
Abstract
Since Salinomycin (Sal) emerged its ability to target breast cancer stem cells in 2009, numerous experiments have been carried out to test Sal’s anticancer effects. What deserve to be mentioned is that Sal can efficiently induce proliferation inhibition, cell death and metastasis suppression against human cancers from different origins both in vivo and in vitro without causing serious side effects as the conventional chemotherapeutical drugs on the body. There may be novel cell death pathways involving the anticancer effects of Sal except the conventional pathways, such as autophagic pathway. This review is focused on how autophagy involves the effects of Sal, trying to describe clearly and systematically why autophagy plays a vital role in predominant anticancer effects of Sal, including its distinctive characteristic. Based on recent advances, we present evidence that a dual role of Sal involving in autophagy may account for its unique anticancer effects - the preference for cancer cells. Further researches are required to confirm the authenticity of this suppose in order to develop an ideal anticancer drug.
Similar content being viewed by others


Background
According to the latest World Health Organization (WHO) data, cancer is the second-leading cause of death globally and accounts for 8.8 million death in 2015 [1]. However, major current tumor therapeutic strategies like operation, radio- and chemo-therapy still exist some defects, failing to cure most tumor patients completely. Cancer stem cells (CSCs), which are resistant to many current anticancer therapies, perhaps account for the failure of treatments. CSCs refer to the subpopulation of cancer cells endowed with self-renewal, multi-lineage differential capacity and innate resistance to conventional radio- and chemo- therapy [2]. CSCs, relying on those capacities, are regarded as the culprit of recurrence and metastasis of cancer [3, 4]. Hence, eradication of CSCs will be the key to the success of cancer treatment.
Of note, with further study of Sal, it stands out as one of the notable landmarks in the progress of chemotherapeutical drugs on CSCs. Sal, isolated from the bacterium Streptomyces albusin 1974 [5](See Fig. 1), exhibits a broad-spectrum antibiotic activity particularly against Gram-positive bacteria, fungi, parasites, protozoa [5, 6]. It is widely used as an anticoccidial drug in animal farming and is fed to ruminants to improve nutrient absorption and promote growth [7]. In 2009, Gupta et al. screened about 16,000 compounds in order to hunt for chemicals that are preferentially toxic to CSCs. The screening identified 32 substances that are able to impair CSCs. Finally, Sal was found to be the most efficient one, having a more than 100-fold efficiency of Sal compared to Paclitaxel to kill breast CSCs in mice [8]. After that, the efficiency of Sal against the CSCs in several malignancies, including breast-, prostrate-, brain-, blood-, liver-, pancreatic-, skeleton- and lung cancers have been further verified [9,10,11,12]. In addition, it has been proved that Sal is able to kill chemotherapeutical agents resistant cancer cells such as Doxorubicin-, Cisplatin-, Gemcitabine-, Temozolamide-, verapamil- and Imatinib- resistant cells and simultaneously sensitize radio-resistant cancer cells [9, 13,14,15]. Besides its predominant anticancer activities, it has been also verified that Sal does not emerge severe adverse effects on human normal tissues like other conventional chemotherapeutical drugs. Sal induces T-cells apoptosis in T-lymphocytic leukemia patients, but not in healthy people [16]. Similar results have been demonstrated in further studies [17, 18]. Furthermore, several successful pilot studies in cancer patients have showed temporary and minor effects while causing the regression of various solid tumors [9, 19].

The structural formula of Sal. Sal is a 750 Da monocarboxylic polyether antibiotic with unique tricyclic ring system, whose molecular formula is C42H70O11
Despite the predominant antitumor effects and fewer adverse effects of Sal, the mechanism by which Sal brings about cancer cell death while non-malignant cells are exempted from the lethal effects remaining poorly understood. According to the studies both in vivo and in vitro, such following mechanisms that mitochondria-dependent cell death [20, 21], Death receptor - mediated cell death [14], increased DNA damage and cell cycle arrest [22, 23], p-glycoprotein inhibition [24, 25] have been reported to involve the predominant anticancer effects of Sal. However, those mechanisms may just play a partial role in the anti-cancer effects of Sal as conventional chemotherapeutical agents rely on them to induce cancer cell death. Further studies have demonstrated that Sal suppresses Wnt/β-catenin signaling pathway conferring CSCs resistance to radiation [26, 27] and to chemotherapeutical agents [28, 29]. Moreover, other studies have showed that Sal blocks the Hedgehog (Hh) pathway, which plays a crucial role in the stemness maintenance of CSCs [30, 31]. Those may elucidate Sal’s preferential toxicity towards CSCs. Besides the classical cell death pathway, alternative cell death pathway has been also observed in the experiments, the result has displayed that Sal is able to mediate apoptosis in cancer cells through a pathway independent of activation of p53, CD95/CD95L system, caspase, and the proteasome [32]. A variety of cell death pathways or the apoptotic signal pathways forming a complex reticular structure involves in the anticancer effects of Sal, but the key component to initiate that process is still unknown, what a role autophagy plays in the reticular structure requires more studies.
Conventional chemotherapeutical drugs cannot efficiently kill CSCs, which led to the recurrence of the cancer [4, 33, 34]. Comparing to the conventional chemotherapeutical drugs, Sal targeting CSCs must have its unique targets or signaling pathways. Therefore, the study of Sal cannot only provide an effective tumor treatment drug, but also can deepen the understanding of the characteristics of CSCs, so as to give a basis for the development of new anticancer drugs, and ultimately overcome this catastrophic disease.
With a further research, scientists have found that autophagy is a homeostatic process that prevents the accumulation of impaired proteins and organelles and limits the production of reactive oxygen species (ROS). Autophagy can simultaneously provide the cell with nutrients and energy that it needs to survive. However, autophagy plays multifarious roles in the initiation, development and progression of cancer. On the one hand, autophagy suppresses malignant transformation by many mechanisms such as maintaining genomic stability via the elimination of dysfunctional mitochondria and the decrease of ROS production, the degradation of onco-proteins, the induction cell death following oncogene activation, anti-inflammatory function and assisting the immune system to eliminate the malignant cells [35]. On the other hand, after a formation of neoplastic lesion, autophagy is believed to promote and maintain tumorigenesis and to mediate resistance to different forms of anticancer therapy [36]. Autophagy is generally considered to play a cyto-protective role for cell survival against apoptosis. However with further study, many literatures have demonstrated that autophagy is primarily a cell survival pathway, but if excessive, it may kill cells [37,38,39]. So the exact role of autophagy playing in the death process of cancer cells requires further study. Additionally, autophagy is dysregulated in various diseases, such as neurodegenerative diseases, immune system diseases and cancers.
Sal and Autophagy
Sal is an inducer of autophagy
Similar to other chemotherapeutical agents, the administration of Sal in the tumor cells induces autophagy. It was firstly discovered that Sal induces autophagy in colon and breast cancer cells [40]. The vast majority of autophagy has a protective effect in all tested cancer cell lines. However, Sal-induced autophagy in SW620 cells is involved in provoking cell death, as inhibiting autophagy through transfection with siRNA against ATG7 as a result of partially preventing autophagy induced cell death in 24 h by inhibiting autophagy. Pre-treatment with the PI3K inhibitor, wortmannin, which is commonly used to inhibit autophagy, gets an isovalent effect [40].
Relevant studies have undertaken to verify the mechanisms and roles of autophagy induced by Sal in other human cancer cell lines. Most of them have found that Sal induces cyto-protective autophagy, facilitating Sal-induced cell death after inhibiting the autophagy [18, 20, 41,42,43]. Combined use of Sal and chloroquine (CQ), which is an autophagy inhibitor, not only improves antitumor effects of Sal but also reduces the dose of Sal, which means that combined use of autophagy inhibitors reduces the side effects of Sal, though for now Sal’s adverse effects are slight and transient [44]. Although autophagy plays a cyto-protective role in most cancer cell lines, autophagy also can be an alternative cell death way in a small part of the cancer cell line as described above. So whether the combined use of Sal and autophagy inhibitors being beneficial or not are deserved to be further studied in various human cancer cell lines before Sal is used formally in clinical anti-tumor therapy.
Sal is an inhibitor of autophagy as well
Autophagy is a homeostatic process that prevents the accumulation of impaired proteins and organelles and limits the production of ROS, lessening the impairment to cells. It was discovered interestingly that Sal can inhibit the autophagic flux in breast CSCs interfering with their stemness maintenance. Moreover, Sal does not impair the fusion of autophagosomes and lysosomes. Besides, Sal has no effect on the pH of lysosomes. Using an in vitro cathepsin substrate assay, it has been further demonstrated that Sal dramatically reduces the activity of cathepsins, resulting in the inhibition of lysosomal activity. Sal’s inhibition on autophagy may account for that Sal can block the degradation of mCherry-GFP-LC3 and long-lived proteins [45]. The study has finally revealed that Sal inhibits the autophagic flux in cancer cells by inhibiting the lysosomal activity via inactivation of cathepsins without altering the integrity of the lysosomal compartment [45]. Furthermore, another study discovered that Sal induces an aberrant autophagic flux via the production of ROS in glioblastoma as the alleviation of ROS production restored the autophagic flux. In addition, the inactivation of cathepsin B, Sal also increases the lysosome membrane permeability (LMP) through lipid oxidation, which is confirmed by the release of cathepsin B from lysosomes (See Fig. 2). Both lead to the blocking of autophagic flux. However, a study has found that Sal can block acidification as a result of that the increase of ROS levels can alter lysosomal lipids, affecting lysosome-autophagosome fusion. However, another study has showed a contradictory result [46].

Diagram illustrates the positions of Sal’s inhibition of autophagic flux. Macroautophagy begins with engulfment of the cytoplasmic materials by the phagophore, which detains the materials into a double-membrane vesicle (i.e. autophagosome). The autophagosome fuses with a lysosome to form an autolysosome, and then the contents are degraded by the lysosome. Initiation of autophagosome formation requires the ULK1-Atg13-FIP200 complexes and Beclin1-class III PI3K complexes. Two conjugation systems, Atg12-Atg5-Atg16 and LC3, are critical to the elongation and enclosure step of the autophagosome formation. Lipid conjugation leads to the conversion of the soluble form of LC3-I to the autophagic vesicle-associated form LC3-II, which is widely used as a marker of autophagy. Sal is found to increase the lysosome membrane permeability (LMP), which affects the acidification and the integrity of lysosomes. Moreover, Sal can also inactivate cathepsins in lysosomes, resulting in the inhibition of lysosomal activity
Moreover, a new study has displayed that Sal suppresses late stages of autophagy, leading to impaired recycling and the accumulation of dysfunctional mitochondria accompanying increased ROS- production. All of which are associated with induction of apoptosis. And that the effects of Sal are dose- and time- dependent manner and could be readily replicated by pharmacological or genetic inhibition of Hepatocellular Carcinoma (HCC)-autophagy alone [47]. It has been reported in different animal models that the tumor extracellular pH (pHe) ranges are between 5.9 and 7.2 [48, 49], with average tumor pHe often reported ~ 6.5 [50, 51]. A study has discovered that unlike CQ and its derivatives whose activity are to different degrees when counteracted by acidosis, the ability of Sal to inhibit autophagic flux is potently enhanced in different conditions of both transient and chronic acidosis [52]. Such activity may be linked to the physicochemical characteristics of Sal, which is a weakly acidic, while lipophilic compound with a reported pKa of 6.4 due to its carboxylic group [53]. Moreover, a study also has demonstrated that tumor-mimicking micro-environments can enhance the action of Sal. Furthermore, hypoxic, low-glucose and low-nutrient conditions in hypo-perfused tumor, actually are able to potentiate Sal’s toxicity, and to increase its preferential anticancer activity [52].
Generally speaking, the ability of Sal to induce or inhibit autophagic flux has been verified in various human cancer cell lines. For example, Sal can induce functional autophagy at lower concentrations and such a dose is cell type-dependent. Interestingly, PC3 cells show active autophagic flux at 10 μM concentration of Sal while murine embryonic fibroblasts already show an inhibition of autophagic flux at such doses. A higher concentration of Sal (i.e. 30 μM) inhibits autophagic flux in both cell types. This result has manifested that Sal is an inducer of autophagy, whereas autophagic flux inhibition is a secondary response [54]. Similarly, 10 μM Sal treatment of MCF7 cells for 16 h downregulated Beclin1 and ATG12, which are involved in the initiation and elongation of autophagy, while showing an increase at initial time points (2~ 8 h) and lower doses [40]. Whether the autophagic inhibition of Sal exists in all cancer cell lines demanding further confirmation. And whether there are additional mechanisms besides the increase of LMP and inactivation of cathepsins equally worthy of study. Therefore, when using Sal, it should be determined its suitable doses when Sal acts as an autophagic inhibitor in various tumors and induces cancer cells death simultaneously so as to provide a rational guide for dose selection of clinical treatment of tumors.
The conceivable interpretation for Sal’s preference for cancer cells or CSCs
Chemotherapeutics currently used kill tumor cells but also cause impairment to human normal tissues, such as myelosuppression, immunosuppression, nephrotoxicity, liver injury. Interestingly, Sal selectively induces cancer cells death while sparing benign cells. This may be related to Sal’s distinct targets or signaling pathways that are lethal to cancer cells while are slighter to human normal tissues. Here we will explore whether the inhibition of autophagy is one of the explanations for Sal’s preference for cancer.
Mitochondria, generally called the powerhouses of the cell, comprises a dynamic interconnected network of tubular structures that are engaged in fission and fusion processes [55]. Mitochondrial fission is mediated by localization of dynamin-related protein (Drp-1) on the mitochondrial site of division whereas fusion is mediated by mitofusin proteins (mitofusins 1 and 2) along with optic atrophy protein 1 (OPA1) [56]. Lack of both mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) results in damaged mitochondrial fusion, but both of them can compensate each other’s deficiency [57,58,59,60]. Facing stress or any event leading to dysfunction of mitochondria, the organelle undergoes an asymmetric and protective fission that aims to spare at least part of stressed mitochondria, by cleaving the organelle into a normally-functional part, and a dysfunctional one. A recent study has described that cancer cells often have much smaller, fragmented mitochondria in comparison to normal cells. In the same study, the results have showed that cancer cells express an increased level of Drp1 and decreased level of Mfn protein, leading to a constant mitochondrial fission with impaired fusion, resulting in a smaller fragmented mitochondria in cancer cells [61]. Another study has displayed that specific toxicity of Sal to cancer cells is through mitochondrial hyperpolarization that is observed preferentially in cancer cells. Moreover, Sal-triggered depletion of cellular ATP, in cancer cells but not in primary cells, contributes to Sal’s preferential anticancer toxicity to cancer [62]. Furthermore, Sal induces depolarization of mitochondria contrasting to Dichloroacetate (DCA), which is also a K+ ion channel modulator, and a molecule that also preferentially targets cancer cells [63, 64]. The observed cancer cells specific deleterious effect of Sal is explained by the fact that cancer cells harbor various abnormalities within mitochondria, such as hyperpolarized mitochondria along with defective mitochondrial fission and fusion mechanisms due to the lack of functionally intact mitofusin proteins, DRP1 and other proteins involved in the mitochondrial dynamics [61, 63]. Similarly, a recent study has pointed out that Sal in concentrations effective against CSCs exerts profound toxicity towards both dorsal root ganglia as well as Schwann cells. With further study, the result has showed that the toxic effect is mediated by elevated cytosolic Na+ concentrations, which in turn causes an increase of cytosolic Ca2+ by means of Na+/Ca2+ exchangers (NCXs) in the plasma membrane as well as the mitochondria membrane. Elevated Ca2+ then activates calpain, which triggers caspase-dependent apoptosis involving caspases-12, − 9 and − 3. Combined inhibition of calpain and the mitochondrial NCXs result in significantly decreased cytotoxicity as compared to caspase-3 inhibition in vitro [65]. Furthermore, co-treatment with an inhibitor of the mitochondrial NCX can significantly reduce structural damage in the peripheral nervous system without impairing Sal’s antineoplastic efficacy in vivo [66]. This may account for the abnormal mitochondria in cancer cells, which is not resistant to the effect of Sal. Fig. 3 shows the conceivable mechanism by which Sal damages peripheral nervous system.

Diagram illustrates the observed effects of Sal-induced toxicity in DRGNs. Sal triggers an increase of cytosolic Na+ concentrations, with a secondary increase in cytosolic Ca2+, which is mediated by NCXs in the plasma membrane and mitochondria. Elevatory intracellular Ca2+ levels activate the protease calpain, which leads to apoptotic cell death by the activation of caspase pathways involving caspase 12, caspase 9 and caspase 3. Caspase 9 can be alternatively activated by cytochrome c released from depolarized mitochondria
Emerging data indicate the crucial role of autophagy in the survival, self-renewal and differentiation of both stem cells and CSCs [67,68,69]. Recent study has found that the basal autophagic flux is higher in the ALDH(+) population derived from the HMLER cell line than in the corresponding ALDH1(−) population, the former owns more CSCs features than the latter. Autophagic flux falls in both ALDH(+) and ALDH(−) populations following Sal treatment. However, the fall of autophagic flux is greater in ALDH(+) population than in the ALDH(−) one (51% vs. 10%, respectively). Consistent with data has showed that apoptotic cell death was higher in the ALDH1(+) population derived from the HMLER cell line than in the ALDH(−) population [45]. Similar results have been acquired by another study that the subline HMLER CD24 low, carrying CSC properties has an increased sensitivity to autophagy inhibition mediated by Sal with respect to the non-CSC subline HMLER CD24(+) [52].
Autophagy is generally considered to be vital for the elimination of dysfunctional mitochondria that may actually consume ATP, and/or generate excessive amounts of harmful ROS, thus contributing to loss of cellular homeostasis [70], thus the inhibition of autophagy will enhance the apoptosis. The elimination of dysfunctional mitochondria is highly dependent on autophagy, however, Sal can induce a profound inhibition on the late stage of autophagic flux. That is, on the one hand to induce deformed mitochondria in cancer cells to be dysfunctional, on the other hand Sal inhibits the elimination of these dysfunctional mitochondria with increased ROS-production and induction of apoptosis.
The mechanisms of autophagy induced by Sal
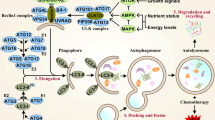
Since the first article, reporting that Sal is able to induce autophagy in cancer cells, was published in 2012 [40], different tumor cell lines have been used to detect the mechanism of autophagy induced by Sal. Sal is a K+ ionophore that interferes with transmembrane K+ potential and facilitates the efflux of K+ from mitochondria [71]. Sal causes the decline of mitochondrial membrane potential (MMP) and the accumulation of ROS [20, 72]. Most of the tumor cells are found to own increased ROS after the treatment with Sal, which plays an important and complex role in autophagic activation. To clarify the role of ROS in Sal-induced autophagy, U2OS cells are pre-treated with N-acetyl-L-cysteine (NAC), a ROS inhibitor. As a result, the sharp decrease of LC3II expression and acidic vesicular organelles (AVO) accumulation is detected [20]. Similar results are also found in osteoblastoma [42], breast cancer [73], prostate cancer [74], glioma [75]. Growing data have manifested that ROS inhibits PI3K/AKT/mTOR signaling [76] and activates the AMP-activated protein kinase (AMPK) signaling [77]. ROS is also reported to initiate mitogen-activated protein kinase (MAPK) signaling, including of C-jun N-terminal kinase (JNK), p38 and extracellular signal-regulated kinases (ERK) [78] (See Fig. 4). Except for the three mechanisms mentioned above, DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is also considered to be required for Sal-induced autophagy activation [18]. Table 1 has summarized the relevant information in recent advances.

Diagram illustrates the dominating signaling pathways involving Sal- induced autophagy. Sal is able to induce intensive autophagic flux than commonly used autophagic inducers such as chloroquine, as many autophagy-initiated pathways being activated by Sal. ROS is deemed to play a key role in it. Pretreating with NAC, an ROS inhibitor, suppresses the autophagic flux being confirmed by different researchers. Sal activates AMPK signaling pathway, MAPK signaling pathway and ER stress initiating autophagy, and inhibits PI3K/AKT/mTORC1 axis simultaneously. (1) AMPK activates autophagy by directly binding and activating ULK1 complex through phosphorylation of Ser 317 [110, 111]. Enhanced stimulatory TSC2 phosphorylation at Ser-1387 by AMPK, and reduced inhibitory TSC2 phosphorylation at Ser-939/Thr-1462 catalyzed by AKT augmented TSC2/TSC1 activity, which led to mTORC1 inhibition. AMPK-mediated raptor phosphorylation further reduced mTOR’s kinase function and mTORC1 activity [42]; (2) Moreover, it has been reported that ROS suppress PI3K/AKT/mTOR signaling [112]. In addition, it’s demonstrated that Sal suppresses AKT1 activity through ATF4-DDIT3/CHOP-TRIB3-AKT1 axis in human cancer cells after activation of ER stress response, resulting in MTOR inhibition and autophagy consequently [43]; (3) ROS inactivates MAPK phosphatases [113]. This subsequently leads to the phosphorylation of JNK, and of its target the transcription factor JUN. JNK activation may promote autophagy through induction of ATG7 [94], or by phosphorylation of BCL2, which leads to dissociation of BCL2 from Beclin-1 [95]
PI3K/AKT/mTOR signaling pathway
Two kind of mammalian target of rapamycin (mTOR) complexes, the mTOR complex 1 (mTORC1) and complex 2 (mTORC2), have been verified. The rapamycin-sensitive mTORC1 is composed of mTOR, regulatory associated protein of mTOR (Raptor), G protein β-subunit-like protein (GβL) and DEP domain-containing mTOR-interacting protein (DEPTOR). The mTOR-interacting protein can stabilize the mTOR dimer and promote mTOR’s substrate specificity [79,80,81,82]. In comparison, mTORC2, which is not as sensitive to rapamycin as the former, is composed of mTOR, GβL, rapamycin-insensitive companion of mTOR (Rictor), protein observed with RICTOR (Protor) and stress-activated protein kinase-interacting protein 1 (SIN1) [80, 81] mTORC1 obtains energy via small GTPases, for instance, Ras-related GTP binding protein (Rag) and Ras homolog enriched in brain (Rheb). Elevated level of amino acid and growth factors can activate mTORC1 through Rag and Rheb, separately. Activation of mTORC1 mainly lead to the phosphorylation of two downstream targets, p70 ribosomal S6 kinase/S6 ribosomal protein (p70S6K/S6RP) and eukaryotic translation-initiation factor 4E–binding protein 1 (4E–BP1), enhancing protein synthesis from increased ribosome biogenesis and messenger RNA translation under nutrient-rich conditions, promoting lipid biogenesis, and inhibiting autophagy [81].. The TSC2/TSC1 tuberous sclerosis complex, acting as downstream of AKT, negatively regulates mTORC1 by inhibiting the GTPase activity of Rheb, which is a positive regulator of mTORC1. TSC2/TSC1 inhibition due to AKT-mediated TSC2 phosphorylation at serine-939 and threonine-1462 leads to mTORC1 activation, whereas mTORC1 inhibition and consequent cell growth arrest in response to nutrient deprivation. Under nutrient-rich conditions, mTORC1 limits autophagy to counter lysosomal engulfment and degradation of cellular organelles and other cytosolic contents by mediating inhibitory phosphorylation of the unc-51-like kinase 1 (ULK1) complex, an obligatory component of autophagosome [83] .
A study has verified that Sal blocks inhibitory phosphorylation of TSC2 by AKT. Treatment of 1 μM Sal in the cells pre-treated with an Akt inhibitor, Triciribine, triggered increased accumulation of the autophagic marker LC3-II. However, under the same conditions in the presence of 10 μM Sal, it cannot observe any change in LC3-II levels compared to Sal treatment alone [84]. Sal’s blocked effect on AKT is further proved that reduced TSC2 phosphorylation at Ser-939 and Thr-1462 (two AKT-targeted sites) within 6 h Sal treatment contributes to mTORC1 inactivation [41]. Furthermore, Sal decreases phosphorylated of AKT and phosphorylated mTOR in prostate cancer cells. In addition, pretreatment with NAC, a general ROS scavenger, inhibits Sal-induced autophagy by suppressing ROS production [74].
AMPK signaling pathway
AMPK activation dictates energy metabolism, gene transcription, cell mitosis and autophagy through regulating its many downstream kinases [85, 86]. In contrast to mTORC1, AMPK is a positive regulator of autophagy under the condition of nutrient/energy depletion. On the one hand, phosphorylation of TSC2 and raptor by AMPK is known to elevate TSC2/TSC1 activity and reduce mTOR’s kinase function, respectively [87, 88]. On the other hand, AMPK stimulates phosphorylation of the ULK1 complex, which is essential for the initiation of autophagy [89].
Sal induces a profound AMPK activation. AMPK inhibition by compound C or by AMPKα RNAi prevents Sal-induced autophagy activation. AMPK activation relies on ROS production in osteoblastoma cells, antioxidant NAC significantly inhibits Sal-induced AMPK activation and autophagic induction [42]. Further, another study has reported that AMPK activation and raptor phosphorylation are detected within 1 h after Sal treatment, whereas increased TSC2 phosphorylation at Ser-1387 and reduced TSC2 phosphorylation at Ser-939/Thr-1462 are detected after 2 h -- changes being more prominent at 20 h [41].
MAPK signaling pathway
MAPK signaling pathway is generally considered to be a vital pathway to initiate the autophagic flux [90, 91].One way to activate the MAPK pathways is through the inactivation of MAPK-phosphatases, of which ROS can oxidize the catalytic site cysteine [92]. Another pathway for the activation of JNK proceeds via apoptotic signal-regulating kinase 1 (ASK1) [93]. This subsequently leads to the phosphorylation of JNK, and its target the transcription factor JUN. JNK activation may promote autophagy through the induction of ATG7 [94], or phosphorylation of BCL2, leading to dissociation of BCL2 from Beclin-1 [95].
Sal can lead to the formation of ROS eliciting JNK activation and induction of the transcription factor JUN. However, JNK inhibition can only partially block autophagy. Other signaling pathways must also play a role after administration of Sal [40]. Moreover, pretreatment with PD98059 and SB203580, an ERK and p38 inhibitors, suppresses the Sal-induced autophagy by reversing the upregulation of ERK and p38. Notably, Sal decreases the phosphorylation of AKT and the phosphorylation of mTOR in prostate cancer cells. In addition, pretreatment with NAC, an antioxidant, inhibits Sal-induced autophagy by suppressing ROS production. Furthermore, Sal activates ERK and p38 MAPK in a time-dependent manner in both PC-3 and LNCaP cells. Autophagy and LC3 expression are significantly restored by a ROS scavenger, NAC, in both PC-3 and LNCaP cells [74].
ER stress
The endoplasmic reticulum (ER) is an organelle where synthesis, folding and maturation of proteins happen. Growing evidence indicate that the ER may act as a source of the membrane of autophagosomes [96, 97]. ER stress activates the unfolded protein response (UPR), which initiates the inhibition of protein translation through phosphorylation of eukaryotic translation initiation factor 2A (EIF2A) [98]. The phosphorylation of EIF2A stimulates the selective translation of transcription factors nuclear protein 1(NUPR1), activating transcription factor 4 (ATF4), and DNA damage inducible transcript 3(DDIT3) as well as the pseudokinase tribbles pseudokinase 3(TRIB3). The pseudokinase TRIB3 inhibits the AKT1-MTORC1 axis to stimulate autophagy [99, 100].
It has been demonstrated that Sal suppresses AKT1 activity through ATF4- DDIT3/CHOP- TRIB3- AKT1 axis in human non-small cell lung cancer (NSCLC) cells after activation of ER stress response, resulting in mTOR inhibition and autophagy consequently. Moreover, Sal upregulates ER stress-related proteins such as phospho-EIF2A, ATF4, DDIT3 in a time- and dose- dependent manner in human NSCLC cells. Furthermore, using RNA interference against ATF4 or DDIT3 and inhibitors of ER stress in combination with Sal, it has been confirmed that the EIF2A-ATF4-DDIT3 axis is the crucial mediator of Sal-induced autophagy. Besides, data have demonstrated that Sal reduces the activation of AKT1 as well as its downstream substrate mTOR, leading to autophagy. Similar study also has demonstrated that Sal induces ROS generation and the ROS scavenger NAC is found to inhibit the Sal-induced apoptosis, ER stress and autophagy. The inhibition of ER stress with 4-phenylbutyric acid depresses Sal-induced apoptosis and autophagy [75].
Other mechanisms
In addition to the mainstream of autophagy activated pathways are mentioned above. DNA-PKcs is also required for Sal-induced autophagic activation [18]. DNA-PKcs is the 460-kDa serine/threonine protein kinase that belongs to PI3K-like protein kinase (PIKK) kinase family [101]. DNA-PKcs will be activated when facing DNA damages, and its normal function is to provoke non-homologous end joining (NHEJ) pathway to repair DNA double strand breaks [102]. DNA-PKcs inhibition, shRNA knockdown or miR-101 expression inhibits Sal-induced Beclin-1 expression and autophagic induction [18].
Sal-induced cancer cell death via other cellular responses
It has been reported that Sal also induces apoptosis, which differs among the diverse cell types, in cancer cells [14, 16, 72, 103, 104] or different originated CSCs [7, 21, 105]. Apoptosis is induced in colorectal CSCs in a caspase-dependent manner, which proved by an increase of cleaved caspase-3, -8 and -9. JC-1 staining further revealed that Sal induces colorectal cancer cell apoptosis via the mitochondrial pathway [21]. Besides, Sal has been reported to induce human lung cancer cell apoptosis through the caspase 3/7-associated cell death pathway [106]. Moreover, Sal triggers apoptosis of PC-3 cells by increasing the intracellular ROS level, which is associated with reduced MMP, translocation of Bax protein to mitochondria, release of cytochrome c to the cytoplasm, activation of the caspase-3, cleavage of PARP-1 as well as the inhibition of the survival protein Bcl-2 [72]. It also has been noted that non-malignant RWPE-1 prostate cells are relatively less sensitive to Sal-induced lethality [72]. Besides the classical apoptotic pathway, Sal has been demonstrated to activate a unique apoptotic pathway that is not accompanied by cell cycle arrest and is independent of tumor suppressor protein p53, caspase activation, the CD95/CD95L system and the proteasome, which has been thought to overcome the high resistance to other anti-cancer drugs of cancer cells by increasing the expression of the Bcl-2 protein, P-gp as well as the 26S proteasome [16].
Uncontrolled cell cycle is an extremely important part of tumorigenesis. Several studies have also found that Sal can play a role in inhibiting cancer cell proliferation by inducing cell cycle arrest. It has been documented that Sal induces apoptosis in NCI/ADR-RES, DXR, and OVCAR-8 ovarian cancer cells via downregulation of S-phase kinase-associated protein-2 (Skp-2) and signal transducer and activator of transcription 3 (Stat3) inactivation. Skp-2 is overexpressed in majority of the CSCs. p27kip1 protein inhibits tumorigenesis through inhibiting cell cycle changes. Sal induces degradation of Skp2 and thus accumulated p27Kip1, ultimately cell cycle being arrested at G1 and apoptosis being induced [25]. Another study has showed that high concentrations of Sal induces a G2 arrest via downregulation cyclin B1, downregulation of survivin and triggering apoptosis. What is an interesting thing is that the administration with low concentrations of Sal induces a transient G1 arrest through downregulation cyclin D1 at an earlier time point and G2 arrest via downregulation cyclin B1 at a later point [107].
Autophagic cell death (ACD), named type II programmed cell death, distinguishes from apoptosis and necrosis, which was first established based on observations of increased autophagic markers in dying cells [108]. There is a big difference between ACD and apoptosis process. The morphological characteristics of apoptosis-related cells are the clearance of cytoskeleton and other proteins by caspases. Cytoskeletal clearance occurs in the early stages of apoptosis, but the organelles are cleared later; On the contrary, ACD is associated with a large accumulation of autophagic vacuoles in the cell, so that the degradation of organelles occurs at an early stage, and the cytoskeleton is cleared at a later stage [109]. In most cases, autophagy can serve as a cell survival promoter, however, autophagy may also act as cell death inducer. The pro-apoptotic effect of autophagy may result from two independent functions of autophagy: the pro-apoptotic function and the induction of ACD [108]. The autophagy regulator Atg5 activates caspase by interacting with the adaptor protein Fas-associated protein with death domain (FADD), a component of the extrinsic apoptotic pathway. Alternatively, cleavage of Atg5 by calpain causes the truncated Atg5 protein to translocate to the mitochondria to induce mitochondrial outer membrane permeabilization (MOMP) leading to cytochrome c release and caspase activation [109]. As a whole, there is a complicated relationship between autophagy and apoptosis. As mentioned above, Sal has been reported to induce cell death mainly via mitochondria-mediated apoptotic pathway, which can be considered as the executor of cancer cell death resulting from the severe damage caused by Sal. Whether the conjecture that the abnormal mitochondria in cancer cells or CSCs, which are not resistant to the effect of Sal, can be impaired by Sal and thus initiates the cell death process are true or not being worthy of further study. Moreover, several studies have showed that Sal both induces intensive autophagy and inhibits the late stage of autophagic flux which may play a crucial role in the ACD or pro-apoptotic process being of equal significance.
Conclusion and perspective
Sal, being a K+ ionophore, can effectively target CSCs, kill chemo-resistant cancer cells and sensitize radio-resistant cancer cells simultaneously. Of note, its side effects are much less than the conventional chemotherapeutical drugs. Although Sal exhibits predominant anticancer effects in vivo and in vitro experiments and a small number of clinical trials, the exact mechanisms are still unknown.
We summarize the autophagy-related researches, providing a new sight into Sal’s preference for CSCs and cancer cells. Cancer cells are deemed to possess abnormal mitochondria, which may be not resistant to ion fluctuations caused by Sal, then leading to the dysfunction of organelles and the production of ROS, which initiates autophagy. Similarly, DCA, which is likewise a K+ ion channel modulator, preferentially targets cancer cells. Meanwhile, Sal can inhibit the late stage of the autophagic flux, which leads the accumulation of dysfunctional organelles and produces excessive ROS. The combined effects of these two processes lead to the death of tumor cells. The combination of a K+ ionophore and autophagic inhibitor may be able to confirm the rationality of this conjecture.
In addition, some studies have showed that the combined use of Sal and CQ, which is an autophagy inhibitor, not only improves antitumor effects of Sal but also reduces the dose of Sal [44]. Whereas, Sal is able to inhibit the late stage of autophagic flux, so whether the combined use of an early stage inhibitor or a late stage inhibitor can amplify the anticancer effects of Sal requires further studies. What’s more, another study has found that co-treatment with an inhibitor of the mitochondrial NCX can significantly reduce structural damage in the peripheral nervous system without impairing Sal’s anticancer efficacy in vivo [65]. Therefore, a large number of further studies should be carried out to provide a basis for future rational combinative therapy.
CSCs are generally considered to be the chief culprit for the initiation, recurrence and metastasis of cancer. Sal preferentially targeting CSCs may bring us closer to find a cure against this devastating disease. Generally speaking, specific and more potent toxicity of Sal towards CSCs and cancer cells without much adverse effect towards normal cells promise Sal may be an effective anticancer agent in the near future. Understanding the biological mechanism of Sal inducing cell death can aid in designing more effective and less toxic therapeutic strategies. As a new anticancer compound, whether Sal could eventually enter into clinical application needs the joint efforts of all researchers. Anyway the in-depth study of Sal’s direct targets or related signaling pathways will open a new chapter in the near future against cancers.
Abbreviations
- ACD:
-
Autophagic cell death
- AMPK:
-
AMP-activated protein kinase
- ASK1:
-
Apoptosis signal-regulating kinase 1
- ATF4:
-
Activating transcription factor 4
- AVO:
-
Acidic vesicular organelles
- CQ:
-
Chloroquine
- CSCs:
-
Cancer stem cells
- DCA:
-
Dichloroacetate
- DDIT3:
-
DNA damage inducible transcript 3
- DEPTOR:
-
DEP domain-containing mTOR-interacting protein
- DNA-PKcs:
-
DNA-dependent protein kinase catalytic subunit
- DRGNs:
-
Dorsal root ganglia neurons
- Drp-1:
-
Dynamin-related protein
- EIF2A:
-
Eukaryotic translation initiation factor 2A
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinases
- FADD:
-
Fas-associated protein with death domain
- GβL:
-
G protein β-subunit-like protein
- HCC:
-
Hepatocellular Carcinoma
- Hh:
-
Hedgehog
- JNK:
-
C-jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- Mfn1:
-
Mitofusin 1.
- Mfn2:
-
Mitofusin 2.
- MMP:
-
Mitochondrial membrane potential.
- MOMP:
-
mitochondrial outer membrane permeabilization.
- mTOR:
-
Mammalian target of rapamycin.
- NAC:
-
N-acetyl-L-cysteine.
- NCXs:
-
Na+/ Ca2+ exchangers.
- NHEJ:
-
Non-homologous end joining.
- NSCLC:
-
Non-small cell lung cancer.
- NUPR1:
-
Nuclear protein 1.
- OPA1:
-
Optic atrophy protein 1.
- pHe:
-
Extracellular pH.
- PIKK:
-
PI3K-like protein kinase.
- Protor:
-
protein observed with RICTOR.
- p70S6K:
-
p70 ribosomal S6 kinase.
- Raptor:
-
regulatory associated protein of mTOR.
- Rag:
-
Ras-related GTP binding protein.
- Rheb:
-
Ras homolog enriched in brain.
- Rictor:
-
rapamycin-insensitive companion of mTOR.
- ROS:
-
Reactive oxygen species.
- Sal:
-
Salinomycin.
- SIN1:
-
stress-activated protein kinase-interacting protein 1.
- Skp-2:
-
S-phase kinase-associated protein-2.
- Stat3:
-
signal transducer and activator of transcription 3.
- S6RP:
-
S6 ribosomal protein.
- TRIB3:
-
Tribbles pseudokinase 3.
- ULK1:
-
Unc-51-like kinase 1.
- UPR:
-
Unfolded protein response.
- 4E–BP1:
-
4E–binding protein 1.
References
Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (London, England). 2012;7(4):597–615.
Shibata M, Shen MM. The roots of cancer: stem cells and the basis for tumor heterogeneity. BioEssays : news and reviews in molecular, cellular and developmental biology. 2013;35(3):253–60.
Chen D, Wu M, Li Y, Chang I, Yuan Q, Ekimyan-Salvo M, Deng P, Yu B, Yu Y, Dong J, et al. Targeting BMI1+ cancer stem cells overcomes Chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell. 2017;20(5):621–634.e626.
Miyazaki Y, Shibuya M, Sugawara H, Kawaguchi O, Hirsoe C. Salinomycin, a new polyether antibiotic. The Journal of antibiotics. 1974;27(11):814–21.
Schafer H, Clauss W, Hornicke H. Cationophore properties of the new polyether antibiotic Salinomycin investigated in distal rabbit colon in vivo and in vitro. Comparative biochemistry and physiology A, Comparative Physiology. 1984;79(3):387–92.
Naujokat C, Fuchs D, Opelz G. Salinomycin in cancer: a new mission for an old agent. Mol Med Rep. 2010;3(4):555–9.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–59.
Antoszczak M, Huczynski A. Anticancer activity of polyether Ionophore-Salinomycin. Anti Cancer Agents Med Chem. 2015;15(5):575–91.
Calzolari A, Saulle E, De Angelis ML, Pasquini L, Boe A, Pelacchi F, Ricci-Vitiani L, Baiocchi M, Testa U. Salinomycin potentiates the cytotoxic effects of TRAIL on glioblastoma cell lines. PLoS One. 2014;9(4):e94438.
He L, Wang F, Dai WQ, Wu D, Lin CL, Wu SM, Cheng P, Zhang Y, Shen M, Wang CF, et al. Mechanism of action of salinomycin on growth and migration in pancreatic cancer cell lines. Pancreatology : official journal of the International Association of Pancreatology (IAP) [et al]. 2013;13(1):72–8.
Larzabal L, El-Nikhely N, Redrado M, Seeger W, Savai R, Calvo A. Differential effects of drugs targeting cancer stem cell (CSC) and non-CSC populations on lung primary tumors and metastasis. PLoS One. 2013;8(11):e79798.
Daman Z, Montazeri H, Azizi M, Rezaie F, Ostad SN, Amini M, Gilani K. Polymeric micelles of PEG-PLA copolymer as a carrier for Salinomycin against gemcitabine-resistant pancreatic cancer. Pharm Res. 2015;32(11):3756–67.
Parajuli B, Shin SJ, Kwon SH, Cha SD, Chung R, Park WJ, Lee HG, Cho CH. Salinomycin induces apoptosis via death receptor-5 up-regulation in cisplatin-resistant ovarian cancer cells. Anticancer Res. 2013;33(4):1457–62.
Kim WK, Kim JH, Yoon K, Kim S, Ro J, Kang HS, Yoon S. Salinomycin, a p-glycoprotein inhibitor, sensitizes radiation-treated cancer cells by increasing DNA damage and inducing G2 arrest. Investig New Drugs. 2012;30(4):1311–8.
Fuchs D, Heinold A, Opelz G, Daniel V, Naujokat C. Salinomycin induces apoptosis and overcomes apoptosis resistance in human cancer cells. Biochem Biophys Res Commun. 2009;390(3):743–9.
Ketola K, Hilvo M, Hyotylainen T, Vuoristo A, Ruskeepaa AL, Oresic M, Kallioniemi O, Iljin K. Salinomycin inhibits prostate cancer growth and migration via induction of oxidative stress. Br J Cancer. 2012;106(1):99–106.
Zhen YF, Li ST, Zhu YR, Wang XD, Zhou XZ, Zhu LQ. Identification of DNA-PKcs as a primary resistance factor of salinomycin in osteosarcoma cells. Oncotarget. 2016;7(48):79417–27.
Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:950658.
Kim SH, Choi YJ, Kim KY, Yu SN, Seo YK, Chun SS, Noh KT, Suh JT, Ahn SC. Salinomycin simultaneously induces apoptosis and autophagy through generation of reactive oxygen species in osteosarcoma U2OS cells. Biochem Biophys Res Commun. 2016;473(2):607–13.
Zhang C, Tian Y, Song F, Fu C, Han B, Wang Y. Salinomycin inhibits the growth of colorectal carcinoma by targeting tumor stem cells. Oncol Rep. 2015;34(5):2469–76.
Zhang Y, Zuo Y, Guan Z, Lu W, Xu Z, Zhang H, Yang Y, Yang M, Zhu H, Chen X. Salinomycin radiosensitizes human nasopharyngeal carcinoma cell line CNE-2 to radiation. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(1):305–11.
Niwa AM, GF DE, Marques LA, Semprebon SC, Sartori D, Ribeiro LR, Mantovani MS. Salinomycin efficiency assessment in non-tumor (HB4a) and tumor (MCF-7) human breast cells. Naunyn Schmiedeberg's Arch Pharmacol. 2016;389(6):557–71.
Hermawan A, Wagner E, Roidl A. Consecutive salinomycin treatment reduces doxorubicin resistance of breast tumor cells by diminishing drug efflux pump expression and activity. Oncol Rep. 2016;35(3):1732–40.
Koo KH, Kim H, Bae YK, Kim K, Park BK, Lee CH, Kim YN. Salinomycin induces cell death via inactivation of Stat3 and downregulation of Skp2. Cell Death Dis. 2013;4:e693.
Sun P, Pavlostathis SG, Huang CH. Photodegradation of veterinary ionophore antibiotics under UV and solar irradiation. Environmental science & technology. 2014;48(22):13188–96.
Yao H, Sun P, Minakata D, Crittenden JC, Huang CH. Kinetics and modeling of degradation of ionophore antibiotics by UV and UV/H2O2. Environmental science & technology. 2013;47(9):4581–9.
Riccioni R, Dupuis ML, Bernabei M, Petrucci E, Pasquini L, Mariani G, Cianfriglia M, Testa U. The cancer stem cell selective inhibitor salinomycin is a p-glycoprotein inhibitor. Blood Cells Mol Dis. 2010;45(1):86–92.
Zhang ZM, Wu JF, Luo QC, Liu QF, Wu QW, Ye GD, She HQ, Li BA. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/beta-catenin pathway. Oncogene. 2016;35(36):4787–97.
Lu Y, Ma W, Mao J, Yu X, Hou Z, Fan S, Song B, Wang H, Li J, Kang L, et al. Salinomycin exerts anticancer effects on human breast carcinoma MCF-7 cancer stem cells via modulation of hedgehog signaling. Chem Biol Interact. 2015;228:100–7.
He M, Fu Y, Yan Y, Xiao Q, Wu H, Yao W, Zhao H, Zhao L, Jiang Q, Yu Z, et al. The hedgehog signalling pathway mediates drug response of MCF-7 mammosphere cells in breast cancer patients. Clinical science (London, England : 1979). 2015;129(9):809–22.
Kim JH, Chae M, Kim WK, Kim YJ, Kang HS, Kim HS, Yoon S. Salinomycin sensitizes cancer cells to the effects of doxorubicin and etoposide treatment by increasing DNA damage and reducing p21 protein. Br J Pharmacol. 2011;162(3):773–84.
Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, Zhao W, Zhang X, Yu J, Wang QE. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci U S A. 2015;112(14):4411–6.
Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017;109:63–73.
Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75.
Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35(1):1–11.
Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94.
Szlosarek PW. Arginine deprivation and autophagic cell death in cancer. Proc Natl Acad Sci U S A. 2014;111(39):14015–6.
Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–10.
Verdoodt B, Vogt M, Schmitz I, Liffers ST, Tannapfel A, Mirmohammadsadegh A. Salinomycin induces autophagy in colon and breast cancer cells with concomitant generation of reactive oxygen species. PLoS One. 2012;7(9):e44132.
Mirkheshti N, Park S, Jiang S, Cropper J, Werner SL, Song CS, Chatterjee B. Dual targeting of androgen receptor and mTORC1 by salinomycin in prostate cancer. Oncotarget. 2016;7(38):62240–54.
Zhu LQ, Zhen YF, Zhang Y, Guo ZX, Dai J, Wang XD. Salinomycin activates AMP-activated protein kinase-dependent autophagy in cultured osteoblastoma cells: a negative regulator against cell apoptosis. PLoS One. 2013;8(12):e84175.
Li T, Su L, Zhong N, Hao X, Zhong D, Singhal S, Liu X. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9(7):1057–68.
Xie F, Zhang S, Liu J, Gong Z, Yang K, Zhang H, Lu Y, Zou H, Yu Y, Chen Y, et al. Codelivery of salinomycin and chloroquine by liposomes enables synergistic antitumor activity in vitro. Nanomedicine (London, England). 2016;11(14):1831–46.
Yue W, Hamai A, Tonelli G, Bauvy C, Nicolas V, Tharinger H, Codogno P, Mehrpour M. Inhibition of the autophagic flux by salinomycin in breast cancer stem-like/progenitor cells interferes with their maintenance. Autophagy. 2013;9(5):714–29.
Xipell E, Gonzalez-Huarriz M, Martinez de Irujo JJ, Garcia-Garzon A, Lang FF, Jiang H, Fueyo J, Gomez-Manzano C, Alonso MM. Salinomycin induced ROS results in abortive autophagy and leads to regulated necrosis in glioblastoma. Oncotarget. 2016;7(21):30626–41.
Klose J, Stankov MV, Kleine M, Ramackers W, Panayotova-Dimitrova D, Jager MD, Klempnauer J, Winkler M, Bektas H, Behrens GM, et al. Inhibition of autophagic flux by salinomycin results in anti-cancer effect in hepatocellular carcinoma cells. PLoS One. 2014;9(5):e95970.
Hashim AI, Zhang X, Wojtkowiak JW, Martinez GV, Gillies RJ. Imaging pH and metastasis. NMR Biomed. 2011;24(6):582–91.
Lindner D, Raghavan D. Intra-tumoural extra-cellular pH: a useful parameter of response to chemotherapy in syngeneic tumour lines. Br J Cancer. 2009;100(8):1287–91.
Gallagher FA, Kettunen MI, Day SE, Hu DE, Ardenkjaer-Larsen JH, Zandt R, Jensen PR, Karlsson M, Golman K, Lerche MH, et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature. 2008;453(7197):940–3.
Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69(6):2260–8.
Pellegrini P, Dyczynski M, Sbrana FV, Karlgren M, Buoncervello M, Hagg-Olofsson M, Ma R, Hartman J, Bajalica-Lagercrantz S, Grander D, et al. Tumor acidosis enhances cytotoxic effects and autophagy inhibition by salinomycin on cancer cell lines and cancer stem cells. Oncotarget. 2016;7(24):35703–23.
Lagas JS, Sparidans RW, van Waterschoot RA, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein limits oral availability, brain penetration, and toxicity of an anionic drug, the antibiotic salinomycin. Antimicrob Agents Chemother. 2008;52(3):1034–9.
Jangamreddy JR, Panigrahi S, Los MJ. Monitoring of autophagy is complicated--salinomycin as an example. Biochim Biophys Acta. 2015;1853(3):604–10.
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science (New York, NY). 2012;337(6098):1062–5.
Benard G, Karbowski M. Mitochondrial fusion and division: regulation and role in cell viability. Semin Cell Dev Biol. 2009;20(3):365–74.
Koshiba T, Detmer SA, Kaiser JT, Chen H, JM MC, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science (New York, NY). 2004;305(5685):858–62.
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1(4):515–25.
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282(15):11521–9.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160(2):189–200.
Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26(5):2175–86.
Jangamreddy JR, Ghavami S, Grabarek J, Kratz G, Wiechec E, Fredriksson BA, Rao Pariti RK, Cieslar-Pobuda A, Panigrahi S, Los MJ. Salinomycin induces activation of autophagy, mitophagy and affects mitochondrial polarity: differences between primary and cancer cells. Biochim Biophys Acta. 2013;1833(9):2057–69.
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra34.
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51.
Boehmerle W, Endres M. Salinomycin induces calpain and cytochrome c-mediated neuronal cell death. Cell Death Dis. 2011;2:e168.
Boehmerle W, Muenzfeld H, Springer A, Huehnchen P, Endres M. Specific targeting of neurotoxic side effects and pharmacological profile of the novel cancer stem cell drug salinomycin in mice. Journal of molecular medicine (Berlin, Germany). 2014;92(8):889–900.
Salemi S, Yousefi S, Constantinescu MA, Fey MF, Simon HU. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012;22(2):432–5.
Oliver L, Hue E, Priault M, Vallette FM. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012;21(15):2779–88.
Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16–27.
Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833(1):205–12.
Mitani M, Yamanishi T, Miyazaki Y, Otake N. Salinomycin effects on mitochondrial ion translocation and respiration. Antimicrob Agents Chemother. 1976;9(4):655–60.
Kim KY, Yu SN, Lee SY, Chun SS, Choi YL, Park YM, Song CS, Chatterjee B, Ahn SC. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem Biophys Res Commun. 2011;413(1):80–6.
Kim KY, Park KI, Kim SH, Yu SN, Lee D, Kim YW, Noh KT, Ma JY, Seo YK, Ahn SC. Salinomycin induces reactive oxygen species and apoptosis in aggressive breast cancer cells as mediated with regulation of autophagy. Anticancer Res. 2017;37(4):1747–58.
Kim KY, Park KI, Kim SH, Yu SN, Park SG, Kim YW, Seo YK, Ma JY, Ahn SC. Inhibition of autophagy promotes Salinomycin-induced apoptosis via reactive oxygen species-mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer cells. Int J Mol Sci. 2017;18(5)
Yu SN, Kim SH, Kim KY, Ji JH, Seo YK, Yu HS, Ahn SC. Salinomycin induces endoplasmic reticulum stress mediated autophagy and apoptosis through generation of reactive oxygen species in human glioma U87MG cells. Oncol Rep. 2017;
Kim GD, Oh J, Park HJ, Bae K, Lee SK. Magnolol inhibits angiogenesis by regulating ROS-mediated apoptosis and the PI3K/AKT/mTOR signaling pathway in mES/EB-derived endothelial-like cells. Int J Oncol. 2013;43(2):600–10.
Ji C, Yang YL, Yang Z, Tu Y, Cheng L, Chen B, Xia JP, Sun WL, Su ZL, He L, et al. Perifosine sensitizes UVB-induced apoptosis in skin cells: new implication of skin cancer prevention? Cell Signal. 2012;24(9):1781–9.
Rubio N, Verrax J, Dewaele M, Verfaillie T, Johansen T, Piette J, Agostinis P. p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic Biol Med. 2014;67:292–303.
Buel GR, Blenis J. CELL SIGNALING. Seeing mTORC1 specificity. Science (New York, NY). 2016;351(6268):25–6.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Sun J, Liu Y, Tran J, O'Neal P, Baudry M, Bi X. mTORC1-S6K1 inhibition or mTORC2 activation improves hippocampal synaptic plasticity and learning in Angelman syndrome mice. Cellular and molecular life sciences : CMLS. 2016;73(22):4303–14.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–71.
Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32.
Jangamreddy JR, Jain MV, Hallbeck AL, Roberg K, Lotfi K, Los MJ. Glucose starvation-mediated inhibition of salinomycin induced autophagy amplifies cancer cell specific cell death. Oncotarget. 2015;6(12):10134–45.
Hardie DG, Ross FA, Hawley SA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012;19(10):1222–36.
Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303.
Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, Manning BD. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463–71.
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–68.
Hardie DG. AMPK and autophagy get connected. EMBO J. 2011;30(4):634–5.
Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X, Sinha S, Gaestel M, Levine B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. elife. 2015;4
Zhou YY, Li Y, Jiang WQ, Zhou LF. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35(3)
Temkin V, Karin M. From death receptor to reactive oxygen species and c-Jun N-terminal protein kinase: the receptor-interacting protein 1 odyssey. Immunol Rev. 2007;220:8–21.
Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct Funct. 2003;28(1):23–9.
Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Correction: simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One. 2016;11(7):e0159352.
Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278(3):403–13.
Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5(8):1180–5.
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–93.
Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69(10):4415–23.
Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–17.
Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, Vazquez P, Blazquez C, Torres S, Garcia S, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119(5):1359–72.
Jin Y, Chen Y, Zhao S, Guan KL, Zhuang Y, Zhou W, Wu X, Xu T. DNA-PK facilitates piggyBac transposition by promoting paired-end complex formation. Proc Natl Acad Sci U S A. 2017;114(28):7408–13.
Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–11.
Kaplan F, Teksen F. Apoptotic effects of salinomycin on human ovarian cancer cell line (OVCAR-3). Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(3):3897–903.
Qin LS, Jia PF, Zhang ZQ, Zhang SM. ROS-p53-cyclophilin-D signaling mediates salinomycin-induced glioma cell necrosis. Journal of experimental & clinical cancer research : CR. 2015;34:57.
Tang QL, Zhao ZQ, Li JC, Liang Y, Yin JQ, Zou CY, Xie XB, Zeng YX, Shen JN, Kang T, et al. Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett. 2011;311(1):113–21.
Arafat K, Iratni R, Takahashi T, Parekh K, Al Dhaheri Y, Adrian TE, Attoub S. Inhibitory effects of Salinomycin on cell survival, Colony growth, migration, and invasion of human non-small cell lung cancer A549 and LNM35: involvement of NAG-1. PLoS One. 2013;8(6):e66931.
Al Dhaheri Y, Attoub S, Arafat K, Abuqamar S, Eid A, Al Faresi N, Iratni R. Salinomycin induces apoptosis and senescence in breast cancer: upregulation of p21, downregulation of survivin and histone H3 and H4 hyperacetylation. Biochim Biophys Acta. 2013;1830(4):3121–35.
Shen HM, Codogno P. Autophagic cell death: loch ness monster or endangered species? Autophagy. 2011;7(5):457–65.
Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis : an international journal on programmed cell death. 2008;13(1):1–9.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–4.
Lee JH, Won YS, Park KH, Lee MK, Tachibana H, Yamada K, Seo KI. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis : an international journal on programmed cell death. 2012;17(12):1275–86.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–61.
Acknowledgments
Not applicable
Funding
This review article was supported by the Natural Science Foundation of China (no. 30772601) and the University Innovation Team Project Foundation of Education Department of Liaoning Province (no. LT2013019).
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
All the authors contributed for the preparation of this manuscript. JJ and ZYT were responsible for confirming the topic, collecting the data, analyzing article and drafting the first copy. HLL and JZ were responsible for searching the literature and editing the manuscript. Eskandar Qaed was responsible for furtherly polishing the manuscript. YSS, RW, XMB and QYW were responsible for the figures and legends, final editing, and preparation of the manuscript for submission. All authors read and approved the final manuscript.
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jiang, J., Li, H., Qaed, E. et al. Salinomycin, as an autophagy modulator-- a new avenue to anticancer: a review. J Exp Clin Cancer Res 37, 26 (2018). https://doi.org/10.1186/s13046-018-0680-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-018-0680-z