
Abstract
Objective
Exposure to hyperoxemia and hypoxemia is common in out-of-hospital cardiac arrest (OHCA) patients following return of spontaneous circulation (ROSC), but its effects on neurological outcome are uncertain, and study results are inconsistent.
Methods
Exploratory post hoc substudy of the Target Temperature Management (TTM) trial, including 939 patients after OHCA with return of spontaneous circulation (ROSC). The association between serial arterial partial pressures of oxygen (PaO2) during 37 h following ROSC and neurological outcome at 6 months, evaluated by Cerebral Performance Category (CPC), dichotomized to good (CPC 1–2) and poor (CPC 3–5), was investigated. In our analyses, we tested the association of hyperoxemia and hypoxemia, time-weighted mean PaO2, maximum PaO2 difference, and gradually increasing PaO2 levels (13.3–53.3 kPa) with poor neurological outcome. A subsequent analysis investigated the association between PaO2 and a biomarker of brain injury, peak serum Tau levels.
Results
Eight hundred sixty-nine patients were eligible for analysis. Three hundred patients (35%) were exposed to hyperoxemia or hypoxemia at some time point after ROSC. Our analyses did not reveal a significant association between hyperoxemia, hypoxemia, time-weighted mean PaO2 exposure or maximum PaO2 difference and poor neurological outcome at 6-month follow-up after correction for co-variates (all analyses p = 0.146–0.847). We were not able to define a PaO2 level significantly associated with the onset of poor neurological outcome. Peak serum Tau levels at either 48 or 72 h after ROSC were not associated with PaO2.
Conclusion
Hyperoxemia or hypoxemia exposure occurred in one third of the patients during the first 37 h of hospitalization and was not significantly associated with poor neurological outcome after 6 months or with the peak s-Tau levels at either 48 or 72 h after ROSC.
Similar content being viewed by others


Background
Survival after out-of-hospital cardiac arrest (OHCA) has improved over the last two decades and patients admitted to critical care are frequently discharged alive with increasingly good neurological outcome [1,2,3,4]. Following OHCA, patients regularly suffer from post cardiac arrest syndrome including symptoms of anoxic brain injury and reperfusion-related damage [5, 6]. In recent years, the optimal oxygen content in the post-cardiac arrest period has been a matter of debate since ventilation with high concentrations of oxygen after return of spontaneous circulation (ROSC) has been linked to worse outcome and an increased degree of cerebral neuronal damage in experimental cardiac arrest models [7,8,9,10]. In healthy volunteers, hyperoxemia decreases cerebral blood flow [11, 12], whilst hypoxemia is associated with the opposite effect [13]. Hyperoxemia also augments the production of reactive oxygen species (ROS), increases lipid oxidation and amplifies the inflammatory reaction in the brain during reperfusion after circulatory arrest [10, 14].
Clinical studies evaluating the impact of hyperoxemia and hypoxemia on neurological outcome after ROSC have shown inconsistent results when compared to the preclinical cardiac arrest models [15,16,17,18,19]. Two large landmark studies published in 2010 and 2011 limit their analysis to single or very few partial pressure of oxygen (PaO2) values in the post cardiac arrest phase [15, 17]. Although more recent studies have analysed multiple PaO2 values over time, so far, human studies continue to differ regarding patient selection, the use of targeted temperature management, outcome measurement, and methods of analysing blood gas and often lack a pre-defined sampling protocol [18,19,20,21,22,23].
We conducted this exploratory substudy of the prospectively collected blood-gas measurements in the Target Temperature Management after Out-of-Hospital Cardiac Arrest (TTM) trial in order to describe the fluctuation in PaO2 in the post-cardiac arrest phase and the association of hyperoxemia and hypoxemia with neurological outcome after 6 months [24]. An analysis of peak levels of serum Tau (s-Tau), a novel marker for neuronal injury, at either 48 or 72 h after ROSC and its association with PaO2 was subsequently performed to validate our results.
Methods
The present study is a post hoc analysis of data acquired from 939 unconscious (Glasgow Coma Scale (GCS) < 8) adult (18 years or older) OHCA patients included in the TTM trial conducted between November 2010 and January 2013. Ethical committees in each participating country approved the TTM trial protocol, and informed consent was waived or obtained from all participants or relatives according to national legislations, in line with the Helsinki declaration.
The TTM trial was a randomized clinical trial recruiting patients in 36 intensive care units in Europe and Australia, designed to evaluate two target temperature regimes, 33 °C (n = 473) and 36 °C (n = 466), in unconscious adult OHCA patients after sustained ROSC [24]. Target temperature management was commenced at inclusion into the study. After 28 h, the patients were rewarmed to 37 °C core temperature over a period of 8 h and mandatory sedation was discontinued at the end of the 36-h intervention period. Resuscitation data was reported according to the Utstein style protocol [25]. Follow-up was obtained by structured face-to-face interview with the patient (86%) or structured telephone interview with the patient, care provider, or relative (14%) by a blinded assessor. The TTM trial did not show a significant difference between the two temperature groups in overall mortality at the end of the trial or in the composite of poor neurologic function or death at 180 days [24].
Baseline, intervention-related and physiological variables as demographic characteristics, comorbidities, pre-hospital and admission data, characteristics of the cardiac arrest, and baseline laboratory analyses were prospectively collected. A complete arterial blood gas analysis was performed in all patients at admission to hospital (T-1), start of intervention (T0), and after 4 (T4), 12 (T12), 20 (T20), 28 (T28), 32 (T32), and 36 (T36) hours post inclusion. In order to include the admission blood gas analysis, obtained after ROSC but before inclusion, we timed this analysis to 1 h before randomization in the statistical analysis of the present study. All arterial blood gases were collected according to an a priori designed protocol and analysed using the alpha-stat method only [26]. Median time from ROSC to inclusion was 133 (interquartile range 83–188) min. For the present study, PaO2 and FiO2 data were assessed and manually corrected for registration shortcomings by two authors in consensus (FE and NN). Details of the correction process are described in Additional file 1: Methods. Patient identification data were pseudomized. Patients who demised before the end of the intervention period were excluded from the present study to allow for a homogenous exposure period to PaO2. We chose to orientate on the STROBE Statement style for the study manuscript [27].
Outcome
The primary outcome was overall neurological function at follow-up 6 months after cardiac arrest, assessed by Cerebral Performance Category (CPC) and dichotomized into good and poor with CPC 1 (good cerebral performance) and CPC 2 (mild neurological impairment) considered as good outcome, and CPC 3 to 5 as poor outcome with CPC 3–4 representing severe neurological impairment or vegetative state and CPC 5 death [28, 29]. In addition, we used the serum levels of Tau as a surrogate marker of neuronal injury in a subgroup analysis.
Definition of hyperoxemia, hypoxemia, and normoxemia
We a priori defined hyperoxemia as a PaO2 > 40 kPa and hypoxemia as a PaO2 < 8 kPa in accordance with previous studies [15, 17, 23]. All values not defined as hyperoxemia or hypoxemia were defined as normoxemia.
Primary analysis
Absolute oxygen levels
We divided our cohort according to the most extreme PaO2 of the individual patient into three groups: hyperoxemia, hypoxemia, and normoxemia. Thereafter, we compared the outcome of the hyperoxemia and hypoxemia group with normoxemia, followed by the comparison of each group’s outcome with the outcome of a composite group of the remaining patients.
Threshold analysis
In order to identify a possible PaO2 threshold value for the onset of the association of PaO2 and poor neurological outcome, we performed multivariable regression models with gradually increasing PaO2 levels.
Secondary analyses
Oxygen exposure over time
To evaluate the cumulative PaO2 exposure over time, we formed a PaO2 over time integral from which we derived the time-weighted mean PaO2 (PaO2-TWM). Primarily, we evaluated the association of the PaO2-TWM from T-1 to T36 with outcome and, secondarily, from T-1 to T12 in order to identify effects of early hyperoxemia or hypoxemia.
Oxygen pressure difference
The difference between the most extreme PaO2 values during the observation time was calculated for each patient. The association of this maximum PaO2 difference with neurological outcome was analysed.
For an illustration of our primary and secondary analyses, see Additional file 1: Figure S1.
Association between PaO2 and s-Tau
We used the cohort of 689 patients of the TTM trial substudy by Mattsson et al. [30] and evaluated the association of our multivariable PaO2 models with the highest level of s-Tau at either 48 or 72 h.
Sensitivity analyses
Sensitivity analyses were performed with a complete case cohort of 468 patients with blood gas samples registered from all measuring points and an all-patient cohort of 922 patients including also those not surviving the full exposure period. Seventeen patients had insufficient data for analysis and were not included in the sensitivity analyses. Subsequently, we performed a sensitivity analysis of our primary analysis cohort including FiO2 as an additional co-variable and an all-cause mortality analysis.
Statistics
Proportions are presented as percentages and continuous variables as mean with standard deviations (SD). Missingness was assumed at random [31]. Since the number of missing values exceeded 5%, we employed multiple imputation to compensate for the missing data [32]. Predictive mean matching, utilizing available non-missing values as well as available TTM trial study variables on the same individual and variables obtained from matching patients, was used. Twenty imputations were generated by chained equations and assessed by graphical methods. For each imputed dataset, PaO2 was evaluated using summary measures and regression models. The estimates from the regression for each imputed sample were combined into one estimate with 95% confidence intervals (CI) including the uncertainty from the multiple imputations based on Rubin’s rule [33]. Missing outcome data and death before end of intervention time entailed exclusion from analysis and was not compensated for.
Logistic regression analysis was used to assess the association between PaO2 and neurological outcome at 6-month follow-up. Results of our multivariable regression models are presented as odds ratios (OR) with 95% CI, OR describing continuous data present changes in one unit; for PaO2 1 kPa, for pH one unit. All regression analyses were adjusted for pre-specified and in the context of OHCA relevant co-variates: age (years), sex (male/female), chronic heart failure (yes/no), asthma/chronic obstructive pulmonary disease (yes/no), cardiac arrest witnessed (yes/no), bystander cardiopulmonary resuscitation (yes/no), time to ROSC (minutes), Glasgow Coma Scale-Motor Score (1 vs 2–5), circulatory shock on admission (yes/no), first rhythm shockable (yes/no), and pH (units). We pooled the two temperature groups (33 °C and 36 °C) as there was no significant interaction between the PaO2 groups and the two temperature groups.
For the s-Tau analysis, multivariable linear regression was used and the depending variables were adjusted for the co-variables and interaction analyses as described above. After transforming the s-Tau values to a logarithmic scale, they were used as dependent variable in the linear regression analyses. The multiplicative change in s-Tau was depicted by the regression coefficients obtained for each independent variable after back transformation. Linear regression results are presented as beta-coefficient estimates with 95% CI.
The primary analyses were performed on a multiple imputation cohort as described above. The complete case and all-patient cohorts were used for sensitivity analysis. We regarded a two-sided P value < 0.05 as significant. Analyses were conducted using IBM SPSS statistics for Windows (version 22.0, Armonk NY) and R: A language and environment for statistical Computing (version 3.3.3 R Foundation for Statistical Computing, Vienna, Austria). The R package mice was used for multiple imputations [34].
Results
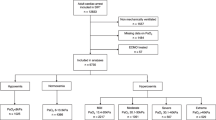
Data for this explorative substudy was derived from 939 patients randomized in the TTM trial. We excluded patients who did not survive the intervention period (n = 62) and patients with no PaO2 data (n = 2) and missing neurological outcome data at 6-month follow-up (n = 6), which left 869 patients (92.5%) eligible for analysis (Fig. 1). Baseline characteristics for all the patients included and the different exposure groups are presented in Table 1. Overall, 441 patients (50.7%) had a good outcome whereas 428 patients (49.3%) had a poor outcome (Table 2). Of 869 patients, 384 (44.2%) died. Nine hundred eighteen of 6952 (13.2%) PaO2-measuring points were missing (Additional file 1: Table S1). At hospital admission, mean PaO2 was 25.1 (SD 17.0) kPa and diminished gradually in the temperature and outcome groups over time (Fig. 2a and b). In our primary analysis, we found that 199 of 869 (22.9%) patients were exposed to hyperoxemia at some point after admission to hospital, 112 (12.9%) were exposed to hypoxemia, 11 (1.3%) experienced both hyper- and hypoxemia, and 569 (65.5%) remained normoxic throughout. One hundred ninety-seven of 199 exposures to hyperoxemia occurred within the first 5 h after admission to hospital. Detailed post ROSC PaO2 data of the primary analysis groups are displayed in Table 3. In our secondary analyses, we found that PaO2-TWM from T-1 to T12 was mean 17.2 (SD 5.5) kPa whilst PaO2-TWM for all measurements was mean 14.5 (SD 3.2) kPa. The median maximum PaO2 difference was 14.3 kPa, with an interquartile range (IQR) of 8.6 to 27.2 kPa. For this study, we pooled the patients from the two TTM trial temperature groups into one cohort, which was feasible since the term of interaction analysis between the PaO2 exposure groups and TTM group affiliation (33 °C or 36 °C) showed no significant results (pinteraction = 0.537–0.972) (Additional file 1: Table S2).

Patient selection pathway. TTM, targeted temperature management. TTM group, 33 or 36 °C core body temperature derived from the TTM trial [24]. n, number of patients. The diagram does not display the selection pathway for the s-Tau analysis

Boxplots depicting the distributional characteristics of PaO2 at 8 measurement points from admission to hospital to the end of intervention time for the TTM 33 and TTM 36 groups (a) and the investigated combined cohort dichotomized into good and poor outcome (b). Boxplot values are displayed as median, 25% quartiles from median and range. TTM, target temperature management. PaO2, partial arterial oxygen pressure. kPa, kilopascal. Core body temperature, 33 °C or 36 °C. White circle denotes the outliers. Asterisk denotes extreme outliers
Primary outcome analyses
The absolute oxygen pressure analysis did not show a significant association between hyperoxemia versus normoxemia OR 1.24 (0.81, 1.89) p = 0.314 or hyperoxemia versus no hyperoxemia OR 1.28 (0.86, 1.91) p = 0.219 and poor neurological outcome. We also found no association with poor outcome in the hypoxemia exposure groups: hypoxemia versus normoxemia OR 1.06 (0.60, 1.85) p = 0.847 and hypoxemia versus no hypoxemia OR 1.13 (0.66, 1.91) p = 0.647. Detailed multivariate models of the hyperoxemia and hypoxemia analyses are presented in Tables 4 and 5. Figure 3 shows the adjusted ORs for poor neurological outcome of the PaO2 threshold analysis. We were not able to identify a PaO2 threshold value significantly associated with the onset of poor neurological outcome across gradually increasing PaO2 levels.

Forrest plot showing the adjusted OR’s (bullet points) with 95% CI’s (horizontal lines) for poor neurological outcome according to Cerebral Performance Category (CPC) for different PaO2 threshold values. OR, odds ratio. CI, confidence interval. PaO2, partial pressure of oxygen. kPa, kilopascal. CPC, cerebral performance category. CPC 1-2, good outcome, CPC 3-5, poor outcome. ORs and CIs are presented on a logarithmic scale. OR above 1.0 indicates worse outcome above the PaO2 threshold and OR below 1.0 indicates better outcome above the PaO2 threshold
Secondary outcome analyses
In our PaO2-TWM analyses, we did not find an association with poor neurological outcome, either for the complete exposure period (T-1 to T36), OR 1.03 (0.97, 1.09) p = 0.375, or for the early exposure period (T-1 to T12), OR 1.02 (0.98, 1.05) p = 0.288. We were also not able to show an association between maximum PaO2 difference and poor neurological outcome OR 1.01 (0.99, 1.02) p = 0.146.
Association between PaO2 and s-Tau
Of the 689 patients in the s-Tau analysis, 64 were excluded as per our eligibility criteria and 36 had missing peak s-Tau levels at 48 or 72 h after ROSC, leaving 589 patients for analysis. Table 6 displays the detailed multivariable models of PaO2 and s-Tau. We did not find statistically significant associations between PaO2 and highest s-Tau at either 48 or 72 h after ROSC (p = 0.198–0.687).
Sensitivity analyses
The analysis of the complete case cohort (n = 468) revealed non-significant results, in line with our multiple imputation cohort used for our primary analyses (p = 0.057–0.811). The all-patient cohort (n = 922), including also the patients dying during the exposure period, showed non-significant results (p = 0.060–0.979). The results with all-cause mortality instead of neurological function as the outcome were non-significant and similar to our primary analyses (p = 0.307–0.969). Adding FiO2 as a confounder to our primary analyses cohort did not significantly alter outcome (p = 0.102–0.793). Details of the sensitivity analyses are displayed in Additional file 1: Tables S3–S6. FiO2 and PaO2 were weakly correlated (r = − 0.23).
Discussion
In this exploratory post hoc substudy of the TTM trial, we investigated the association of PaO2 outside normal ranges in the post cardiac arrest phase with poor neurological outcome 6 months after OHCA and peak s-Tau at either 48 or 72 h after ROSC. We found that 35% of patients were exposed to hyperoxemia or hypoxemia following ROSC. We did not find statistically significant associations between exposure to hyperoxemia or hypoxemia with poor neurological outcome at 6-month follow-up. The PaO2-TWM and maximum PaO2 difference analyses did not show an association with neurological outcome. Our findings did not indicate a PaO2 threshold value associated with the onset of poor neurological outcome. We were not able to detect an association between PaO2 and peak s-Tau at either 48 or 72 h after ROSC.
Our data shows that exposure to PaO2 outside the normal range, especially hyperoxemia, was common and most pronounced in the first hours after admission. This probably reflects clinical practice to continue ventilation with high FiO2 after ROSC, thus causing a propensity for early hyperoxemia, despite recommendations to titrate FiO2 to a target SpO2 of 94–98% to avoid hyperoxemia [35].
A recent observational multicentre study by Roberts et al. [23] found that exposure to early hyperoxemia, defined by two protocol-directed blood gas samples within the first 6 h after ROSC, was independently associated with poor neurological outcome at hospital discharge, corroborating results from a previous retrospective study by the same group [15]. Additionally, they identified PaO2 ≥ 40kPa as the threshold value for the association between poor neurological outcome and PaO2. Our primary hyperoxemia analyses are comparable since hyperoxemia exposure occurred almost exclusively early and the cut-off values of our threshold analysis are akin, but we did not confirm the findings by Roberts et al. In contrast to our study, their cohort was smaller, including in- and out-of-hospital cardiac arrest patients, follow-up was shorter, and exposure to hyperoxemia was more common (38% versus 23%).
Oxygen exposure over the first 24 h in a cohort of 409 OHCA patients treated with hypothermia to 33 °C was investigated in a prospective observational study by Vaahersalo et al. [22], showing in agreement with our time-weighted mean analyses, no association between oxygen exposure over time and neurological outcome. However, several aspects of this study make direct comparison with our study difficult; in the study by Vaahersalo et al., only 6% of patients were exposed to a PaO2 > 40 kPa, and blood gases were not obtained by protocol and analysed by pH and alpha-stat methods instead of alpha-stat only.
The physiological cerebral vascular response to hyperoxemia is vasoconstriction, alteration of cerebral blood flow (CBF), and subsequently reduced regional oxygen delivery [36, 37]. A study by Voicu et al. indicates that this mechanism might be impaired in a proportion of OHCA patients [38]. Two blood gas management strategies, alpha-stat versus pH-stat, were investigated and revealed an absence of change in CBF velocities between the two modalities in non-survivors compared to survivors. Furthermore, the non-survivor group also showed no difference in jugular vein oxygen saturation, arteriojugular oxygen content, and cerebral oxygen extraction. This study identifies a subgroup of OHCA patients with failed cerebral vascular autoregulation and increased risk for secondary brain injury due to possible cerebral hyperperfusion and unregulated hyperoxemia exposure. Similar subgroups have been described in previous studies [39, 40], highlighting the heterogeneity of OHCA cohorts.
In a hypoxic state, neurons are unable to utilize oxidative phosphorylation and are forced to resort to glycolysis for ATP production which is a short lived rescue mechanism before the onset of neuronal cell injury and death [41]. This primary, hypoxic injury occurs during OHCA and is together with the secondary, reperfusion injury that begins immediately after ROSC regarded as one of the major contributors to the post cardiac arrest syndrome [6, 13], whilst the effects of prolonged hypoxemia after ROSC are undetermined. In a non-OHCA cohort, well-trained acclimatized climbers were fully functional at PaO2 levels as low as 2.54 kPa [42], and in a canine model EEG readings were normal at PaO2 values of 2.6 kPa, provided that no CBF impairment was present [43]. Mechanisms of physiological acclimatization to hypoxia and unimpaired CBF are implausible in adult OHCA patients due to increased age and co-morbidities. Hence, the PaO2 threshold for the onset of hypoxic neuronal demise is presumably above the presented extreme values, but also likely to be significantly lower than the 8 kPa cut off employed in our and previous investigations, which provides an explanation for the deviating results of studies investigating hypoxemia [15, 17, 20, 21].
In the present study, we did not find an association between PaO2 and peak s-Tau levels, which supports the lack of association between PaO2 and neurological outcome. S-Tau is in the context of OHCA, a novel biomarker for neuronal injury, and the 48- and 72-h peak level is a significantly better clinical predictor for neurological outcome after 6 months than neuron-specific enolase (NSE) or clinical information alone [30].
In summary, clinical observational studies investigating hyperoxemia and hypoxemia differ in outcome, possibly due to patient selection, sampling and analysis methodology, and random error. Observational studies are not showing associations with a better outcome after hyperoxemia or hypoxemia exposure, but clinical randomized trials randomizing patients to PaO2 values outside normal ranges are lacking and would be a plausible next step to further investigate the influence of PaO2 on outcome.
Study limitations and strengths
In this study, we employed different analytic approaches to test the association of serial PaO2 measurements in resuscitated comatose patients after OHCA with functional parameters and biomarkers as outcome. The present study was conceived after completion of the TTM trial, and due to the nature of this exploratory, post hoc substudy, all results must be regarded as hypothesis generating and we cannot make causality statements from our findings. Considering the direction of the ORs and the widths of the CIs of our analyses, we cannot rule out possible associations. Threshold values for the detrimental effects of hyperoxemia or hypoxemia are undetermined; therefore, we accepted values in keeping with previous studies. For this study, we hypothesized that oxygen pressure in-between PaO2 measurement was linear and we were not able to account for short-term variations of PaO2. FiO2 management in the primary study was not protocolized and at the physician’s discretion. Our study nevertheless has considerable strengths. The investigated cohort of 939 OHCA was homogenous and large, and patients were selected from a multicenter randomized clinical trial with liberal inclusion criteria and a trial protocol reflecting standard practice. All physiological and biochemical parameters were collected prospectively, according to a pre-defined time-based protocol, eliminating measurement bias. Blood gases were analysed by a uniform method. Our results were adjusted for in the context of OHCA important confounders. The findings of this study were strengthened by an all-patient, a complete case, and an all-cause mortality sensitivity analysis, additionally supported by using a biomarker. The association of PaO2 outside normal ranges after OHCA with a biomarker of neurological injury has to our knowledge not previously been investigated.
Follow-up data was acquired using a structured protocol, with a majority performed face-to-face, and a minimal loss of patients in the follow-up period [24].
Conclusion
Although exposure to hyperoxemia and hypoxemia following OHCA was common in this study, we found hyperoxemia, hypoxemia, time-weighted mean oxygen exposure, and maximum partial pressure of oxygen difference not to be independently associated with neurological outcome at 6-month follow-up or with s-Tau at either 48 or 72 h after ROSC. Our findings did not indicate a PaO2 threshold value for the onset of poor neurological outcome.
Abbreviations
- AUC:
-
Area under the curve
- BE:
-
Base excess
- CABG:
-
Coronary artery bypass graft
- CBF:
-
Cerebral blood flow
- COPD:
-
Chronic obstructive pulmonary disease
- CPC:
-
Cerebral performance category
- CPR:
-
Cardio pulmonary resuscitation
- FiO2 :
-
Fraction of inspired oxygen
- GCS:
-
Glasgow Coma Scale
- ICU:
-
Intensive care unit
- kPa:
-
Kilopascal
- NSE:
-
Neuron-specific enolase
- OHCA:
-
Out-of-hospital cardiac arrest
- OR:
-
Odds ratio
- PaO2 :
-
Partial pressure of oxygen
- PCI:
-
Percutaneous coronary intervention
- ROSC:
-
Return of spontaneous circulation
- SD:
-
Standard deviation
- S-Tau:
-
Serum Tau
- TIA:
-
Transient ischemic attack
- TTM:
-
Target temperature management
- TWM:
-
Time-weighted mean
References
Kragholm K, Wissenberg M, Mortensen RN, Hansen SM, Malta Hansen C, Thorsteinsson K, Rajan S, Lippert F, Folke F, Gislason G, et al. Bystander efforts and 1-year outcomes in out-of-hospital cardiac arrest. N Engl J Med. 2017;376(18):1737–47.
Stromsoe A, Svensson L, Axelsson AB, Claesson A, Goransson KE, Nordberg P, Herlitz J. Improved outcome in Sweden after out-of-hospital cardiac arrest and possible association with improvements in every link in the chain of survival. Eur Heart J. 2015;36(14):863–71.
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135(10):e146–603.
Hassager C, Nagao K, Hildick-Smith D. Out-of-hospital cardiac arrest: in-hospital intervention strategies. Lancet. 2018;391(10124):989–98.
Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A scientific statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008;79(3):350–79.
Sekhon MS, Ainslie PN, Griesdale DE. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: a “two-hit” model. Crit Care. 2017;21(1):90.
Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R. The effect of hyperoxia following cardiac arrest - a systematic review and meta-analysis of animal trials. Resuscitation. 2012;83(4):417–22.
Brucken A, Kaab AB, Kottmann K, Rossaint R, Nolte KW, Weis J, Fries M. Reducing the duration of 100% oxygen ventilation in the early reperfusion period after cardiopulmonary resuscitation decreases striatal brain damage. Resuscitation. 2010;81(12):1698–703.
Balan IS, Fiskum G, Hazelton J, Cotto-Cumba C, Rosenthal RE. Oximetry-guided reoxygenation improves neurological outcome after experimental cardiac arrest. Stroke. 2006;37(12):3008–13.
Dell'Anna A, Lamanna I, Vincent JL, Taccone F. How much oxygen in adult cardiac arrest? Crit Care. 2014;18(5):555.
Watson NA, Beards SC, Altaf N, Kassner A, Jackson A. The effect of hyperoxia on cerebral blood flow: a study in healthy volunteers using magnetic resonance phase-contrast angiography. Eur J Anaesthesiol. 2000;17(3):152–9.
Kety SS, Schmidt CF. The effects of altered arterial TENSIONS of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal YOUNG men. J Clin Invest. 1948;27(4):484–92.
Xu F, Liu P, Pascual JM, Xiao G, Lu H. Effect of hypoxia and hyperoxia on cerebral blood flow, blood oxygenation, and oxidative metabolism. J Cereb Blood Flow Metab. 2012;32(10):1909–18.
Liu Y, Rosenthal RE, Haywood Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological outcome. Stroke. 1998;29(8):1679–86.
Kilgannon JH, Jones AE, Shapiro NI, Angelos MG, Milcarek B, Hunter K, Parrillo JE, Trzeciak S. Emergency Medicine Shock Research Network I: association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303(21):2165–71.
Janz DR, Hollenbeck RD, Pollock JS, McPherson JA, Rice TW. Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med. 2012;40(12):3135–9.
Bellomo R, Bailey M, Eastwood GM, Nichol A, Pilcher D, Hart GK, Reade MC, Egi M, Cooper DJ. Study of Oxygen in Critical Care G: arterial hyperoxia and in-hospital mortality after resuscitation from cardiac arrest. Crit Care. 2011;15(2):R90.
Ihle JF, Bernard S, Bailey MJ, Pilcher DV, Smith K, Scheinkestel CD. Hyperoxia in the intensive care unit and outcome after out-of-hospital ventricular fibrillation cardiac arrest. Crit Care Resusc. 2013;15(3):186–90.
Elmer J, Scutella M, Pullalarevu R, Wang B, Vaghasia N, Trzeciak S, Rosario-Rivera BL, Guyette FX, Rittenberger JC, Dezfulian C. The association between hyperoxia and patient outcomes after cardiac arrest: analysis of a high-resolution database. Intensive Care Med. 2015;41(1):49–57.
Johnson NJ, Dodampahala K, Rosselot B, Perman SM, Mikkelsen ME, Goyal M, Gaieski DF, Grossestreuer AV. The association between arterial oxygen tension and neurological outcome after cardiac arrest. Ther Hypothermia Temp Manag. 2017;7(1):36–41.
Wang HE, Prince DK, Drennan IR, Grunau B, Carlbom DJ, Johnson N, Hansen M, Elmer J, Christenson J, Kudenchuk P, et al. Post-resuscitation arterial oxygen and carbon dioxide and outcomes after out-of-hospital cardiac arrest. Resuscitation. 2017;120:113–8.
Vaahersalo J, Bendel S, Reinikainen M, Kurola J, Tiainen M, Raj R, Pettila V, Varpula T, Skrifvars MB. For the FSG: arterial blood gas Tensions after resuscitation from out-of-hospital cardiac arrest: associations with long-term neurological outcome. Crit Care Med. 2014;42(6):1463–70.
Roberts BW, Kilgannon JH, Hunter BR, Puskarich MA, Pierce L, Donnino M, Leary M, Kline JA, Jones AE, Shapiro NI, et al. Association between early Hyperoxia exposure after resuscitation from cardiac arrest and neurological disability: prospective multicenter protocol-directed cohort study. Circulation. 2018;137(20):2114–24.
Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, et al. Targeted temperature management at 33 degrees C versus 36 degrees C after cardiac arrest. N Engl J Med. 2013;369(23):2197–206.
Jacobs I, Nadkarni V, Bahr J, Berg RA, Billi JE, Bossaert L, Cassan P, Coovadia A, D'Este K, Finn J, et al. Cardiac arrest and cardiopulmonary resuscitation outcome reports: update and simplification of the Utstein templates for resuscitation registries: a statement for healthcare professionals from a task force of the International Liaison Committee on Resuscitation (American Heart Association, European Resuscitation Council, Australian Resuscitation Council, New Zealand Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation councils of southern Africa). Circulation. 2004;110(21):3385–97.
Tallman RD Jr. Acid-base regulation, alpha-stat, and the emperor’s new clothes. J Cardiothorac Vasc Anesth. 1997;11(3):282–8.
von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med. 2007;147(8):573–7.
Phelps R, Dumas F, Maynard C, Silver J, Rea T. Cerebral performance category and long-term prognosis following out-of-hospital cardiac arrest. Crit Care Med. 2013;41(5):1252–7.
Jennett B, Bond M. Assessment of outcome after severe brain damage. Lancet. 1975;1(7905):480–4.
Mattsson N, Zetterberg H, Nielsen N, Blennow K, Dankiewicz J, Friberg H, Lilja G, Insel PS, Rylander C, Stammet P, et al. Serum tau and neurological outcome in cardiac arrest. Ann Neurol. 2017;82(5):665–75.
Bhaskaran K, Smeeth L. What is the difference between missing completely at random and missing at random? Int J Epidemiol. 2014;43(4):1336–9.
Bennett DA. How can I deal with missing data in my study? Aust N Z J Public Health. 2001;25(5):464–9.
Marshall A, Altman DG, Holder RL, Royston P. Combining estimates of interest in prognostic modelling studies after multiple imputation: current practice and guidelines. BMC Med Res Methodol. 2009;9:57.
van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate imputation by chained equations in R. Jstatsoft. 2011;45(3):67.
Nolan JP, Berg RA, Callaway CW, Morrison LJ, Nadkarni V, Perkins GD, Sandroni C, Skrifvars MB, Soar J, Sunde K, et al. The present and future of cardiac arrest care: international experts reach out to caregivers and healthcare authorities. Intensive Care Med. 2018;44(6):823–32.
Sjoberg F, Gustafsson U, Eintrei C. Specific blood flow reducing effects of hyperoxaemia on high flow capillaries in the pig brain. Acta Physiol Scand. 1999;165(1):33–8.
Bulte DP, Chiarelli PA, Wise RG, Jezzard P. Cerebral perfusion response to hyperoxia. J Cereb Blood Flow Metab. 2007;27(1):69–75.
Voicu S, Deye N, Malissin I, Vigue B, Brun PY, Haik W, Champion S, Megarbane B, Sideris G, Mebazaa A, et al. Influence of alpha-stat and pH-stat blood gas management strategies on cerebral blood flow and oxygenation in patients treated with therapeutic hypothermia after out-of-hospital cardiac arrest: a crossover study*. Crit Care Med. 2014;42(8):1849–61.
Sundgreen C, Larsen FS, Herzog TM, Knudsen GM, Boesgaard S, Aldershvile J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke. 2001;32(1):128–32.
Pham P, Bindra J, Chuan A, Jaeger M, Aneman A. Are changes in cerebrovascular autoregulation following cardiac arrest associated with neurological outcome? Results of a pilot study. Resuscitation. 2015;96:192–8.
Neumar RW. Optimal oxygenation during and after cardiopulmonary resuscitation. Curr Opin Crit Care. 2011;17(3):236–40.
Grocott MP, Martin DS, Levett DZ, McMorrow R, Windsor J, Montgomery HE. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med. 2009;360(2):140–9.
Brierley JB. Experimental hypoxic brain damage. J Clin Pathol Suppl (R Coll Pathol). 1977;11:181–7.
Acknowledgements
None.
Funding
F.E. received an independent research grant from Stig och Ragna Gorthons Stiftelse and European Regional Development Fund through the Interreg IV A OKS program.
The TTM trial was funded by independent research grants from the Swedish Research Council, the Swedish Heart-Lung Foundation; Arbetsmarknadens försäkringsaktiebolag (AFA) Insurance Foundation; the TRYG foundation, Regional research support, Region Skåne; Government funding of clinical research within the Swedish National Health Services (ALF); Thelma Zoega Foundation; Krapperup Foundation; Thure Carlsson Foundation; Hans-Gabriel and Alice Trolle-Wachtmeister Foundation for Medical Research; Skåne University Hospital, Sweden; the European Clinical Research Infrastructures Network; the European Critical Care Research Network; the Ministry of Higher Education and Research of Luxembourg; and the National Research Fund, Luxembourg. No commercial funding was received. The funding organizations did not have any access to the data, nor did they have any influence on data analysis or interpretation.
Availability of data and materials
Data analyzed during the present study are currently stored in the TTM trial database. Their availability is regulated by the authorization of the TTM trial steering committee.
Author information
Authors and Affiliations
Contributions
FE and NN conceived this study. NN, HF, CH, JK, TC, JW, and FE obtained the funding. NN, FE, and SU designed the statistical analyses. SU performed the statistical analyses. FE and NN wrote the first draft of the manuscript; FE, AÅ, PP, MK, JU, HF, MPW, TC, NM, JK, CH, and NN actively recruited patients and contributed to the data acquisition. JW reviewed and modified the final manuscript. All authors read, critically reviewed, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The TTM trial protocol was approved by ethics committees in the following institutions: St George Hospital, Sydney. North Shore Hospital, Sydney. Liverpool Hospital, Sydney. The George Institute of Global Health, Sydney. General University Hospital in Prague, Prague. The Heart Center, Copenhagen University Hospital Rigshospitalet, Copenhagen. Ospedale Universitario di Cattinara, Trieste. Santa Maria degli Angeli Hospital, Pordenone. San Martino, Genoa. Medical Centre, Luxembourg. Amsterdam Medical Centre, Amsterdam. Leeuwarden Hospital, Leeuwarden. Rijnstaate Hospital, Arnhem. Onze Lieuwe Vrouwe Gasthuis, Amsterdam. Oslo University Hospital, Oslo. Haukeland University Hospital, Bergen. Helsingborg Hospital, Helsingborg. Karlstad Hospital, Karlstad. Kungälv Hospital, Kungälv: Linköping University Hospital, Linköping. Skåne University Hospital, Lund. Skåne University Hospital, Malmö. Norra Älvsborgs Län Hospital. Vrinnevi Hospital, Norrköping.
Sahlgrenska University Hospital, Gothenburg. Örebro University Hospital, Örebro. Geneva University Hospital, Geneva. Hospital St Gallen, St Gallen. Hospital La Chaux de Fonds. University Hospital of Wales, Cardiff. Royal Berkshire Hospital, Reading. Royal Bournemouth Hospital, Bournemouth. Guy’s and St Thomas’ NHS Trust, London. St George’s Hospital, London. Informed consent was waived or was obtained according to national legislation, in line with the Helsinki declaration.
Consent for publication
Not applicable.
Competing interests
H.F. has received lecture fees from Bard Medical and is scientific advisor at QuickCool.
M.P.W. has done an advisory board and educational meeting for Bard Medical, N.N. has received lecture fees from Bard Medical. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Associations between partial pressure of oxygen and neurological outcome in out-of-hospital cardiac arrest patients: an explorative analysis of a randomized trial. Additional details on study methods, explanatory figure and tables depicting detailed information on missing patients, interaction analysis, and sensitivity analyses. (DOCX 82 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ebner, F., Ullén, S., Åneman, A. et al. Associations between partial pressure of oxygen and neurological outcome in out-of-hospital cardiac arrest patients: an explorative analysis of a randomized trial. Crit Care 23, 30 (2019). https://doi.org/10.1186/s13054-019-2322-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-019-2322-z