carp GI tract. The major group of obtained sequences belonged to the phylum Fusobacteria. Bacteroidetes, Planctomycetes and Gammaproteobacteria were other well represented groups of micro-organisms. Verrucomicrobiae, Clostridia and Bacilli (the latter two belonging to the phylum Firmicutes) had fewer representatives among the analyzed sequences. Many of these bacteria might be of high physiological relevance for carp as these groups have been implicated in vitamin production, nitrogen cycling and (cellulose) fermentation.
Similar content being viewed by others

Introduction
The intestine is a multifunctional organ system involved in the digestion and absorption of food, electrolyte balance, endocrine regulation of food metabolism and immunity against pathogens (Ringo et al. 2003). The gastrointestinal (GI) tract is inhabited by many different micro-organisms. As in mammals, this dynamic population of micro-organisms is of key importance for the health of the piscine host (Ringo et al. 2003; Rawls et al. 2004). The gut is also a potential route for pathogens to invade and infect their host. The micro-organisms in the GI tract are involved in the protection against these pathogens by the production of inhibitory compounds and competition for nutrients and space. As in mammals, the intestinal microbiota of fish can influence the expression of genes involved in epithelial proliferation, nutrient metabolism and innate immunity (Rawls et al. 2004). Due to their importance in animal health, the investigation of the intestinal microbiota of fish is highly relevant for aquaculture practice. We investigated the diversity of the microbiota in common carp (Cyprinus carpio), one of the most cultivated freshwater fish species worldwide (FAO, 2011).
The morphology of the GI tract of fishes varies greatly among species. Common carp belong to the family of Cyprinidae, which are herbivorous, stomachless fish. These fish lack pyloric caeca, the finger-like blind sacs in the proximal intestine that increase the absorptive surface of the intestines in many fish (Ringo et al. 2003; Buddington and Diamond 1987). The composition of the gut microbiota of common carp has previously been investigated using culture-dependent methods (Sugita et al. 1990; Namba et al. 2007; Tsuchiya et al. 2008). Most bacterial species found in these studies were aerobes and facultative anaerobes. Two studies demonstrated a high abundance of Aeromonas species (Namba et al. 2007; Sugita et al. 1990). Other bacteria isolated were Enterobacteriaceae (Sugita et al. 1990; Namba et al. 2007), Pseudomonas (Sugita et al. 1990; Namba et al. 2007), Bacteriodetes (Sugita et al. 1990; Tsuchiya et al. 2008), Plesiomonas (Sugita et al. 1990), Moraxella (Sugita et al. 1990; Namba et al. 2007), Acinetobacter (Sugita et al. 1990; Namba et al. 2007), Flavobacterium (Sugita et al. 1990), Staphylococcus (Sugita et al. 1990), Micrococcus (Sugita et al. 1990; Namba et al. 2007), Streptococcus (Sugita et al. 1990), Bacillus (Sugita et al. 1990), Clostridium (Sugita et al. 1990), Vibrio (Namba et al. 2007) and Cetobacterium (Tsuchiya et al. 2008). However, these studies only reveal the microbes that can be cultured and these most likely do not reflect the complete microbial composition of the carp gut since studies on mammals have shown that most members of the microbiota in the GI tract cannot be cultured when removed from the gut (Suau et al. 1999; Moya et al. 2008). The use of culture-independent studies such as molecular screening of the 16S rRNA gene may be a more reliable method to estimate microbial diversity in the GI tract of fish (Wu et al. 2010). Next generation sequencing is a powerful technique to investigate the composition of complex microbial communities in different environments (Hong et al. 2010; Qin et al. 2010; Vahjen et al. 2010; Moya et al. 2008; Kip et al. 2011; Roeselers et al. 2011). The combination of 16S rRNA gene amplification using multiple primer sets and the subsequent sequencing of the PCR products by Roche 454 pyrosequencing should therefore be a powerful method to assess the diversity of the microbiota in the GI tract of common carp. Obtained 16S rRNA gene sequences were used to classify the different microorganisms present in the fish gut and here we will also discuss the possible functions of these bacteria in the carp gut.
Materials and methods
Fish and system configuration
Common carp (Cyprinus carpio L.) were kept in 140 L tanks in a closed recirculating aquaculture system with a total volume of 3000 L at the Radboud University Nijmegen (The Netherlands). Fish were fed commercial food (Trouvit, at a daily ration of 1% estimated body weight), containing 45% protein. Water quality of the system was maintained by a biofilter and a weekly water replacement of 10% of the total volume. Ten fish (male and female) weighing 60 to 158 gram were used. All experimental procedures were performed with permission of the local ethical review committee (Radboud University Nijmegen).
DNA extraction, PCR amplification and sequence analysis
Ten fish were euthanized using 0.1% ethyl-m-aminobenzoate methane sulfonate salt (MS-222, MP Biomedicals, Illkirch, France, pH adjusted to 7) followed by decapitation. The body surface of the fish was washed with 70% ethanol and the GI tract was removed aseptically. The whole content of the GI tract was removed by carefully flushing with PBS and DNA was extracted from this material using a cetyltrimethylammoniumbromide (CTAB)-based extraction method (Zhou et al. 1996). Briefly, samples were mixed with CTAB-extraction buffer (100 mM Tris-HCl (pH 8.0), 100 mM EDTA, 100 mM sodium phosphate (pH 8.0), 1.5 M NaCl, 1% CTAB, 675 μl per 250 mg sample) and protease K (10 mg/ml) and incubated for 30 min at 37°C. After protease treatment 10% SDS was added, followed by incubation at 65°C for 2 h. DNA was recovered by phenol/chloroform extraction and ethanol precipitation and the resulting DNA pellet was resuspended in 1 ml ultrapure water. Before additional purification, DNA was treated with RNAse. The DNA thus obtained was purified using Sephadex beads (Amersham Bioscience, USA) according to the manufacturer's protocol and its integrity was checked on agarose gel. DNA concentrations were estimated spectophotometrically using NanoDrop® technology (Thermoscientific, USA).
Retrieval of 16S RNA gene sequences
Obtained DNA (20 ng) was used for amplification in 20 μl reactions using Phusion Flash enzymes (Finnzymes, Finland). In order to target as many bacterial taxa as possible, the Pla46 F primer was combined with EubI R, EubII R or EubIII R and for the 616 F primer the same set of reverse primers was used (Table 1). This resulted in 6 different combinations. All reactions were done for individual fish separately. PCR reactions were started by an initial denaturation at 98°C for 1 min followed by 35 amplification cycles (98°C for 6 s, 10 s at annealing temperature, 72°C for 20 s) and a final extension step for 1 min at 72°C. PCR products were examined for size and yield using agarose gel in TAE buffer (20 mM Tris-HCl, 10 mM sodium acetate, 0.5 mM Na2EDTA, pH 8.0). After successful amplification, obtained products of different reactions were pooled and 9.2 μg PCR product was used for pyrosequencing using the Roche 454 GS FLX Titanium sequencer (Roche, Switzerland). A problem with 454 pyrosequencing is 'blinding' of the camera due to flashing caused by incorporation of the same nucleotide in many spots, which can occur when many similar DNA templates are sequenced (Kip et al. 2011). This was circumvented by mixing 16S rRNA gene products in a 1:1 ratio with pmoA PCR products (targeting a subunit of the particulate methane monooxygenase) from a non-related experiment (Kip et al. 2011).
Phylogenetic analysis
A Megablast search (using default parameters) of all sequenced reads larger than 100 nt against the Silva SSURef database (version 102) was done to extract all 17,892 16S rRNA gene sequences (average length 314 nt). The taxonomic annotations available in the Silva SSURef database were used to classify the sequenced reads. Each read was assigned to the taxonomic clade of its highest scoring Megablast hit, when a sequence was assigned to more than one clade, its vote was divided equally. Furthermore, obtained sequences were processed using the Classifier tool (Wang et al. 2007) of the RDP pyrosequencing pipeline http://pyro.cme.msu.edu/. The confidence threshold used was 50%. The sequence reads are available at the MG-Rast Metagenome analysis server http://metagenomics.anl.gov/ under Project ID 4449604.3 and from the Sequence Read Archive (SRA) at http://www.ebi.ac.uk/ena/data/view/ under accession number ERP000995.
Results
The use of next generation sequencing technologies for sequencing of a mixture of 16S rRNA amplicons amplified with primer sets targeting as many phyla as possible will give a much broader taxonomic overview compared to the use 16S rRNA hypervariable regions (Kysela et al. 2005). To avoid missing a certain group of bacteria, different primer sets (Table 1) were used targeting as much species as possible. Obtained amplicons from all different reactions were mixed and sequenced using Roche 454 titanium technology and this revealed a high microbial diversity in the GI tract of common carp (Cyprinus carpio). It should be noted that the use of multiple primer sets biases the number of sequences belonging to the identified taxa. The number of obtained sequences belonging to a specific group may not be representative for their abundances in vivo; therefore no quantitative statements could be made.
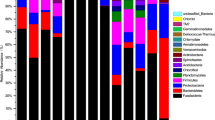
Figure 1 displays the taxonomic classification derived from mapping the pyrosequencing reads to the Silva SSURef database, which classified 17,641 reads (99%). Similar results were obtained when the RDP database pyrosequencing pipeline was used, which classified 16,768 reads (94%, Additional file 1). Almost half of the obtained sequences, i.e. 46%, found belonged to the Fusobacteria (Additional file 2). Other well represented groups within the retrieved sequences were the Bacteroidetes (21%), Planctomycetes (12%), and Gammaproteobacteria (7%); less retrieved sequences belonged to the Clostridia (3%), Verrucomicrobiae (1%), and Bacilli (1%). Furthermore, a few sequences (< 1%) were identified as Opitutae, Chlamydiae. Verrucomicrobiae subdivision 3, Betaproteobacteria and Nitrospira were also detected (Additional file 2). 77 sequences were classified as cyanobacteria-like, probably these are chloroplast sequences that originate from the plant components of the food (Additional file 2). Interestingly, most of the retrieved sequences belong to bacterial taxa that are known to be involved in vitamin production and food digestion (Table 2).

Phylogenetic diversity of 16S rRNA sequences retrieved from the GI tract content of common carp. Clasification the 17,641 reads was performed using the taxonomic annotations available in the Silfva SSURef database. The number of sequences (10log-transformed) belonging to each clade is indicated by the red circles.
Discussion
Almost all Fusobacterial 16S rRNA sequences, 8081 out of 8085, from the carp GI tract belonged to the genus Cetobacterium. Cetobacteria were not observed in most culture-dependent studies done on the GI tract microbiota of common carp (Sugita et al. 1990; Namba et al. 2007), only Tsuchiya et al. (2008) described the isolation and characterization of Cetobacterium somerae from the GI tract of five different fresh water fish, including carp. Cetobacterium was also shown to be present in the gut of zebrafish (Rawls et al. 2006), a cyprinid species closely related to common carp. Furthermore, Cetobacterium isolated from human faeces performed fermentation of peptides and carbohydrates (Finegold et al. 2003). It has also been shown that Cetobacterium can produce vitamin B12 (Tsuchiya et al. 2008). This can wel explain why carp do not have a dietary vitamin B12 requirement (Sugita et al. 1991). The combination of a fermentative metabolism together with vitamin production may explain the relevance of Cetobacterium sp. in the GI tract of carp.
Another well represented group within the obtained sequences were the Bacteroidetes (22% of obtained sequences), a phylum known for a fermentative metabolism and degradation of oligosaccharides derived from plant material (Van der Meulen et al. 2006). The Bacteroidetes sequences found could be divided into 4 major groups (Additional file 1): Marinilabiaceae (or Cytophaga, 13%), Porphyromonadaceae (39%), Bacteroidaceae (15%) and Bacteroidales_incertae_sedis (33%). All Marinilabiaceae sequences belonged to the same group: the Anaerophaga. This relatively newly discovered group of bacteria includes strictly anaerobic, chemo-organotrophic, fermentative bacteria (Denger et al. 2002). These bacteria may play an important role in the fermentation of food in the GI tract of herbivorous carp since anaerobic fermentation is generally an important step in the digestion of plant material. Porphyromonadaceae are present in the GI tract of several organisms including human and pigs (Mulder et al. 2009). These bacteria can be pathogens but in this niche they are most probably involved in fermentation. By using labelled glucose, it has been shown that these bacteria are involved in saccharide fermentation (Li et al. 2009). Also the Sphingobacteria present could also be involved in oligosaccharide degradation since Sphingobacterium sp. TN19, an endosymbiont in insects, contains a xylanase encoding gene (Zhou et al. 2009). Xylanases are involved in the breakdown of xylan, a polysaccharide found in plant material. The presence of fermenting microorganisms is not suprising, since it has been shown that the GI microbiota of carp is able to ferment different oligosacharides (Kihara and Sakata 2002).
The obtained Planctomycete sequences (13% of classified sequences) could be divided into 9 groups (Additional file 1); Gemmata, Pirellula, Schlesneria and Zavarzinella were the most abundantly found groups. Gemmata and Pirellula are aerobic chemo-heterotrophs, Schlesneria are chemo-organotrophic facultative aerobes and Zavarzinella are aerobic heterotrophs. The presence of Planctomycetes has been shown before in gut microbiota of fish and other organisms (Ley et al. 2008; Rawls et al. 2006). The exact function of these bacteria in the GI tract is not clear, possibly these bacteria live from products of the metabolism of other bacteria. However, the relatively high abundance of Planctomycetes in close association with other organisms such as kelp, marine sponges and prawn (Bengtsson and Ovreas 2010; Pimental-Elardo 2003; Fuerst et al. 1997; Lahav et al. 2009) suggests a more important role. Possibly, these bacteria are involved in the metabolism of complex compounds. In a recent study, in which the close association of Planctomycetes with the brown seeweed kelp (Laminaria hyperborea) was investigated, it was hypothesized that these bacteria are degraders of sulfated polysacharides produced by kelp (Bengtsson and Ovreas 2010). The organisms found in the biofilm at the plant's surface were mainly members of the lineage Pirellulae (which includes Pirellula, Rhodopirellula and Blastopirellula). The genome sequence of Rhodopirellula baltica SH1 revealed many genes involved in the breakdown of sulfated polysaccharides (Glockner et al. 2003). Possibly, the heterotrophic Planctomycetes found in carp gut confer a similar ability of polysaccharide breakdown to the host. Furthermore, a separate lineage within the Planctomycetes, the anammox bacteria, were present in the carp gut (Figure 1). These anaerobic bacteria, described before in fish gut (Lahav et al. 2009), are involved in nitrogen cycling. Together with the Nitrosomonas and Nitrospira species (also present within the obtained sequences, Figure 1), ammonium can be converted into dinitrogen gas. The removal of nitrogenous compounds from aquaculture systems is one of the most important challenges in aquaculture. The presence of nitrogen cycling bacteria in fishes could offer new in situ solutions for the removal of nitrogen from aquaculture systems.
The Gammaproteobacteria sequences found could be classified as bacteria that are known members of the GI microbiota of many organisms including fish (Wu et al. 2010; Lee et al. 2009). Most Gammaproteobacteria (Additional file 1) found in carp belonged to the Aeromonas group. Members of the genus Aeromonas are mainly distributed in freshwater and sewage, often in association with aquatic animals (Cahill 1990; Sugita et al. 1995). They can cause a diverse spectrum of diseases in both warm- and cold-blooded animals but they also appear to be aquatic envrionments including in fish intestines (Sugita et al. 1995). Other abundantly present members among the Gammaproteobacterial sequences were the genera Enterobacterium and Vibrio. Enterobacterium spp. are widespread in GI tracts of various organisms (Wu et al. 2010), whereas Vibrio sp. are commonly found in aquaeous environments, aquaculture systems and in association with eukaryotes (Wu et al. 2010; Thompson et al. 2004). This phylum also contains Plesiomonas and Acinetobacter species that have been found in carp before (Sugita et al. 1991; Cahill 1990). Furthermore, the presence of high number Proteobacteria has also been shown for zebrafish, which is closely related to carp (Rawls et al. 2006). Also in other fish belonging to the Cyprinidae members of the Gammaproteobacteria (Enterobacter and Citrobacter species) were found (Ray et al. 2010). Enterobacter and Citrobacter species isolated from the GI tract of Indian carp (Cyprinidae) were shown to produce amylase, cellulase and protease (Ray et al. 2010), which indicates that these bacteria can be actively involved in the digestion of food in carp guts.
Another abundant phylum within our amplicon sequences were the Verrucomicrobiae (including subdivision 3 and 4 (Optitiae)). Verrucomicrobiae species are most commonly found in aquatic environments but are also known members of the gut microbiota in different organisms including seacucumbers (Echinodermata), termites and humans (Wagner and Horn 2006). These bacteria seem to be well adapted to live with eukaryotes, since the genome of some verrucomicrobial species contain a protein secretion system which mediates interactions between eukaryotic and bacterial cells (Wagner and Horn 2006). Verrucomicrobiae usually have an aerobic or obligate anaerobic fermentative metabolism (Schlesner et al. 2006) and could also play a role in the metabolism of plant beta glycans in carp GI tract. Indeed, Pedosphaera parvula Ellin514 (Verrucomicrobia subdivision 3) contains a cellulase in its genome (Kant et al. 2011). Ruminants and postgastric fermenters depend on bacteria containing this gene for the fermentation of plant material in which cellulose is converted to β-glucose. Various fish species do have a cellulase activity in their guts (Saha and Ray 1998; Saha et al. 2006; Ray et al. 2010) which decreases after antibiotic treatments (Saha and Ray 1998), indicating that the GI microbiota is responsible for this activity.
Clostridia and Bacilli, both present in the microbiota of the sampled fish (Figure 1), are members of the phylum Firmicutes. Representative genera of this phylum, including Clostridium, Bacillus, Streptococcus and Staphylococcus spp., have been shown in the microbiota of fish before (Navarrete et al. 2009; Rawls et al. 2006; Ray et al. 2010; Sugita et al. 1990). Gut isolates belonging to the Firmicutes fermented various carbon sources (Ray et al. 2010), again implicating a role in the utilization of plant materials.
To our knowledge, this is the first detailed analysis of the microbiota of common carp by high throughput sequencing. Our culture independent investigation of the microbial flora of the GI tract gives a more reliable and more complete characterization of the diversity of compared to other studies. Furthermore, great similarities between the microbiota in carp and zebrafish (a closely related fish species) were shown (Roeselers et al. 2011). The GI microbiota is important for the health of the animal and therefore this study could be relevant for aquaculture. Furthermore, the presence of different nitrogen cycling bacteria in the GI tract of fish could offer new possibilities in the removal of nitrogen compounds in aquaculture. The microbiota of the GI tract plays an important role in the digestion and chemical processing of the food as exemplified by the large number of bacteria involved in vitamin production and fermentation of saccharides and beta-glycans (cellulose, hemicellulose) (Table 2). The presence of many different types of bacteria in the herbivorous carp could be predicted since it has been shown that eukaryotes with an herbivorous diet have a higher microbial diversity (Ley et al. 2008). However, the carp in our study were fed commercially available food with high protein and low plant content. According to their GI microbiota, these fish are very well able to adapt to a more herbivorous diet and this is probably also the case for other cultured fish. Therefore it could be possible to lower the amount of fish meal, one of the major components of fish food, in the food for these fish. Furthermore, it shows that the gut microbes are probably important in the protection of the host against pathogens which should be taken into consideration in aquaculture where a lot of antibiotics are used (Cabello 2006). It is known that antibiotics have a negative effect on the microbial community in the gut of human (Dethlefsen et al. 2008) and this is possibly also the case for fish. The routinely use of antibiotics may be harmful for the animal. A better knowledge about the microbiota in fish guts is important; it can lead to a better health of cultured fish and therefore to a more efficient fish culture.
References
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA: Combination of 16S Ribosomal-RNA-targeted oligonucleotide probes with flow-cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 1990, 56: 1919–1925.
Bengtsson MM, Ovreas L: Planctomycetes dominate biofilms on surfaces of the kelp Laminaria hyperborea . BMC Microbiol 2010, 10: 1–12. 10.1186/1471-2180-10-1
Buddington RK, Diamond JM: Pyloric ceca of fish - a new absorptive organ. Am J Physiol 1987, 252: G65-G76.
Cabello FC: Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Micobiol 2006, 8: 1137–1144.
Cahill MM: Bacterial flora of fishes - a review. Microb Ecol 1990, 19: 21–41. 10.1007/BF02015051
Daims H, Bruhl A, Amann R, Schleifer KH, Wagner M: The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 1999, 22: 434–444. 10.1016/S0723-2020(99)80053-8
Denger K, Warthmann R, Ludwig W, Schink B: Anaerophaga thermohalophila gen. nov., sp nov., a moderately thermohalophilic, strictly anaerobic fermentative bacterium. Int J Syst Evol Microbiol 2002, 52: 173–178.
Dethlefsen L, Huse S, Sogin ML, Relman DA: The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. Plos Biol 2008, 6: 2383–4000.
Detkova EN, Zaichikova MV, Kevbrin VV: Physiological and biochemical properties of the alkaliphilic anaerobic hydrolytic bacterium Alkaliflexus imshenetskii . Microbiol 2009, 78: 273–279. 10.1134/S0026261709030035
Finegold SM, Vaisanen ML, Molitoris DR, Tomzynski TJ, Song Y, Liu C, Collins MD, Lawson PA: Cetobacterium somerae sp nov from human feces and emended description of the genus Cetobacterium . Syst Appl Microbiol 2003, 26: 177–181. 10.1078/072320203322346010
Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA: Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nature Rev Microbiol 2008, 6: 121–131. 10.1038/nrmicro1817
Fuerst JA, Gwilliam HG, Lindsay M, Lichanska A, Belcher C, Vickers JE, Hugenholtz JE: Isolation and molecular identification of planctomycete bacteria from postlarvae of the giant tiger prawn. Penaeus monodon . Appl Environ Microbiol 1997, 63: 254–262.
Glockner FO, Kube M, Bauer M, Teeling H, Lombardot T, Ludwig W, Gade D, Beck A, Borzym K, Heitmann K, Rabus R, Schlesner H, Amann R, Reinhardt R: Complete genome sequence of the marine Planctomycete Pirellula sp strain 1. Proc Natl Acad Sci USA 2003, 100: 8298–8303. 10.1073/pnas.1431443100
Hong PY, Hwang C, Ling F, Andersen GL, Lechevallier MW, Liu WT: Pyrosequencing analysis of bacterial biofilm communities in water meters of a drinking water distribution system. Appl Environ Microbiol 2010, 76: 5631–5635. 10.1128/AEM.00281-10
Huber I, Spanggaard B, Appel KF, Rossen L, Nielsen T, Gram L: Phylogenetic analysis and in situ identification of the intestinal microbial community of rainbow trout ( Oncorhynchus mykiss , Walbaum). J Appl Microbiol 2004, 96: 117–132. 10.1046/j.1365-2672.2003.02109.x
Jiang Y, Xie CX, Yang GG, Gong XL, Chen XJ, Xu LX, Bao BL: Cellulase-producing bacteria of Aeromonas are dominant and indigenous in the gut of Ctenopharyngodon idellus (Valenciennes). Aquac Res 2011, 42: 499–505. 10.1111/j.1365-2109.2010.02645.x
Juretschko S, Timmermann G, Schmid M, Schleifer KH, Pommerening-Roser A, Koops HP, Wagner M: Combined molecular and conventional analyses of nitrifying bacterium diversity in activated sludge Nitrosococcus mobilis and Nitrospira -like bacteria as dominant populations. Appl Environ Microbiol 1998, 64: 3042–3051.
Kant R, Van Passel MWJ, Sangwan P, Palva A, Lucas S, Copeland A, Lapidus A, Glavina Del Rio T, Dalin E, Tice H, Bruce D, Goodwin L, Pitluck S, Chertkov O, Larimer FW, Land ML, Hauser L, Brettin TS, Detter JC, Han S, de Vos WM, Janssen PH, Smidt H: Genome sequence of 'Pedosphaera parvula ' Ellin514, an aerobic verrucomicrobial isolate from pasture soil. J Bacteriol 2011, 193: 2900–2901. 10.1128/JB.00299-11
Kihara M, Sakata T: Production of short-chain fatty acids and gas from various oligosaccharides by gut microbes of carp ( Cyprinus carpio L.). in micro-scale batch culture. Comp Biochem Phys A 2002, 132: 333–340. 10.1016/S1095-6433(02)00029-6
Kip N, Dutilh BE, Pan Y, Bodrossy L, Neveling K, Kwint MP, Jetten MSM, Op den Camp HJM: Ultra deep pyrosequencing of pmoA amplicons confirms the prevalence of Methylomonas and Methylocystis in Sphagnum mosses from a Dutch peat bog. Environ Microbiol Reports 2011.
Kulichevskaya IS, Baulina OI, Bodelier PLE, Rijpstra WIC, Sinninghe Damste JS, Dedysh SN: Zavarzinella formosa gen. nov., a novel stalked, Gemmata -like planctomycete from a Siberian peat bog. Int J Syst Evol Microbiol 2009, 59: 357–364. 10.1099/ijs.0.002378-0
Kulichevskaya IS, Ivanova AO, Belova SE, Baulina OI, Bodelier PLE, Rijpstra WIC, Damste JSS, Zavarzin GA, Dedysh SN: Schlesneria paludicola gen. nov., sp nov., the first acidophilic member of the order Planctomycetales , from Sphagnum-dominated boreal wetlands. Int J Syst Evol Microbiol 2007, 57: 2680–2687. 10.1099/ijs.0.65157-0
Kysela DT, Palacios C, Sogin ML: Serial analysis of V6 ribosomal sequence tags (SARST-V6): a method for efficient, high-throughput analysis of microbial community composition. Environ Microbiol 2005, 7: 356–364. 10.1111/j.1462-2920.2004.00712.x
Lahav O, Bar Massada I, Yackoubov D, Zelikson R, Mozes N, Tal Y, Tarre S: Quantification of anammox activity in a denitrification reactor for a recirculating aquaculture system. Aquaculture 2009, 288: 76–82. 10.1016/j.aquaculture.2008.11.020
Lee C, Kim J, Hwang K, Hwang S: Fermentation and growth kinetic study of Aeromonas caviae under anaerobic conditions. Appl Microbiol Biotechnol 2009, 83: 767–773. 10.1007/s00253-009-1983-y
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schleger ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI: Evolution of mammals and their gut microbes. Science 2008, 320: 1647–1651. 10.1126/science.1155725
Li TL, Mazeas L, Sghir A, Leblon G, Bouchez T: Insights into networks of functional microbes catalysing methanization of cellulose under mesophilic conditions. Environ Microbiol 2009, 11: 889–904. 10.1111/j.1462-2920.2008.01810.x
Moya A, Pereto J, Gil R, Latorre A: Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat Rev Genet 2008, 9: 218–229. 10.1038/nrg2319
Mulder IE, Schmidt B, Stokes CR, Lewis M, Baily M, Aminov RI, Prosser JI, Gill BP, Pluske JR, Mayer CD, Musk CC, Kelly D: Environmentally-acquired bacteria influence microbial diversity and natural inate immune responses at gut surfaces. BMC Biology 2009, 7.
Namba A, Mano N, Hirose H: Phylogenetic analysis of intestinal bacteria and their adhesive capability in relation to the intestinal mucus of carp. J Appl Microbiol 2007, 102: 1307–1317. 10.1111/j.1365-2672.2006.03204.x
Navarrete P, Espejo RT, Romero J: Molecular analysis of microbiota along the digestive tract of juvenile Atlantic salmon ( Salmo salar L.). Microb Ecol 2009, 57: 550–561. 10.1007/s00248-008-9448-x
Neef A, Amann R, Schlesner H, Schleifer KH: Monitoring a widespread bacterial group: in situ detection of planctomycetes with 16S rRNA-targeted probes. Microbiol 1998, 144: 3257–3266. 10.1099/00221287-144-12-3257
Pimental-Elardo S, Wehrl M, Friedrich AB, Jensen PR, Hentschel U: Isolation from Planctomycetes from Aplysina sponges. Aquat Microb Ecol 2003, 33: 239–243.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, et al.: A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464: 59–65. 10.1038/nature08821
Rawls JF, Mahowald MA, Ley RE, Gordon JI: Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006, 127: 423–433. 10.1016/j.cell.2006.08.043
Rawls JF, Samuel BS, Gordon JI: Gnotobiotic zebrafish reveal evolutionarily conserved responses to the gut microbiota. Proc Natl Aca Sci USA 2004, 101: 4596–4601. 10.1073/pnas.0400706101
Ray AK, Roy T, Mondal S, Ringo E: Identification of gut-associated amylase, cellulase and protease-producing bacteria in three species of Indian major carps. Aquac Res 2010, 41: 1462–1469.
Ringo E, Gatesoupe FJ: Lactic acid bacteria in fish: a review. Aquaculture 1998, 160: 177–203. 10.1016/S0044-8486(97)00299-8
Ringo E, Olsen RE, Mayhew TM, Myklebust R: Electron microscopy of the intestinal microflora of fish. Aquaculture 2003, 227: 395–415. 10.1016/j.aquaculture.2003.05.001
Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF: Evidence for a core gut microbiota in the zebrafish. ISME J 2011, 1–14.
Saha AK, Ray AK: Cellulase activity in rohu fingerlings. Aquac Int 1998, 6: 281–291. 10.1023/A:1009210929594
Saha S, Roy RN, Sen SK, Ray AK: Characterization of cellulase-producing bacteria from the digestive tract of tilapia, Oreochromis mossambica (Peters) and grass carp, Ctenopharyngodon idella (Valenciennes). Aquac Res 2006, 37: 380–388. 10.1111/j.1365-2109.2006.01442.x
Schlesner H, Jenkins C, Staley JT: The phylum Verrucomicrobia : A phylogenetically heterogenous bacterial group. Prokaryotes 2006, 7: 881–896.
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J: Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 1999, 65: 4799–4807.
Sugita H, Miyajima C, Deguchi Y: The vitamin B12 producing ability of the intestinal microflora of fresh-water fish. Aquaculture 1991, 92: 267–276.
Sugita H, Miyajima C, Kobayashi H, Deguchi Y: Distribution of microflora in the intestinal tract of Carp Cyprinus carpio . Nippon Suisan Gakkaishi 1990, 56: 1133–1138. 10.2331/suisan.56.1133
Sugita H, Tanaka K, Yoshinami M, Deguchi Y: Distribution of Aeromonas Species in the intestinal tracts of river fish. Appl Environ Microbiol 1995, 61: 4128–4130.
Sugita H, Shibuya K, Shimooka H, Deguchi Y: Antibacterial abilities of intestinal bacteria in freshwater cultured fish. Aquaculture 1997, 145: 195–203.
Thompson FL, Iida T, Swings J: Biodiversity of Vibrios . Microbiol Mol Biol Rev 2004, 68: 403–431. 10.1128/MMBR.68.3.403-431.2004
Tsuchiya C, Sakata T, Sugita H: Novel ecological niche of Cetobacterium somerae , an anaerobic bacterium in the intestinal tracts of freshwater fish. Lett Appl Microbiol 2008, 46: 43–48.
Vahjen W, Pieper R, Zentek J: Bar-coded pyrosequencing of 16S rRNA gene amplicons reveals changes in ileal porcine bacterial communites due to high dietary zinc intake. Appl Environ Microbiol 2010, 76: 6689–6691. 10.1128/AEM.03075-09
Van der Meulen R, Makras L, Verbrugghe K, Adriany T, De Vuyst L: In vitro kinetic analysis of oligofructose consumption by Bacteroides and Bifidobacterium spp. indicates different degradation mechanisms. Appl Environ Microbiol 2006, 72: 1006–1012. 10.1128/AEM.72.2.1006-1012.2006
Wagner M, Horn M: The Planctomycetes, Verrucomicrobia, Chlamydiae and sister phyla comprise a superphylum with biotechnological and medical relevance. Curr Opinion Biotechnol 2006, 17: 241–249. 10.1016/j.copbio.2006.05.005
Wang J, Jenkins C, Webb RI, Fuerst JA: Isolation of Gemmata -like and Isosphaera -like Planctomycete bacteria from soil and freshwater. Appl Environ Microbiol 2002, 68: 417–422. 10.1128/AEM.68.1.417-422.2002
Wang Q, Garrity GM, Tiedje JM, Cole JR: Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007, 73: 5261–5267. 10.1128/AEM.00062-07
Wu SG, Gao TH, Zheng YZ, Wang WW, Cheng YY, Wang GT: Microbial diversity of intestinal contents and mucus in yellow catfish ( Pelteobagrus fulvidraco ). Aquaculture 2010, 303: 1–7. 10.1016/j.aquaculture.2009.12.025
Zhou JP, Huang HQ, Meng K, Shi PJ, Wang YR, Luo HY, Yang PL, Bai YG, Zhou ZG, Yao B: Molecular and biochemical characterization of a novel xylanase from the symbiotic Sphingobacterium sp TN19. Appl Microbiol Biotechnol 2009, 85: 323–333. 10.1007/s00253-009-2081-x
Zhou JZ, Bruns MA, Tiedje JM: DNA recovery from soils of diverse composition. Appl Environ Microbiol 1996, 62: 316–322.
Acknowledgements
We would like to thank Alexander Hoischen and Nienke Wieskamp from the Department of Human Genetics (Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands) for their help with the Roche 454 pyrosequencing. Bas E. Dutilh is supported by the Dutch Science foundation (NWO) Horizon project (050-71-058) and by NWO Veni grant (016.111.075). Mike Jetten and Maartje van Kessel are supported by an ERC grant (232937). Roche 454 pyrosequencer was obtained with a grant from the Dutch Science Foundation (911-08-025).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Electronic supplementary material
13568_2011_35_MOESM2_ESM.PDF
Additional file 2: Details of the phylogenetic composition of the bacterial sequences. Supplemental Table S1. (PDF 21 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
van Kessel, M.A., Dutilh, B.E., Neveling, K. et al. Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Expr 1, 41 (2011). https://doi.org/10.1186/2191-0855-1-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2191-0855-1-41