
Abstract
Hypoxic ischemic brain injury (HIBI) after cardiac arrest (CA) is a leading cause of mortality and long-term neurologic disability in survivors. The pathophysiology of HIBI encompasses a heterogeneous cascade that culminates in secondary brain injury and neuronal cell death. This begins with primary injury to the brain caused by the immediate cessation of cerebral blood flow following CA. Thereafter, the secondary injury of HIBI takes place in the hours and days following the initial CA and reperfusion. Among factors that may be implicated in this secondary injury include reperfusion injury, microcirculatory dysfunction, impaired cerebral autoregulation, hypoxemia, hyperoxia, hyperthermia, fluctuations in arterial carbon dioxide, and concomitant anemia.
Clarifying the underlying pathophysiology of HIBI is imperative and has been the focus of considerable research to identify therapeutic targets. Most notably, targeted temperature management has been studied rigorously in preventing secondary injury after HIBI and is associated with improved outcome compared with hyperthermia. Recent advances point to important roles of anemia, carbon dioxide perturbations, hypoxemia, hyperoxia, and cerebral edema as contributing to secondary injury after HIBI and adverse outcomes. Furthermore, breakthroughs in the individualization of perfusion targets for patients with HIBI using cerebral autoregulation monitoring represent an attractive area of future work with therapeutic implications.
We provide an in-depth review of the pathophysiology of HIBI to critically evaluate current approaches for the early treatment of HIBI secondary to CA. Potential therapeutic targets and future research directions are summarized.
Similar content being viewed by others


Background
Cardiac arrest (CA) is a major cause of mortality and neurologic disability. The incidence of out-of-hospital CA is approximately 80 patients per 100,000 persons annually [1]. Despite advances in resuscitation, outcomes remain dismal, with 10% of patients surviving until hospital discharge and 5% experiencing full neurologic recovery [1].
The primary determinant of outcome after CA is hypoxic ischemic brain injury (HIBI). HIBI is the primary cause of death in 68% of inpatient CA and in 23% of out-of-hospital CA [2]. HIBI is associated with significant neurologic disability, ranging from mild cognitive deficits to minimally conscious and persistent vegetative states [2, 3]. Consequently, considerable effects on quality of life and incidence of psychiatric comorbidities, such as depression, anxiety, and posttraumatic stress disorder, are highly prevalent in HIBI survivors [4, 5]. The vast spectrum of acute and chronic HIBI phenotypes requires detailed understanding of cerebral physiologic perturbations that occur after CA and make clarifying the pathophysiology essential.
Management of HIBI is focused on limiting secondary injury [3] by optimizing the balance between cerebral oxygen delivery (CDO2) and use. Despite rigorous research, HIBI outcomes have not appreciably changed over 20 years [6, 7]. This stagnation is in contrast with improved outcomes in other critical care diseases [8].
Considerable opportunities remain to delineate the pathophysiology of HIBI. HIBI pathophysiology is a “two-hit” model, being determined by primary injury from immediate cessation of CDO2 during CA and secondary injury occurring after resuscitation. We present a narrative review of a two-hit model of HIBI pathophysiology as it pertains to physiologic parameters involved in maintaining the balance of CDO2 and use. We highlight advances pertaining to cerebral autoregulation, optimal hemoglobin, carbon dioxide, cerebral edema, normobaric hyperoxia, and targeted temperature management.
Primary injury
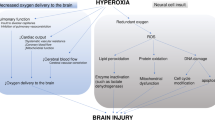
During CA, cessation of CDO2 occurs with resultant neuron ischemia and cell death within minutes [9] (Fig. 1). The cerebrum consumes 20% to 25% of cardiac output to maintain function [3]. The brain is devoid of nutrient stores, and consequently neuroglycopenia and metabolic crisis occur within minutes after CA [10], leading to cell death.

A schematic demonstrating the various microvascular and cellular pathophysiologic consequences which occur during the primary and secondary injury in hypoxic ischemic brain injury (HIBI). Decreased cerebral oxygen delivery manifests as reduced neuronal aerobic metabolism, causing reduced cellular adenosine triphosphate (ATP) production. Intracellular calcium accumulation leads to mitochondrial toxicity and further reduced ATP production. Inability to sustain cellular respiration results in cell death and apoptosis. Additionally, in the microvasculature, endothelial dysfunction leads to a porous blood-brain barrier, formation of cerebral edema, formation of microthrombi and limitation of cerebral blood flow with exacerbation of cellular ischemia. AQP 4 Aquaporin-4, RBC Red blood cells, WBC White blood cells
As CDO2 decreases, adenosine triphosphate production halts, causing cessation of energy-dependent ion channel function [11]. Subsequent intracellular Na+ accumulation results in cytotoxic edema. Depletion of adenosine triphosphate leads to anaerobic metabolism, cerebral lactate accumulation, and intracellular acidosis [12]. Additionally, cellular ischemia causes intracellular Ca2+ influx through N-methyl-d-aspartate channels, which activates lytic enzymes [13] and mitochondrial dysfunction, thereby depleting adenosine triphosphate further [14]. Finally, excitatory neurotransmitter release activates lipases and proteases, which leads to apoptosis [15].
Clinically, loss of neurologic function is manifested by a decreased level of consciousness after global cerebral ischemia. Historically, Rossen et al. demonstrated that cessation of cerebral blood flow (CBF) by neck cuff insufflation to 600 mmHg in humans precipitated acute decreased level of consciousness within 10 seconds [16]. Decreased level of consciousness after CA occurs within 20 seconds after onset of ventricular fibrillation [17]. Loss of neurologic function has been demonstrated by isoelectric electroencephalography in observational studies [16]. Pana et al. identified human studies demonstrating isoelectric electroencephalography rhythms within 15 seconds and 30 seconds of asystole and ventricular fibrillation, respectively [16]. These findings are corroborated by animal studies establishing a similar timeline of 10 to 30 seconds from the onset of cerebral ischemia to isoelectric electroencephalography [18].
Although primary injury causes substantial neuronal loss, the ensuing postresuscitation additive cerebral injury accounts for significant cerebral ischemia and cellular death. The key pathophysiologic factors that are implicated in secondary injury are physiologic modifiers involved in maintaining the balance between CDO2 and use. We next discuss secondary injury and physiologic determinants that are targets of therapeutic interventions after HIBI.
Secondary injury
Secondary injury is the additive cerebral injury characterized by an imbalance in postresuscitation CDO2 and use, ultimately culminating in neuronal death. It begins immediately after return of spontaneous circulation (ROSC). Structures especially susceptible include the hippocampi, thalami, cerebral cortex, corpus striatum, and cerebellar vermi [3] (Fig. 2), owing to highly metabolically active tissue. Aside from hypothermia, there are limited studies examining physiologic variables that exacerbate secondary injury. Table 1 summarizes the mechanisms of secondary injury.

Magnetic resonance imaging sequences show focal hypoxic ischemic brain injury (HIBI) within the hippocampi and basal ganglia bilaterally. The images shown represent the acute changes after HIBI within the first week after resuscitation. In the top row, T2-weighted sequences reveal abnormal signaling in the hippocampi and basal ganglia as highlighted by the red arrows. In the bottom row, restricted diffusion-weighted imaging confirms HIBI in the affected regions of the hippocampi and basal ganglia as highlighted by the red arrows
Microcirculation and reperfusion injury
After ROSC, microcirculatory perturbations lead to further neuron dysfunction. The cerebrovascular endothelium plays a critical role in maintaining blood-brain barrier integrity, regulation of microcirculatory blood flow, and release of autoanticoagulant mediators [19]. Endothelial functions are compromised, and biomarkers of cerebrovascular endothelial injury are associated with adverse outcomes in HIBI [20].
Following ROSC, reperfusion injury causes neuronal dysfunction despite restoration of CDO2 [21]. An initial period of cerebral hyperemia is followed by hypoperfusion, resulting in a “no-reflow” [22] state that exacerbates secondary injury. Mechanisms implicated in the no-reflow state include impaired vasomotor regulation, decreased nitric oxide production, and resultant vasoconstriction [3, 19, 20]. Extravasation of intravascular water through a porous blood-brain barrier with perivascular edema leads to increased intravascular viscosity and cerebrovascular resistance [22]. Other mechanisms implicated in reperfusion injury include free radical release, glutamate production, and intracellular Ca2+ accumulation [23].
Endothelial autoanticoagulant dysfunction causes diffuse microthrombi in the cerebrovasculature [24]. Concomitant impaired vasodilation causes increased cerebrovascular resistance and reduces CBF [3, 22]. Interventional studies demonstrate that heparin and tissue plasminogen activator improve microcirculatory flow [25]. These findings have not translated into improved outcomes when evaluated prospectively, however [24, 26]. Finally, intravenous prostacyclin is suggested to promote endothelial function through vasodilatory and antiplatelet effects [19], but clinical studies are not yet available. Table 2 summarizes mechanisms involved in reperfusion injury.
Hemoglobin
Hemoglobin is a major determinant of arterial oxygen content. In animal studies of traumatic brain injury, concomitant anemia exacerbates secondary injury from apoptosis [27]. However, physiologic benefits of improved CDO2 from transfusion must be balanced by risks associated with exogenous red blood cells. Although hemoglobin <70 g/L is the accepted transfusion threshold for nonbleeding critical care patients [28], it remains unclear if a liberal threshold is appropriate for patients with brain injury, who are susceptible to secondary injury from anemia [29].
Evidence of anemia in contributing to secondary injury in HIBI is limited to observational studies. Nakao et al. conducted a retrospective study of 137 subjects with witnessed CA and established that higher admission hemoglobin was an independent predictor of a 28-day favorable neurologic outcome (OR 1.26, 95% CI 1.00–1.58) [30]. These findings were corroborated by Wang et al., who demonstrated an association with adverse outcome and lower admission hemoglobin [31]. Recently, Johnson et al. conducted a multicenter observational study of 598 patients and found that favorable outcome patients had significantly higher hemoglobin (126 g/L versus 106 g/L, p < 0.001), a finding that persisted after adjustment [32].
Despite regression adjustment, admission anemia may be subject to strong residual or unmeasured confounding. It is unclear if admission hemoglobin captures the magnitude of effect that anemia has on secondary injury. Wormsbecker et al. accounted for this by investigating the relationship between mean hemoglobin over 7 days and neurologic outcome. They established that patients with a favorable outcome had significantly higher 7-day mean hemoglobin (115 g/L versus 107 g/L, p = 0.05) [33]. Furthermore, multivariable regression demonstrated that lower 7-day mean hemoglobin was associated with adverse outcome (OR 0.75 per 10 g/L change in hemoglobin, 95% CI 0.57–0.97) [33]. Importantly, Ameloot et al. established a link between hemoglobin and a measure of brain oxygenation in an observational study of 82 patients. They found a linear association between hemoglobin and brain regional saturation of oxygen (rSO2) using near-infrared spectroscopy [34], with hemoglobin <100 g/L being identified as a cutoff for lower rSO2 [34]. Additionally, they demonstrated that mean hemoglobin concentration <123 g/L was associated with worse neurologic outcome, particularly in patients with rSO2 < 62.5% (OR 2.88, 95% CI 1.02–8.16) [34]. Further research is required to establish an association between anemia with simultaneous brain hypoxia and investigate the effect of transfusion thresholds on outcome in HIBI.
Carbon dioxide
Partial pressure of arterial carbon dioxide (PaCO2) modulates cerebrovascular resistance and CBF via its effects on vascular smooth muscle [35]. Specifically, hypocapnia (PaCO2 < 35 mmHg) induces cerebrovascular vasoconstriction and decreases CBF by about 2% to 3% for every 1 mmHg of PaCO2 [35]. Clinically, hypocapnia reduces intracranial pressure (ICP) by reducing cerebrovascular volume [35]. However, sustained hypocapnia can decrease CBF, increase cerebral oxygen extraction, and induce ischemia [36, 37]. Conversely, hypercapnia (PaCO2 > 45 mmHg) is a cerebrovascular vasodilator that causes hyperemia, exacerbates ICP [38], and reduces CBF [38]. Hypercapnia is also associated with excitotoxicity and increased cerebral oxygen demand [39]. Importantly, PaCO2 vascular reactivity is preserved after HIBI, making regulation of PaCO2 clinically significant and a crucial determinant of CDO2 [40]. The optimal PaCO2 in individual patients is not known but presents a unique opportunity for advanced neurophysiologic monitoring using transcranial Doppler ultrasonography to evaluate CBF, ICP, and cerebrovascular resistance with varying PaCO2 levels in HIBI.
Perturbations in PaCO2 in HIBI have been evaluated in observational studies of HIBI. Roberts et al. conducted a retrospective study of 193 patients and investigated the effects of hypocapnia and hypercapnia compared with normocapnia (PaCO2 35–45 mmHg) on outcome. They demonstrated a relationship between adverse neurologic outcome and both hypocapnia (OR 2.43, 95% CI 1.04–5.65) and hypercapnia (OR 2.20, 95% CI 1.03–4.71) [35]. Exposure of hypocapnia and hypercapnia occurred 36% and 42% of the time after CA [35], respectively, making the exposure of CO2 fluctuation significant. The authors followed that study with an analysis of a prospective registry of patients with HIBI and found a significant association between normocapnia and good neurologic outcome (OR 4.44, 95% CI 1.33–14.85) [41]. Schneider et al. conducted a large multicenter database study of 16,542 patients with HIBI and investigated the effects of hypocapnia in HIBI, and they demonstrated a significant association between hospital mortality and hypocapnia (OR 1.12, 95% CI 1.00–1.24) compared with normocapnia [42]. Given the sound biological plausibility and available clinical data, regulation of PaCO2 warrants further systematic study to determine the precise optimal therapeutic strategy after HIBI. Critical links with intracranial physiologic parameters pertaining to ICP, CBF, and brain oxygenation and fluctuations in PaCO2 are logical future goals in this field.
Cerebral edema
After HIBI, cerebral edema is a recognized complication that causes secondary injury. Because of a fixed overall intracranial volume, an increase in the parenchymal bulk from cerebral edema in HIBI can cause intracranial hypertension [43] with resultant decreases in cerebral perfusion pressure, CBF, and CDO2 [3]. This vicious cycle of cerebral edema precipitating increased ICP causes transtentorial herniation and brain death.
The origin of cerebral edema occurs as a result of either vasogenic or cytotoxic mechanisms. In the early stages, vasogenic edema emanates from fluid shifts from the intravascular to the cerebral interstitial space. Key to this process, aquaporin-4 is a membrane protein that transports water across cell membranes in the central nervous system. Aquaporin-4 proteins are located in perivascular astrocytic endfeet, processes, and ependyma [44]. The aquaporin-4 perivascular pool is identified as the predominant cluster involved in the pathophysiology of cerebral edema after HIBI, with increased aquaporin-4 expression occurring within 48 h after the onset of cerebral ischemia [44]. Interestingly, Nakayama et al. showed that 7.5% hypertonic saline attenuated cerebral edema in a wild-type mouse model of HIBI but had no effect in an aquaporin-4-knockout model, thereby demonstrating the importance of aquaporin-4 in the pathophysiology of cerebral edema and highlighting its therapeutic potential [44]. Hypertonic saline administration also restores blood-brain barrier integrity mediated by aquaporin-4 in the hippocampi, cerebellum, cortex, and basal ganglia [44]. Furthermore, Nakayama et al. established that achieving serum osmolality >350 mOsm/L with continuous infusion of conivaptan, a V1 and V2 antagonist, attenuated cerebral edema [45], thereby demonstrating that the effect of aquaporin-4 to decrease cerebral edema occurs through osmotic gradients, as opposed to a specific intravenous osmotic agent itself (e.g., 7.5% hypertonic saline).
Alternatively, cytotoxic edema originates from cellular metabolic crisis and intracellular energy depletion. Decreased adenosine triphosphate (Fig. 1) leads to energy-dependent ion channel failure and intracellular sodium and water retention. Rungta et al. established that the Na+Cl− receptor SLC26A11 is a critical modulator of intracellular transport of chloride and subsequent cerebral edema after ischemia [46]. The authors showed that blockade of this receptor attenuated cytotoxic cerebral edema [46] after HIBI. The role of Na+Cl− receptor antagonism after HIBI is yet to be clarified but represents a future therapeutic target.
Furthermore, sulfonylurea receptors are also implicated in the pathophysiology of cerebral edema after ischemia. Glyburide, a sulfonylurea receptor inhibitor, attenuates malignant cerebral edema after acute middle cerebral infarction [47]. These findings are corroborated by animal studies that demonstrate sulfonylurea receptor antagonism decreases cerebral edema after neuronal ischemia [48].
Cerebral autoregulation
The brain has an innate ability to regulate blood flow to match metabolic demands. This phenomenon, termed cerebral autoregulation, allows the cerebrovasculature to undergo vasoconstriction and vasodilation over a range of mean arterial pressure (MAP) to maintain stable CBF [49]. Cerebral autoregulation mitigates the effects of hypoperfusion (ischemia) and hyperperfusion [49].
The identification of individualized MAP targets after HIBI using cerebral autoregulation monitoring is an attractive concept that has garnered significant interest. Initially, Nishizawa et al. demonstrated a linear relationship between MAP and CBF (as indexed by jugular venous oximetry) [50], suggesting complete dysfunctional cerebral autoregulation after HIBI. Thereafter, Sundgreen et al. constructed cerebral autoregulation curves for patients with HIBI by performing stepwise increases in MAP with norepinephrine and simultaneously estimating CBF with middle cerebral artery velocity on the basis of transcranial Doppler ultrasonography [51]. Of the 18 patients studied by Sundgreen et al., cerebral autoregulation was absent in 8 and present in 10 patients. In five of ten patients with preserved cerebral autoregulation, the lower limit of autoregulation was right-shifted with a median MAP 114 mmHg (range 80–120 mmHg) [51]. This sentinel study demonstrated the heterogeneous nature of cerebral autoregulation in patients with HIBI and suggested that the lower limit of autoregulation may be significantly higher than traditional MAP targets after HIBI.
Recently, monitoring with near-infrared spectroscopy has garnered significant interest as a noninvasive method of optimal MAP identification and assessment of cerebral autoregulation after HIBI. Near-infrared spectroscopy measures the rSO2 in the outermost 2 cm of the frontal lobe, represents the state of oxygenated hemoglobin in the microvasculature, and approximates CBF [52]. Therefore, continually integrating fluctuations between MAP and rSO2, a Pearson’s product-moment correlation coefficient is generated. This correlation coefficient (COx) varies between −1 and +1. Positive COx values, where there is a positive and linear correlation between MAP and rSO2, indicate dysfunctional autoregulation [53]. Near-zero and negative COx values indicate intact autoregulation (i.e., rSO2 remains relatively constant despite varying MAP). The optimal MAP is identified as the MAP with the lowest value of COx, as shown in Fig. 3. Lee et al. demonstrated that COx identified the lower limit of autoregulation in a swine model of pediatric HIBI [53]. Recently, Ameloot et al. retrospectively calculated COx using MAP and rSO2 to indicate that autoregulation was intact in 33 of 51 subjects with HIBI. Thereafter, Pham et al. showed that COx was significantly higher in nonsurvivors of HIBI than in survivors [54]. Although higher COx was associated with nonsurvivors, there was no association between rSO2 and mortality. Recently, our research team demonstrated feasibility of monitoring COx in real time and identification of optimal MAP prospectively in 20 patients after CA [55]. Subjects spent approximately 50% of time outside a ±5 mmHg range from the optimal MAP, and, importantly, the optimal MAP was consistently identified in 19 of 20 subjects. The concept of individualized perfusion pressures is emerging as an attractive therapeutic target and improved clinical outcome is associated if actual MAP is maintained within proximity of the identified optimal MAP. It is imperative to recognize the downsides of targeting significantly right-shifted optimal MAP, particularly in patients with compromised left ventricular function after CA. Increasing afterload on a decompensated left ventricle can dramatically reduce stroke volume and cardiac output, placing the injured brain at increased risk of ischemia. Therefore, increased MAP targets in HIBI should be weighed against concurrent myocardial function. Considerable work remains to further delineate if individualized perfusion targets decrease brain hypoxia and secondary injury and are associated with improved neurologic outcome.

The zone of preserved autoregulation after hypoxic ischemic brain injury appears to be narrowed and right-shifted after cardiac arrest. Within the zone of autoregulation, regional saturation of oxygen (rSO2) is stable owing to the innate vasoconstriction and vasodilation of the cerebral vasculature to maintain stable cerebral blood flow. Outside the zone of autoregulation, a linear relationship exists between rSO2 and mean arterial pressure (MAP). By continually integrating the fluctuations of MAP and rSO2 with one another, a correlation coefficient (COx) can be generated. The COx approaches negative values or near-zero within the preserved zone of autoregulation, resulting in a U-shaped curve. The nadir of the U-shaped curve represents the optimal MAP (MAPOPT) for each individual patient
Temperature
Targeted temperature management has historically been the focus of considerable HIBI research. It is a mainstay in the management of HIBI by mitigating secondary injury after CA [56]. At the cellular level, the beneficial effects of hypothermia are well documented. Cerebral metabolism is reduced by 5% to 10% per 1 °C decrease in core body temperature. In addition, global carbon dioxide production and oxygen consumption are decreased proportionally to reductions in core body temperature [57]. By decreasing cerebral metabolism, hypothermia avoids excessive intracellular anaerobic metabolism, which leads to increased lactate production. Hypothermia also improves cerebral glucose use and allows available cellular energy stores to be used for necessary cellular functions in keeping with neuronal survival [56]. Additional benefits of hypothermia include prevention of apoptosis by decreasing proapoptotic mediators such as p53, tumor necrosis factor α, and caspase enzymes while increasing expression of antiapoptotic proteins such as Bcl-2 [56, 57]. Hypothermia also prevents mitochondrial dysfunction, a key pathway involved in the promotion of apoptosis by release of cytochrome c oxidase into the cellular cytoplasm [56]. Finally, hypothermia decreases inflammatory mediators such as the interleukin-1 family of cytokines [58] as well as chemotaxis of leukocytes into cerebral interstitial tissue [56], reduces excitotoxic neurotransmitter release (glutamate and glycine) [57], and decreases free radical production after HIBI [57]. Sustained hypothermia also has detrimental physiologic effects pertaining to immune suppression, hemoconcentration, coagulopathy, arrhythmias, electrolyte disturbances, and hemodynamic instability, which must be weighed against the possible benefits [56]. Furthermore, unintentional hypothermia can occur after CA, indicating possible severe damage to the key centers of thermoregulation, including the hypothalamus [56].
Hyperthermia is associated with numerous pathophysiologic sequelae that are potentially harmful after HIBI. Specifically, hyperthermia may increase blood-brain barrier permeability, leading to worsening cerebral edema, ICP,, and cerebral ischemia. Furthermore, hyperthermia increases glutamate production, which in turn causes intracellular Ca2+ influx, leading to neuronal cell death, seizures, and further secondary injury [3]. Increased cerebral metabolism, hyperemic blood flow, and increased ICP are additional downstream consequences of uncontrolled hyperthermia in HIBI [3]. Recently, we showed that hyperthermia is associated with dysfunctional autoregulation in patients with HIBI [55].
Clinical studies have established a firm link between hypothermia and improved outcome after CA. In 2002, two randomized controlled trials demonstrated marked improvement in clinical outcomes in patients with CA after ventricular fibrillation or ventricular tachycardia who were treated with hypothermia compared with standard of care [6, 59]. A persistent criticism of both studies was that the standard-of-care groups maintained core body temperatures >37 °C, thereby exposing patients to the harmful effects of hyperthermia. This prompted a third recent randomized controlled trial comparing core body temperature control of 36 °C (normothermia) versus 33 °C (hypothermia) after CA [7]. This pragmatic trial included patients with HIBI with all initial cardiac rhythms and ultimately did not demonstrate an appreciable benefit of hypothermia versus normothermia [7]. Importantly, it must be stated that the maintenance of normothermia at 36 °C after CA requires active cooling. The negative effects of sustained hyperthermia and adverse outcomes after CA are well established [60, 61], thereby reinforcing the importance of aggressive core body temperature control in patients following CA. It is possible that individualized temperature targets exist within patients with HIBI, and the inability of current studies to concurrently monitor cerebral metabolism, ICP, and biomarkers of neuron degeneration has limited our ability to make these patient-specific distinctions.
Normobaric hyperoxia
The dissolved portion of oxygen in plasma is a minor contributor to overall oxygen content. However, in disease states, this portion may have a pivotal role in ensuring adequate hemoglobin saturation for CDO2 and overcome diffusion barriers to restore normal cellular metabolism. Augmenting arterial oxygen content is touted as a crucial modifiable factor in optimizing CDO2 after HIBI, with normobaric hyperoxia being suggested to achieve this goal.
Upon ROSC, reperfusion injury occurs as a result of oxygen free radical production, which leads to intracellular oxidation [62]. Examples include superoxide (O2 −), hydrogen peroxide (H2O2), hydroxyl anion (OH−), and nitrite (NO2 −). Endogenous antioxidants balance the generation of free radicals and stabilize cellular function. Inadvertent normobaric hyperoxia in HIBI may tip this balance in favor of free radical production, cellular oxidation, and neuronal death [62]. Although a systematic review of animal studies of HIBI suggested that increased neuron dysfunction occurs after normobaric hyperoxia, there was significant between-study heterogeneity with respect to ventilation strategies, timing and dose of normobaric hyperoxia, concomitant use of hypothermia, and the chosen primary outcomes [63]. There are also several reported adverse effects associated with normobaric hyperoxia, including increased vascular resistance (cerebral, myocardial, and systemic), decreased CBF, seizures, and increased release neuronal degeneration biomarkers such as neuron-specific enolase [57, 62, 64, 65].
Researchers in several studies have evaluated normobaric hyperoxia in HIBI, with conflicting results. Kuisma et al. conducted a randomized study of patients who were given 21% or 100% inspired oxygen after ROSC [66]. The group that received 21% inspired oxygen exhibited lower serum levels of neuron-specific enolase than the normobaric hyperoxia group that did not undergo concomitant hypothermia. Kilgannon et al. interrogated the Project IMPACT database with more than 400,000 patients [67]. They included patients with nontraumatic CA and cardiopulmonary resuscitation within 24 h prior to intensive care admission. Their objective was to examine the association between hyperoxia and mortality. Compared with the subjects in the normoxia group, subjects with normobaric hyperoxia (partial pressure of arterial oxygen [PaO2] >300 mmHg) had higher associated in-hospital mortality (OR 1.8, 95% CI 1.5–2.2). Compared with normoxia, hypoxia (PaO2 < 60 mmHg) was also associated with increased in-hospital mortality (OR 1.3, 95% CI 1.1–1.5). Spindelboeck et al. studied normobaric hyperoxia and hypoxemia during CA and found that both were associated with increased mortality [68], suggesting that the deleterious effects of normobaric hyperoxia may occur in early stages of HIBI. Finally, Bellomo et al. conducted a retrospective analysis of patients with CA and demonstrated that normobaric hyperoxia and hypoxemia were associated with increased mortality; however, after adjustment, this relationship was no longer significant [69]. Importantly, significant limitations in methodology should be noted, particularly the retrospective nature of these studies, the limitation of using mortality as a primary outcome in a brain injury population, and the fact that the definition of normobaric hyperoxia with a single PaO2 > 300 mmHg does not capture the true biological exposure of patients to normobaric hyperoxia after CA. Furthermore, hypothermia was not routinely used in the aforementioned studies.
Additional retrospective analyses investigating the use of normobaric hyperoxia with concomitant hypothermia have addressed this shortcoming. Janz et al. demonstrated an association between adverse neurologic outcome and normobaric hyperoxia administration [70]. These results are contrasted by those reported by Ihle et al. and Lee et al., who failed to show an association between normobaric hyperoxia and adverse neurologic outcome with concomitant hypothermia [71, 72]. Thereafter, a prospective study revealed an association between favorable neurologic outcome and higher mean PaO2 [73]. Thus, concomitant hypothermia may play a role in modifying the deleterious effects of normobaric hyperoxia in HIBI.
Conclusions
HIBI pathophysiology is complex, with a significant contribution attributable to secondary injury. Researchers have investigated the effects of interventions aimed at preventing secondary injury, most notably hypothermia. Future targets of research include individualized perfusion targets, normobaric hyperoxia, transfusion triggers, and PaCO2 goals.
Abbreviations
- ATP:
-
Adenosine triphosphate
- CA:
-
Cardiac arrest
- CBF:
-
Cerebral blood flow
- CDO2 :
-
Cerebral oxygen delivery
- CMRO2 :
-
Cerebral metabolic rate of oxygen uptake
- Cox:
-
Correlation coefficient
- CPP:
-
Cerebral perfusion pressure
- HIBI:
-
Hypoxic ischemic brain injury
- ICP:
-
Intracranial pressure
- MAP:
-
Mean arterial pressure
- PaCO2 :
-
Partial pressure of arterial carbon dioxide
- PaO2 :
-
Partial pressure of arterial oxygen
- RBC:
-
Red blood cells
- ROSC:
-
Return of spontaneous circulation
- rSO2 :
-
Regional saturation of oxygen
- WBC:
-
White blood cells
References
Gräsner JT, Lefering R, Koster RW, Masterson S, Böttiger BW, Herlitz J, et al. EuReCa ONE—27 Nations, ONE Europe, ONE Registry: a prospective one month analysis of out-of-hospital cardiac arrest outcomes in 27 countries in Europe. Resuscitation. 2016;105:188–95.
Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. 2004;30:2126–8.
Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Böttiger BW, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Stroke. Resuscitation. 2008;79:350–79.
Wilder Schaaf KP, Artman LK, Peberdy MA, Walker WC, Ornato JP, Gossip MR, et al. Anxiety, depression, and PTSD following cardiac arrest: a systematic review of the literature. Resuscitation. 2013;84:873–7.
Bunch TJ, White RD, Smith GE, Hodge DO, Gersh BJ, Hammill SC, et al. Long-term subjective memory function in ventricular fibrillation out-of-hospital cardiac arrest survivors resuscitated by early defibrillation. Resuscitation. 2004;60:189–95.
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–63.
Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, et al. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. 2013;369:2197–206.
Zimmerman JE, Kramer AA, Knaus WA. Changes in hospital mortality for United States intensive care unit admissions from 1988 to 2012. Crit Care. 2013;17:R81.
Imberti R, Bellinzona G, Riccardi F, Pagani M, Langer M. Cerebral perfusion pressure and cerebral tissue oxygen tension in a patient during cardiopulmonary resuscitation. Intensive Care Med. 2003;29:1016–9.
Wagner SR, Lanier WL. Metabolism of glucose, glycogen, and high-energy phosphates during complete cerebral ischemia: a comparison of normoglycemic, chronically hyperglycemic diabetic, and acutely hyperglycemic nondiabetic rats. Anesthesiology. 1994;81:1516–26.
Hoxworth JM, Xu K, Zhou Y, Lust WD, LaManna JC. Cerebral metabolic profile, selective neuron loss, and survival of acute and chronic hyperglycemic rats following cardiac arrest and resuscitation. Brain Res. 1999;821:467–79.
Hossmann KA, Lechtape-Grüter H, Hossmann V. The role of cerebral blood flow for the recovery of the brain after prolonged ischemia. Z Neurol. 1973;204:281–99.
Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–24.
Xiong W, Hoesch RE, Geocadin RG. Post-cardiac arrest encephalopathy. Semin Neurol. 2011;31:216–25.
Kiessling M, Stumm G, Xie Y, Herdegen T, Aguzzi A, Bravo R, et al. Differential transcription and translation of immediate early genes in the gerbil hippocampus after transient global ischemia. J Cereb Blood Flow Metab. 1993;13:914–24.
Pana R, Hornby L, Shemie SD, Dhanani S, Teitelbaum J. Time to loss of brain function and activity during circulatory arrest. J Crit Care. 2016;34:77–83.
Aminoff MJ, Scheinman MM, Griffin JC, Herre JM. Electrocerebral accompaniments of syncope associated with malignant ventricular arrhythmias. Ann Intern Med. 1988;108:791–6.
van Rijn CM, Krijnen H, Menting-Hermeling S, Coenen AML. Decapitation in rats: latency to unconsciousness and the “wave of death”. PLoS One. 2011;6:e16514.
Adams JA. Endothelium and cardiopulmonary resuscitation. Crit Care Med. 2006;34(12 Suppl):S458–65.
Bro-Jeppesen J, Johansson PI, Hassager C, Wanscher M, Ostrowski SR, Bjerre M, et al. Endothelial activation/injury and associations with severity of post-cardiac arrest syndrome and mortality after out-of-hospital cardiac arrest. Resuscitation. 2016;107:71–9.
Madathil RJ, Hira RS, Stoeckl M, Sterz F, Elrod JB, Nichol G. Ischemia reperfusion injury as a modifiable therapeutic target for cardioprotection or neuroprotection in patients undergoing cardiopulmonary resuscitation. Resuscitation. 2016;105:85–91.
Böttiger BW, Krumnikl JJ, Gass P, Schmitz B, Motsch J, Martin E. The cerebral “no-reflow” phenomenon after cardiac arrest in rats—influence of low-flow reperfusion. Resuscitation. 1997;34:79–87.
Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998;43:1382–96-7.
Böttiger BW, Bode C, Kern S, Gries A, Gust R, Glätzer R, et al. Efficacy and safety of thrombolytic therapy after initially unsuccessful cardiopulmonary resuscitation: a prospective clinical trial. Lancet. 2001;357:1583–5.
Fischer M, Böttiger BW, Popov-Cenic S, Hossmann KA. Thrombolysis using plasminogen activator and heparin reduces cerebral no-reflow after resuscitation from cardiac arrest: an experimental study in the cat. Intensive Care Med. 1996;22:1214–23.
Spöhr F, Arntz HR, Bluhmki E, Bode C, Carli P, Chamberlain D, et al. International multicentre trial protocol to assess the efficacy and safety of tenecteplase during cardiopulmonary resuscitation in patients with out-of-hospital cardiac arrest: the Thrombolysis in Cardiac Arrest (TROICA) Study. Eur J Clin Invest. 2005;35:315–23.
Hare GMT, Mazer CD, Hutchison JS, McLaren AT, Liu E, Rassouli A, et al. Severe hemodilutional anemia increases cerebral tissue injury following acute neurotrauma. J Appl Physiol. 2007;103:1021–9.
Hébert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. N Engl J Med. 1999;340:409–17.
LeRoux P. Haemoglobin management in acute brain injury. Curr Opin Crit Care. 2013;19:83–91.
SOS-KANTO study group. Relationship between the hemoglobin level at hospital arrival and post–cardiac arrest neurologic outcome. Am J Emerg Med. 2012;30:770–4.
Wang CH, Huang CH, Chang WT, Tsai MS, Yu PH, Wang AY, et al. Association between hemoglobin levels and clinical outcomes in adult patients after in-hospital cardiac arrest: a retrospective cohort study. Intern Emerg Med. 2016;11:727–36.
Johnson NJ, Rosselot B, Perman SM, Dodampahala K, Goyal M, Gaieski DF, et al. The association between hemoglobin concentration and neurologic outcome after cardiac arrest. J Crit Care. 2016;36:218–22.
Wormsbecker A, Sekhon MS, Griesdale DE, Wiskar K, Rush B. The association between anemia and neurological outcome in hypoxic ischemic brain injury after cardiac arrest. Resuscitation. 2017;112:11–6.
Ameloot K, Genbrugge C, Meex I, Janssens S, Boer W, Mullens W, et al. Low hemoglobin levels are associated with lower cerebral saturations and poor outcome after cardiac arrest. Resuscitation. 2015;96:280–6.
Roberts BW, Kilgannon JH, Chansky ME, Mittal N, Wooden J, Trzeciak S. Association between postresuscitation partial pressure of arterial carbon dioxide and neurological outcome in patients with post-cardiac arrest syndrome. Circulation. 2013;127:2107–13.
Coles JP, Fryer TD, Coleman MR, Smielewski P, Gupta AK, Minhas PS, et al. Hyperventilation following head injury: effect on ischemic burden and cerebral oxidative metabolism. Crit Care Med. 2007;35:568–78.
Diringer MN, Videen TO, Yundt K, Zazulia AR, Aiyagari V, Dacey RG, et al. Regional cerebrovascular and metabolic effects of hyperventilation after severe traumatic brain injury. J Neurosurg. 2002;96:103–8.
Brian JE. Carbon dioxide and the cerebral circulation. Anesthesiology. 1998;88:1365–86.
Huttunen J, Tolvanen H, Heinonen E, Voipio J, Wikström H, Ilmoniemi RJ, et al. Effects of voluntary hyperventilation on cortical sensory responses: electroencephalographic and magnetoencephalographic studies. Exp Brain Res. 1999;125:248–54.
Yundt KD, Diringer MN. The use of hyperventilation and its impact on cerebral ischemia in the treatment of traumatic brain injury. Crit Care Clin. 1997;13:163–84.
Roberts BW, Kilgannon JH, Chansky ME, Trzeciak S. Association between initial prescribed minute ventilation and post-resuscitation partial pressure of arterial carbon dioxide in patients with post-cardiac arrest syndrome. Ann Intensive Care. 2014;4:9.
Schneider AG, Eastwood GM, Bellomo R, Bailey M, Lipcsey M, Pilcher D, et al. Arterial carbon dioxide tension and outcome in patients admitted to the intensive care unit after cardiac arrest. Resuscitation. 2013;84:927–34.
Gueugniaud PY, Garcia-Darennes F, Gaussorgues P, Bancalari G, Petit P, Robert D. Prognostic significance of early intracranial and cerebral perfusion pressures in post-cardiac arrest anoxic coma. Intensive Care Med. 1991;17:392–8.
Nakayama S, Migliati E, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A. Osmotherapy with hypertonic saline attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Crit Care Med. 2016;44:e702–10.
Nakayama S, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A. Conivaptan, a selective arginine vasopressin V1a and V2 receptor antagonist attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Neurocrit Care. 2016;24:273–82.
Rungta RL, Choi HB, Tyson JR, Malik A, Dissing-Olesen L, Lin PJC, et al. The cellular mechanisms of neuronal swelling underlying cytotoxic edema. Cell. 2015;161:610–21.
Greer DM. Mechanisms of injury in hypoxic-ischemic encephalopathy: implications to therapy. Semin Neurol. 2006;26:373–9.
Xie Y, Zacharias E, Hoff P, Tegtmeier F. Ion channel involvement in anoxic depolarization induced by cardiac arrest in rat brain. J Cereb Blood Flow Metab. 1995;15:587–94.
Czosnyka M, Brady K, Reinhard M, Smielewski P, Steiner LA. Monitoring of cerebrovascular autoregulation: facts, myths, and missing links. Neurocrit Care. 2009;10:373–86.
Nishizawa H, Kudoh I. Cerebral autoregulation is impaired in patients resuscitated after cardiac arrest. Acta Anaesthesiol Scand. 1996;40:1149–53.
Sundgreen C, Larsen FS, Herzog TM, Knudsen GM, Boesgaard S, Aldershvile J. Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke. 2001;32:128–32.
Storm C, Leithner C, Krannich A, Wutzler A, Ploner CJ, Trenkmann L, et al. Regional cerebral oxygen saturation after cardiac arrest in 60 patients—a prospective outcome study. Resuscitation. 2014;85:1037–41.
Lee JK, Yang ZJ, Wang B, Larson AC, Jamrogowicz JL, Kulikowicz E, et al. Noninvasive autoregulation monitoring in a swine model of pediatric cardiac arrest. Anesth Analg. 2012;114:825–36.
Pham P, Bindra J, Chuan A, Jaeger M, Aneman A. Are changes in cerebrovascular autoregulation following cardiac arrest associated with neurological outcome? Results of a pilot study. Resuscitation. 2015;96:192–8.
Sekhon MS, Smielewski P, Bhate TD, Brasher PM, Foster D, Menon DK, et al. Using the relationship between brain tissue regional saturation of oxygen and mean arterial pressure to determine the optimal mean arterial pressure in patients following cardiac arrest: a pilot proof-of-concept study. Resuscitation. 2016;106:120–5.
Polderman KH. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet. 2008;371:1955–69.
Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37(7 Suppl):S186–202.
Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury—an inflammatory disease? Brain Res Brain Res Rev. 2005;48:388–99.
Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–56.
Zeiner A, Holzer M, Sterz F, Schörkhuber W, Eisenburger P, Havel C, et al. Hyperthermia after cardiac arrest is associated with an unfavorable neurologic outcome. Arch Intern Med. 2001;161:2007–12.
Takino M, Okada Y. Hyperthermia following cardiopulmonary resuscitation. Intensive Care Med. 1991;17:419–20.
Dell’Anna AM, Lamanna I, Vincent JL, Taccone FS. How much oxygen in adult cardiac arrest? Crit Care. 2014;18:555.
Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R. The effect of hyperoxia following cardiac arrest—a systematic review and meta-analysis of animal trials. Resuscitation. 2012;83:417–22.
Floyd TF, Clark JM, Gelfand R, Detre JA, Ratcliffe S, Guvakov D, et al. Independent cerebral vasoconstrictive effects of hyperoxia and accompanying arterial hypocapnia at 1 ATA. J Appl Physiol. 2003;95:2453–61.
Morimoto Y, Kemmotsu O, Kitami K, Matsubara I, Tedo I. Acute brain swelling after out-of-hospital cardiac arrest: pathogenesis and outcome. Crit Care Med. 1993;21:104–10.
Kuisma M, Boyd J, Voipio V, Alaspää A, Roine RO, Rosenberg P. Comparison of 30 and the 100% inspired oxygen concentrations during early post-resuscitation period: a randomised controlled pilot study. Resuscitation. 2006;69:199–206.
Kilgannon JH, Jones AE, Shapiro NI, Angelos MG, Milcarek B, Hunter K, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303:2165–71.
Spindelboeck W, Schindler O, Moser A, Hausler F, Wallner S, Strasser C, et al. Increasing arterial oxygen partial pressure during cardiopulmonary resuscitation is associated with improved rates of hospital admission. Resuscitation. 2013;84:770–5.
Bellomo R, Bailey M, Eastwood GM, Nichol A, Pilcher D, Hart GK, et al. Arterial hyperoxia and in-hospital mortality after resuscitation from cardiac arrest. Crit Care. 2011;15:R90.
Janz DR, Hollenbeck RD, Pollock JS, McPherson JA, Rice TW. Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med. 2012;40:3135–9.
Lee BK, Jeung KW, Lee HY, Lee SJ, Jung YH, Lee WK, et al. Association between mean arterial blood gas tension and outcome in cardiac arrest patients treated with therapeutic hypothermia. Am J Emerg Med. 2014;32:55–60.
Ihle JF, Bernard S, Bailey MJ, Pilcher DV, Smith K, Scheinkestel CD. Hyperoxia in the intensive care unit and outcome after out-of-hospital ventricular fibrillation cardiac arrest. Crit Care Resusc. 2013;15:186–90.
Vaahersalo J, Bendel S, Reinikainen M, Kurola J, Tiainen M, Raj R, et al. Arterial blood gas tensions after resuscitation from out-of-hospital cardiac arrest: associations with long-term neurologic outcome. Crit Care Med. 2014;42:1463–70.
Acknowledgements
We acknowledge our colleagues in the intensive care unit at Vancouver General Hospital for their insightful guidance.
Funding
MSS is funded by Vancouver Coastal Health Research Institute. DEG is funded by the VGH & UBC Hospital Foundation Best of Health Fund.
Availability of data and materials
Not applicable.
Authors’ contributions
MSS contributed the majority of the manuscript preparation and background research. PNA contributed to the manuscript preparation. DEG contributed to the manuscript preparation. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Authors’ information
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sekhon, M.S., Ainslie, P.N. & Griesdale, D.E. Clinical pathophysiology of hypoxic ischemic brain injury after cardiac arrest: a “two-hit” model. Crit Care 21, 90 (2017). https://doi.org/10.1186/s13054-017-1670-9
Published:
DOI: https://doi.org/10.1186/s13054-017-1670-9