
Abstract
Although experimental studies have suggested that a high arterial oxygen pressure (PaO2) might aggravate post-anoxic brain injury, clinical studies in patients resuscitated from cardiac arrest (CA) have given conflicting results. Some studies found that a PaO2 of more than 300 mm Hg (hyperoxemia) was an independent predictor of poor outcome, but others reported no association between blood oxygenation and neurological recovery in this setting. In this article, we review the potential mechanisms of oxygen toxicity after CA, animal data available in this field, and key human studies dealing with the impact of oxygen management in CA patients, highlighting some potential confounders and limitations and indicating future areas of research in this field. From the currently available literature, high oxygen concentrations during cardiopulmonary resuscitation seem preferable, whereas hyperoxemia should be avoided in the post-CA care. A specific threshold for oxygen toxicity has not yet been identified. The mechanisms of oxygen toxicity after CA, such as seizure development, reactive oxygen species production, and the development of organ dysfunction, need to be further evaluated in prospective studies.
Similar content being viewed by others

Introduction
Sudden cardiac arrest (CA) is the leading cause of death among adults worldwide [1],[2]. In most patients, attempts at cardiopulmonary resuscitation (CPR) remain ineffective and spontaneous cardiac activity cannot be restored [3]. Among those patients who do achieve return of spontaneous circulation (ROSC), there are two key periods when death may occur: early (during the first three days), usually because of recurrent CA or severe cardiovascular failure resulting in multiple organ failure (MOF), and late (beyond day 3), usually secondary to withdrawal of life-sustaining therapies in the absence of neurological recovery [4]. Although several interventions, including target temperature management (TTM), have been introduced into the post-CA care of these patients [5],[6], conflicting results have been obtained [7], and these approaches are not sufficient to prevent the deleterious consequences of brain ischemia in all patients. During the post-CA care, secondary brain insult must be avoided [8] and optimization of brain oxygenation is likely to be an important component of brain recovery. The restoration of adequate systemic hemodynamics is a prerequisite to provide adequate cerebral blood flow in CA patients [9],[10], but brain oxygenation is also determined by the arterial oxygen content. Arterial oxygen pressure (PaO2) itself may influence brain cellular oxygen supply; if hypoxemia (that is, PaO2 of less than 60 mm Hg) is associated with poor outcomes after CA [11], a high PaO2 may also be detrimental in a vulnerable brain, as suggested in patients with traumatic brain injury or stroke [12],[13]. The aims of this article are to review the potential mechanisms of oxygen toxicity after CA and to discuss the clinical impact of oxygen management on post-CA care.
Post-cardiac arrest syndrome: the role of oxygen
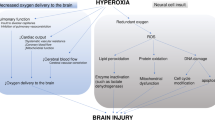
Post-cardiac arrest syndrome (PCAS) is a complex phenomenon, which shares several features with septic shock [7],[14]. In particular, PCAS includes a systemic inflammatory response that can be triggered by the ischemia-reperfusion injury and also specific precipitating events, such as concomitant infections or heart disease. Moreover, PCAS can contribute to brain injury and myocardial dysfunction and can rapidly lead to MOF. The primary ischemia-reperfusion injury [15] activates various intracellular pathways, promoting ion concentration disequilibrium with increased intracellular levels of inorganic phosphate, lactate, and H+, and resulting in an influx of calcium into the cell [16], which aggravates mitochondrial dysfunction and eventually leads to programmed cellular death (apoptosis). After reperfusion has occurred, other mediators, including superoxide (O2-), peroxynitrite (NO2-), hydrogen peroxide (H2O2), and hydroxyl radicals (OH-), contribute to worsen cellular function by oxidizing and damaging numerous cellular components [17] (Figure 1). These reactive oxygen species (ROS) then have a central role in initiating and enhancing the post-ischemic damage [15]. Indeed, supra-normal oxygen concentrations in this context may further stimulate ROS production and contribute to worsen cellular function in a setting of impaired mitochondrial function and impaired oxygen utilization. Moreover, some other systemic detrimental effects of hyperoxemia have been known for many years [18]-[20]. Hyperoxemia causes systemic and coronary vasoconstriction, which can decrease cardiac output and induce myocardial ischemia. In some experimental models of global cerebral ischemia, hyperoxemia has been shown to be detrimental to the brain, probably also because of its vasoconstrictor effects [21],[22]. Hyperoxemia may also provoke or exacerbate seizures, which could aggravate brain injury [23],[24].

Summary of cellular and systemic effects of high oxygen (O
2
) concentrations. H2O, water; H2O2, hydrogen peroxide; NO, nitric oxide; NOS, nitric oxide synthase; superoxide ion; •OH, hydroxide ion; ONOO-, peroxynitrite ion.
superoxide ion; •OH, hydroxide ion; ONOO-, peroxynitrite ion.
Oxygen therapy after cardiac arrest: animal studies
Several studies have assessed the effects of administering high oxygen concentrations in experimental models of CA. Pilcher and colleagues [25] recently reviewed studies that evaluated the role of different oxygenation strategies - that is, one with 100% inspired oxygen fraction (FiO2) and the other with lower FiO2 as a control - after ROSC. Six studies including 95 animals of different species were included in their final meta-analysis; in general, administration of high FiO2 (100%) for 1 hour after ROSC resulted in a worse neurological outcome, as assessed by a neurological deficit score, than other FiO2 values. Four of the five studies that assessed histological damage reported a significantly higher neuronal injury with high FiO2; cerebral metabolic function was also more altered in the high FiO2 group. The extrapolation of such findings to humans, however, may be misleading. First, in all of these experimental studies, the animals were already mechanically ventilated before CA, and this is not usually the case in humans; also, control animals did not all receive the same FiO2 (ranging from 21% to higher levels based on PaO2 values), and the intervention period was rather short (that is, 1 hour). Moreover, different models of CA were employed (that is, asphyxia versus electrical-induced ventricular fibrillation), with shorter durations of CPR than in humans, and in some articles clear PaO2 targets were aimed at, whereas in others only FiO2 was modified without looking at PaO2 values. In addition, no potential neuroprotective therapy, such as TTM, was provided, so that its influence cannot be ascertained. In summary, animal data have highlighted a clear correlation between the application of high FiO2 after CA and poor neurological recovery. Nevertheless, experimental models are quite remote from the human setting, so that the observations cannot be readily translated to humans in the post-CA management.
Oxygen after cardiac arrest: human studies
Two studies reported conflicting results regarding oxygen management during the early phase of CA (Table 1). In 145 out-of-hospital CA patients, Spindelboeck and colleagues [26] observed that high PaO2 (more than 300 mm Hg) levels during CPR were associated with higher rates of ROSC and intact neurological survival when compared with low (PaO2 of less than 60 mm Hg) and normal PaO2. In contrast, Kuisma and colleagues [27] randomly assigned 28 patients to receive either 30% or 100% FiO2 for 1 hour after ROSC; five patients (36%) in the group with lower FiO2 required an increase in FiO2 to 40% because oxygen saturation fell below 95%, according to a standardized protocol. The primary endpoints were biomarker levels of brain injury - that is, neuron-specific enolase (NSE) and S100B - at 24 and 48 hours after ROSC, with no differences in absolute values between the two groups overall. However, in the subgroup of patients who did not undergo TTM, those receiving lower FiO2 had lower NSE levels at 24 hours than the other patients (7.6 ± 4.2 versus 13.5 ± 9.6 μg/mL, P = 0.049). Thus, one may argue that high PaO2 during CPR reflects better lung function or better quality CPR or both, with higher blood flow and better tissue oxygenation (indeed all patients received FiO2 of 100%), whereas administration of high FiO2 immediately after ROSC could enhance brain injury. Importantly, the retrospective nature of the study by Spindelboeck and colleagues [26] may have limited the analysis of all factors possibly affecting outcome (that is, quality of CPR, comorbidities, and so on). Also, blood gas analysis samples were drawn within 60 minutes from CPR initiation, and it is difficult to compare patients analyzed in the early CPR with those included in the later phase, who may have developed pulmonary injury with reduced PaO2 due to prolonged resuscitation itself. It is difficult to determine the role of oxygen levels during CPR per se on neurological outcome of CA patients because they may be a surrogate marker of resuscitation performance or better cardiorespiratory status or both. Moreover, brain injury is a continuous process, and the time course of PaO2 may more significantly influence neurological outcome in these patients.
Few clinical studies have evaluated the role of hyperoxemia during the ICU stay on outcome of CA patients. Kilgannon and colleagues published two different analyses on the same large database from a cohort of patients from 131 US hospitals (Increase Minority Participation and Awareness of Clinical Trials (IMPACT) Database). In the first study [28], including 6,326 patients, hospital mortality was higher in patients with hypoxemia (defined as a PaO2 of less than 60 mm Hg or altered gas exchange with a PaO2/FiO2 ratio of less than 300) or hyperoxemia (PaO2 of more than 300 mm Hg) detected in the first arterial blood gas (ABG) available within 24 hours after admission, compared with patients with normal oxygen levels (hypoxemia 53% versus hyperoxemia 67% versus normoxemia 45%, P < 0.001). In a multivariable logistic regression analysis, hyperoxemia was independently associated with in-hospital mortality: odds ratio (OR) 1.8, 95% confidence interval (CI) 1.5 to 2.2. In the second study, after exclusion of patients with hypoxemia, the investigators wanted to evaluate the association between PaO2, considered as a continuous variable, and in-hospital mortality [29]. In the 4,459 patients studied, the median PaO2 was 231 (interquartile range 149 to 349) mm Hg and in-hospital mortality was 54%. Multivariable analysis yielded a significant 6% increase in mortality for any PaO2 increase by 25 mm Hg (OR 1.06, 95% CI 1.05 to 1.07). Although these two studies support the hypothesis that hyperoxemia is associated with worse outcome after CA, some limitations must be acknowledged. First, only a small proportion of patients were treated with hypothermia. Second, the use of the PaO2/FiO2 ratio does not really reflect hypoxemia, because it is a surrogate of lung failure rather than of low arterial oxygen content. Third, data were not collected following the Utstein style [30] or corrected for severity of disease (for example, Acute Physiology and Chronic Health Evaluation III, or APACHE III). Finally, and even more importantly, only the first ABG was taken into account in the final analysis instead of a mean value over the first 24 hours, which might be more representative of real oxygen exposure during this crucial period.
In a large Australian database (Australian and New Zealand Intensive Care Society-Adult Patient Database, or ANZICS-APD), Bellomo and colleagues [31] evaluated the correlation between different PaO2 levels and hospital mortality in 12,108 patients. Although these authors used the same definitions of hypoxemia and hyperoxemia used in previous studies [28],[29], they reported a lower percentage of patients with hyperoxemia (11% [31] versus 18% [28]) but still a higher mortality rate for patients with hyperoxemia (59%) and hypoxemia (60%) when compared with normoxic patients (47%). In a multivariable model including some major confounding factors, hyperoxemia was significantly associated with mortality (OR 1.2, 95% CI 1.1 to 1.6); however, in a Cox proportional hazards regression model, after adjustment for other relevant covariates (year of admission, treatment limitations, patient’s lowest glucose level in the first 24 hours, patient’s indigenous status, and hospital source from home), PaO2 was no longer statistically associated with poor outcome (hazard ratio 1.1, 95% CI 1.0 to 1.2; P = 0.20). Despite the limitation of a retrospective analysis, this study highlights that some confounders not taken into account in previous studies may have influenced the statistical association between high PaO2 and poor outcome. Importantly, other issues may further limit the power of these findings. The PaO2 from the ABG analysis associated with the worst alveolo-arterial oxygen difference was used, and this PaO2 correlated only fairly with the mean PaO2 (as shown in the analysis of a subgroup of patients) and may not have adequately reflected the exposure of patients to high oxygen concentrations. This observation was also suggested by the lower PaO2 values recorded in this study when compared with previous publications (112 versus 231 mm Hg) [29]. Also, no data on neurological outcomes were reported. Finally, as in previous studies, only a minority of patients underwent hypothermia, but cooling is considered to mitigate ROS production and possibly influence hyperoxemia-mediated brain injury [8].
The first article to include patients treated with hypothermia [32] showed that those with a poor outcome had higher PaO2 values than others (254 versus 198 mm Hg; P = 0.022). A multivariable regression model, including factors known to be associated with poor outcome after CA, confirmed an independent correlation of PaO2 with mortality (OR for a PaO2 increment of every 100 mm Hg above 54 mm Hg 1.48, 95% CI 1.03 to 2.01) and worse neurological outcome (OR 1.48, 95% CI 1.03 to 2.14) at hospital discharge; however, neither FiO2 nor APACHE III score was included in the final analysis. Unfortunately, no specific cutoff of PaO2 was identified to predict poor outcome, although a PaO2 of more than 228 mm Hg was associated with a lower likelihood of neurological recovery. More recently, Ihle and colleagues [33] reviewed 584 patients selected from the ANZICS-APD database, for whom variables could be found following the Utstein model [30]. Unadjusted in-hospital mortality did not differ across different PaO2 ranges (51% hypoxia, 41% normoxia, and 47% hyperoxia; P = 0.28). In a multivariable model including CA characteristics, neither hypoxemia nor hyperoxemia was an independent predictor of in-hospital mortality. In a retrospective cohort of 213 adult patients with CA, Lee and colleagues [34] reported that PaO2 obtained during the first 24 hours was not related to hospital mortality or neurological outcome. In a multivariable model adjusted for established confounding factors after CA, the authors showed a V-shaped relationship between PaO2 and neurological outcome, with the highest probability of good neurologic outcome at PaO2 around 130 mm Hg. Finally, in a prospective observational study (n = 409), Vaahersalo and colleagues [35] calculated the proportion of time spent in different oxygen categories (less than 75 mm Hg, 75 to 150 mm Hg, 150 to 225 mm Hg, and more than 225 mm Hg) during the first 24 hours after CA. The proportion of time spent with a PaO2 of more than 225 mm Hg was similar between groups, as was the association of time within different PaO2 categories and outcome. Mean and highest PaO2 values were higher in patients with good neurological outcome than in those with poor neurological outcome (120 versus 113 mm Hg and 173 versus 150 mm Hg, respectively). Interestingly, the proportion of patients with good neurological outcome was higher in patients with the combination of high mean PaO2 and PaCO2 values.
Perspectives and conclusions
The International Liaison Committee on Resuscitation states that oxygen administration should be titrated to obtain an oxygen saturation of 94% to 96% after ROSC [36]. Thus, routine administration of an FiO2 of 100% is no longer recommended after CPR [36],[37]; however, it still seems prudent to use 100% oxygen during CPR, although the impact of high PaO2 on survival needs to be further evaluated. Although some studies have suggested that hyperoxemia after CA should be considered a cost-free, potentially modifiable risk factor for poor outcome, many potential confounders have been identified and strongly challenge this concept. Moreover, it is very difficult to identify a specific threshold of toxicity, as most of the studies used a PaO2 of more than 300 mm Hg to define hyperoxemia, while some brain injury may also potentially occur at lower values. Also, when a single ABG value was used, the proportion of patients with hyperoxemia ranged from 3% to 25% in the different studies [27]-[29],[31]-[34]. Nevertheless, when all ABG values are considered, the incidence of hyperoxemia may exceed 40% [38], so that the real impact of exposure to high oxygen concentrations has probably been underestimated. Other parameters obtained from the ABG, such as acidemia or hypocapnia, may also be important determinants of poor outcome after CA [39],[40]; however, these variables were not considered in these studies. Importantly, it is worthwhile to prospectively evaluate the mechanisms of oxygen toxicity after CA, such as seizure development, ROS production, impaired microcirculation, or the development of organ dysfunction. Finally, further studies are needed to help understand whether an absolute PaO2 value (that is, `peak’) could be more detrimental than a continuous exposure above a specific threshold and to assess the impact of the timing of hyperoxemia occurrence (early versus late phase after arrest) or of variability in oxygen levels (that is, from hypoxemia to hyperoxemia) in this setting.
Key messages
-
Hyperoxemia may potentially exacerbate or aggravate brain injury after experimental cardiac arrest.
-
Hyperoxemia has been associated with controversial results in humans.
-
Administering high FiO2 (100%) during CPR still seems to be advisable because it may facilitate ROSC.
-
Because of the limited benefit of maintaining potentially harmful supra-normal oxygen levels in such patients, mechanical ventilation should be titrated to maintain an oxygen saturation between 94% and 96% in most patients after ROSC.
Abbreviations
- ABG:
-
Arterial blood gas
- ANZICS-APD:
-
Australian and New Zealand Intensive Care Society-Adult Patient Database
- APACHE III:
-
Acute Physiology and Chronic Health Evaluation III
- CA:
-
Cardiac arrest
- CI:
-
Confidence interval
- CPR:
-
Cardiopulmonary resuscitation
- FiO2:
-
Inspired oxygen fraction
- MOF:
-
Multiple organ failure
- NSE:
-
Neuron-specific enolase
- OR:
-
Odds ratio
- PaO2:
-
Arterial oxygen pressure
- PCAS:
-
Post-cardiac arrest syndrome
- ROS:
-
Reactive oxygen species
- ROSC:
-
Return of spontaneous circulation
- TTM:
-
Target temperature management
References
Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I: Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA. 2008, 300: 1423-1431. 10.1001/jama.300.12.1423.
Atwood C, Eisenberg MS, Herlitz J, Rea TD: Incidence of EMS-treated out-of-hospital cardiac arrest in Europe. Resuscitation. 2005, 67: 75-80. 10.1016/j.resuscitation.2005.03.021.
Goto Y, Maeda T, Nakatsu-Goto Y: Neurological outcomes in patients transported to hospital without a prehospital return of spontaneous circulation after cardiac arrest. Crit Care. 2013, 17: R274-10.1186/cc13121.
Lemiale V, Dumas F, Mongardon N, Giovanetti O, Charpentier J, Chiche JD, Carli P, Mira JP, Nolan J, Cariou A: Intensive care unit mortality after cardiac arrest: the relative contribution of shock and brain injury in a large cohort. Intensive Care Med. 2013, 39: 1972-1980. 10.1007/s00134-013-3043-4.
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K: Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002, 346: 557-563. 10.1056/NEJMoa003289.
Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002, 346: 549-556. 10.1056/NEJMoa012689.
Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, Pellis T, Stammet P, Wanscher M, Wise MP, Åneman A, Al-Subaie N, Boesgaard S, Bro-Jeppesen J, Brunetti I, Bugge JF, Hingston CD, Juffermans NP, Koopmans M, Køber L, Langørgen J, Lilja G, Møller JE, Rundgren M, Rylander C, Smid O, et al: Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. 2013, 369: 2197-2206. 10.1056/NEJMoa1310519.
Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez-Nunez A, Sellke FW, Spaulding C, Sunde K, Vanden Hoek T: Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008, 79: 350-379. 10.1016/j.resuscitation.2008.09.017.
Bisschops LL, van der Hoeven JG, Hoedemaekers CW: Effects of prolonged mild hypothermia on cerebral blood flow after cardiac arrest. Crit Care Med. 2012, 40: 2362-2367. 10.1097/CCM.0b013e318255d983.
Lee JK, Brady KM, Mytar JO, Kibler KK, Carter EL, Hirsch KG, Hogue CW, Easley RB, Jordan LC, Smielewski P, Czosnyka M, Shaffner DH, Koehler RC: Cerebral blood flow and cerebrovascular autoregulation in a swine model of pediatric cardiac arrest and hypothermia. Crit Care Med. 2011, 39: 2337-2345. 10.1097/CCM.0b013e318223b910.
Yan EB, Hellewell SC, Bellander BM, Agyapomaa DA, Morganti-Kossmann MC: Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J Neuroinflamm. 2011, 8: 147-10.1186/1742-2094-8-147.
Rincon F, Kang J, Maltenfort M, Vibbert M, Urtecho J, Athar MK, Jallo J, Pineda CC, Tzeng D, McBride W, Bell R: Association between hyperoxia and mortality after stroke: a multicenter cohort study. Crit Care Med. 2014, 42: 387-396. 10.1097/CCM.0b013e3182a27732.
Rincon F, Kang J, Vibbert M, Urtecho J, Athar MK, Jallo J: Significance of arterial hyperoxia and relationship with case fatality in traumatic brain injury: a multicentre cohort study. J Neurol Neurosurg Psychiatry. 2014, 85: 799-805. 10.1136/jnnp-2013-305505.
Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, Fraisse F, Dinh-Xuan AT, Carli P, Spaulding C, Dhainaut JF, Cavaillon JM: Successful cardiopulmonary resuscitation after cardiac arrest as a `sepsis-like’ syndrome. Circulation. 2002, 106: 562-568. 10.1161/01.CIR.0000023891.80661.AD.
Polderman KH: Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009, 37: S186-S202. 10.1097/CCM.0b013e3181aa5241.
Small DL, Morley P, Buchan AM: Biology of ischemic cerebral cell death. Prog Cardiovasc Dis. 1999, 42: 185-207. 10.1016/S0033-0620(99)70002-2.
Globus MY, Busto R, Lin B, Schnippering H, Ginsberg MD: Detection of free radical activity during transient global ischemia and recirculation: effects of intraischemic brain temperature modulation. J Neurochem. 1995, 65: 1250-1256. 10.1046/j.1471-4159.1995.65031250.x.
Cornet AD, Kooter AJ, Peters MJ, Smulders YM: The potential harm of oxygen therapy in medical emergencies. Crit Care. 2013, 17: 313-10.1186/cc12554.
Neumar RW: Optimal oxygenation during and after cardiopulmonary resuscitation. Curr Opin Crit Care. 2011, 17: 236-240. 10.1097/MCC.0b013e3283454c8c.
Meyhoff CS, Staehr AK, Rasmussen LS: Rational use of oxygen in medical disease and anesthesia. Curr Opin Anaesthesiol. 2012, 25: 363-370. 10.1097/ACO.0b013e328352b402.
Hazelton JL, Balan I, Elmer GI, Kristian T, Rosenthal RE, Krause G, Sanderson TH, Fiskum G: Hyperoxic reperfusion after global cerebral ischemia promotes inflammation and long-term hippocampal neuronal death. J Neurotrauma. 2010, 27: 753-762. 10.1089/neu.2009.1186.
Floyd TF, Clark JM, Gelfand R, Detre JA, Ratcliffe S, Guvakov D, Lambertsen CJ, Eckenhoff RG: Independent cerebral vasoconstrictive effects of hyperoxia and accompanying arterial hypocapnia at 1 ATA. J App Physiol. 2003, 95: 2453-2461.
Morimoto T, Fukuda M, Aibara Y, Nagao H, Kida K: The influence of blood gas changes on hyperthermia-induced seizures in developing rats. Brain Res Dev Brain Res. 1996, 92: 77-80. 10.1016/0165-3806(95)00205-7.
Pilla R, Landon CS, Dean JB: A potential early physiological marker for CNS oxygen toxicity: hyperoxic hyperpnea precedes seizure in unanesthetized rats breathing hyperbaric oxygen. J App Physiol. 2013, 114: 1009-1020. 10.1152/japplphysiol.01326.2012.
Pilcher J, Weatherall M, Shirtcliffe P, Bellomo R, Young P, Beasley R: The effect of hyperoxia following cardiac arrest - a systematic review and meta-analysis of animal trials. Resuscitation. 2012, 83: 417-422. 10.1016/j.resuscitation.2011.12.021.
Spindelboeck W, Schindler O, Moser A, Hausler F, Wallner S, Strasser C, Haas J, Gemes G, Prause G: Increasing arterial oxygen partial pressure during cardiopulmonary resuscitation is associated with improved rates of hospital admission. Resuscitation. 2013, 84: 770-775. 10.1016/j.resuscitation.2013.01.012.
Kuisma M, Boyd J, Voipio V, Alaspaa A, Roine RO, Rosenberg P: Comparison of 30 and the 100% inspired oxygen concentrations during early post-resuscitation period: a randomised controlled pilot study. Resuscitation. 2006, 69: 199-206. 10.1016/j.resuscitation.2005.08.010.
Kilgannon JH, Jones AE, Shapiro NI, Angelos MG, Milcarek B, Hunter K, Parrillo JE, Trzeciak S: Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010, 303: 2165-2171. 10.1001/jama.2010.707.
Kilgannon JH, Jones AE, Parrillo JE, Dellinger RP, Milcarek B, Hunter K, Shapiro NI, Trzeciak S: Relationship between supranormal oxygen tension and outcome after resuscitation from cardiac arrest. Circulation. 2011, 123: 2717-2722. 10.1161/CIRCULATIONAHA.110.001016.
Cummins RO, Chamberlain DA, Abramson NS, Allen M, Baskett PJ, Becker L, Bossaert L, Delooz HH, Dick WF, Eisenberg MS: Recommended guidelines for uniform reporting of data from out-of-hospital cardiac arrest: the Utstein Style. A statement for health professionals from a task force of the American Heart Association, the European Resuscitation Council, the Heart and Stroke Foundation of Canada, and the Australian Resuscitation Council. Circulation. 1991, 84: 960-975. 10.1161/01.CIR.84.2.960.
Bellomo R, Bailey M, Eastwood GM, Nichol A, Pilcher D, Hart GK, Reade MC, Egi M, Cooper DJ: Arterial hyperoxia and in-hospital mortality after resuscitation from cardiac arrest. Crit Care. 2011, 15: R90-10.1186/cc10090.
Janz DR, Hollenbeck RD, Pollock JS, McPherson JA, Rice TW: Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med. 2012, 40: 3135-3139. 10.1097/CCM.0b013e3182656976.
Ihle JF, Bernard S, Bailey MJ, Pilcher DV, Smith K, Scheinkestel CD: Hyperoxia in the intensive care unit and outcome after out-of-hospital ventricular fibrillation cardiac arrest. Crit Care Resusc. 2013, 15: 186-190.
Lee BK, Jeung KW, Lee HY, Lee SJ, Jung YH, Lee WK, Heo T, Min YI: Association between mean arterial blood gas tension and outcome in cardiac arrest patients treated with therapeutic hypothermia. Am J Emerg Med. 2014, 32: 55-60. 10.1016/j.ajem.2013.09.044.
Vaahersalo J, Bendel S, Reinikainen M, Kurola J, Tiainen M, Raj R, Pettilä V, Varpula T, Skrifvars MB: Arterial blood gas tensions after resuscitation from out-of-hospital cardiac arrest: associations with long-term neurologic outcome. Crit Care Med. 2014, 42: 1463-1470. 10.1097/CCM.0000000000000228.
Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, Gabrielli A, Silvers SM, Zaritsky AL, Merchant R, Vanden Hoek TL, Kronick SL: Part 9: post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010, 122: S768-S786. 10.1161/CIRCULATIONAHA.110.971002.
Deakin CD, Nolan JP, Soar J, Sunde K, Koster RW, Smith GB, Perkins GD: European Resuscitation Council Guidelines for Resuscitation 2010 Section 4. Adult Adv Life Support Resusc. 2010, 81: 1305-1352.
Nelskyla A, Parr MJ, Skrifvars MB: Prevalence and factors correlating with hyperoxia exposure following cardiac arrest - an observational single centre study. Scand J Trauma Resusc Emerg Med. 2013, 21: 35-10.1186/1757-7241-21-35.
Schneider AG, Eastwood GM, Bellomo R, Bailey M, Lipcsey M, Pilcher D, Young P, Stow P, Santamaria J, Stachowski E, Suzuki S, Woinarski NC, Pilcher J: Arterial carbon dioxide tension and outcome in patients admitted to the intensive care unit after cardiac arrest. Resuscitation. 2013, 84: 927-934. 10.1016/j.resuscitation.2013.02.014.
Ganga HV, Kallur KR, Patel NB, Sawyer KN, Gowd PB, Nair SU, Puppala VK, Manandhi AR, Gupta AV, Lundbye JB: The impact of severe acidemia on neurologic outcome of cardiac arrest survivors undergoing therapeutic hypothermia. Resuscitation. 2013, 84: 1723-1727. 10.1016/j.resuscitation.2013.07.006.
Acknowledgments
This article is based on a lecture given by FST during the 26th Annual Congress of the European Society of Intensive Care Medicine (Paris, October 2013).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FST and AMDA helped to conceive the manuscript, participated in the data collection, and helped to draft the manuscript. IL participated in the data collection and helped to critically revise the manuscript. JLV helped to critically revise the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Dell’Anna, A.M., Lamanna, I., Vincent, JL. et al. How much oxygen in adult cardiac arrest?. Crit Care 18, 555 (2014). https://doi.org/10.1186/s13054-014-0555-4
Published:
DOI: https://doi.org/10.1186/s13054-014-0555-4