
Abstract
Snail1 is the founding member of the Snail superfamily of zinc-finger transcription factors, which also includes Snail2 (Slug) and Snail3 (Smuc). The superfamily is involved in cell differentiation and survival, two processes central in cancer research. Encoded by the SNAI1 gene located on human chromosome 20q13.2, Snail1 is composed of 264 amino acids and usually acts as a transcriptional repressor. Phosphorylation and nuclear localization of Snail1, governed by PI3K and Wnt signaling pathways crosstalk, are critical in Snail1’s regulation. Snail1 has a pivotal role in the regulation of epithelial-mesenchymal transition (EMT), the process by which epithelial cells acquire a migratory, mesenchymal phenotype, as a result of its repression of E-cadherin. Snail1-induced EMT involves the loss of E-cadherin and claudins with concomitant upregulation of vimentin and fibronectin, among other biomarkers. While essential to normal developmental processes such as gastrulation, EMT is associated with metastasis, the cancer stem cell phenotype, and the regulation of chemo and immune resistance in cancer. Snail1 expression is a common sign of poor prognosis in metastatic cancer, and tumors with elevated Snail1 expression are disproportionately difficult to eradicate by current therapeutic treatments. The significance of Snail1 as a prognostic indicator, its involvement in the regulation of EMT and metastasis, and its roles in both drug and immune resistance point out that Snail1 is an attractive target for tumor growth inhibition and a target for sensitization to cytotoxic drugs.
Similar content being viewed by others


Introduction
The Snail superfamily of transcription factors includes Snail1, Slug, and Scratch proteins, all of which share a SNAG domain and at least four functional zinc fingers [1]. Snail1 has four zinc fingers, located from amino acids 154 to 259, whereas Scratch and Slug each have five [2],[3]. The comparison of these zinc-finger sequences has further subdivided the superfamily into Snail and Scratch families, with Slug acting as a subfamily within the Snail grouping. The Snail superfamily has been implicated in various processes relating to cell differentiation and survival [1].
First characterized in Drosophila melanogaster in 1984, Snail1 also has well-documented homologs in Xenopus, C. elegans, mice, chicks, and humans [4],[5]. In humans, Snail1 is expressed in the kidney, thyroid, adrenal gland, lungs, placenta, lymph nodes, heart, brain, liver, and skeletal muscle tissues [6],[7]. Snail1 is a C2H2 zinc-finger protein composed of 264 amino acids, with a molecular weight of 29.1 kDa [7] (Figure 1). The SNAI1 gene, which is 2.0 kb and contains 3 exons, has been mapped to chromosome 20q.13.2 between markers D20S886 and D20S109 [7]. A Snail1 retrogene (SNAI1P) exists on human chromosome 2 [8].

Amino acid sequences: human and mouse. This figure provides the human Snail1 amino acid sequence. The second representation of the sequence has important features such as phosphorylation sites and zinc fingers highlighted in various colors. 1) Purple indicates nuclear localization signals 2) blue is motif 1 for GSK-3β-mediated phosphorylation 3) green is motif 2 for GSK-3β-mediated phosphorylation 4) yellow is the PAK1 phosphorylation site and 5) gray denotes the zinc-finger region. In addition, human Snail2 (Slug) and mouse Snail1 amino acid sequences are shown for comparison to the human Snail1 sequence. Human Slug is 48% identical to human Snail1, and mouse Snail1 is 88% identical to human Snail1. The sequence alignments were run through BLAST [9].
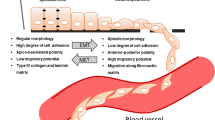
Epithelial-to-mesenchymal transition (EMT) is the process by which epithelial cells lose their apical polarity and adopt a mesenchymal phenotype, thereby, increasing migratory properties, invasiveness and apoptotic resistance. The expression of mesenchymal markers, like vimentin and fibronectin, replaces that of the usual epithelial markers, including E-cadherin, cytokeratins and Mucin-1 [10]. EMT is fundamental to both normal developmental processes and metastatic cancer. The induction of epithelial-to-mesenchymal transition (EMT) is Snail1’s most studied function, as this process is crucial for the formation of the mesoderm and the neural crest [1]. Snail1 knockout in mice is lethal because gastrulation does not occur [11]. The primary mechanism of Snail1-induced EMT is the repression of E-cadherin, which causes reduced cell adhesion and promotes migratory capacity [12]. The further elucidation of Snail1’s role in EMT provides a critical insight into the development of metastatic cancer. In addition, Snail1 has been recently implicated in the regulation of drug/immune resistance and the cancer stem cell (CSC) phenotype [13] [16].
Regulation of Snailexpression
Transcriptional regulation
The Notch intracellular domain, LOXL2, NF-κB, HIF-1α, IKKα, SMAD, HMGA2, Egr-1, PARP-1, STAT3, MTA3, and Gli1 all interact directly with the Snail1 promoter to regulate Snail1 at the transcriptional level [17][29]. Hypoxic stress, caused by insufficient oxygen, prompts a transcriptional response mediated by hypoxia-inducible factors (HIFs) [17]. Notch increases HIF-1α recruitment to the LOX promoter, and LOXL2 oxidizes K98 and/or K127 on the Snail1 promoter, leading to a conformational change in shape [18]. Under hypoxic conditions, HIF-1α binds to HRE2, contained within -750 to -643 bp of the Snail1 promoter, and increases Snail1 transcription. Knockdown of HIF-1α results in the repression of both Snail1 and EMT [19]. NF-κB also binds to the Snail1 promoter, between -194 and -78 bp, and increases its transcription [20]. SMAD2 and IKKα bind concurrently to the Snail1 promoter between -631 and -506 bp, resulting in Snail1’s upregulation [21]. HMGA2 cooperates in this complex as well, as the binding of HMGA2 to the Snail1 promoter increases SMAD binding [22].
In addition, ILK promotes PARP-1 binding, and STAT3 binds as a final result of an IL-6/JAK/STAT pathway [23],[24]. In mice, a pathway beginning with HB-EGF and progressing through the MEK/ERK pathway has also induced STAT3 binding to the Snail1 promoter [25]. Gli1 and Snail1 interact through a positive feedback loop: Shh and Wnt crosstalk results in the upregulation of both [26]. MTA3, a subunit of the Mi-2/NuRD complex, transcriptionally represses Snail1 in an ER-dependent manner. Snail1, in turn, binds to the ER promoter to complete the negative feedback loop [27],[28]. In a similar fashion, Egr-1 and Snail1 relate via a negative feedback loop. Egr-1, another zinc-finger transcription factor, binds to the Snail1 promoter at four sites between -450 and -50 bp. This process necessitates the presence of HGF and is mediated by the MAPK pathway, and it ultimately results in Snail1 upregulation. Snail1, in turn, represses Egr-1 [29].
YY1 and Snail1 itself are two special instances of transcriptional Snail1 regulation. YY1 binds to the3’ enhancer, rather than the promoter, and knockdown of YY1 has been shown to decrease Snail1 expression [30]. Furthermore, Snail1 is capable of binding to its own promoter and upregulating itself [31]. Snail1 binds to the E box region within the Snail ILK Responsive Element (SIRE); PARP-1 also binds to the SIRE, which is located between -134 and -69 bp, when induced by ILK [23] (Figure 2).

Regulation at the Snail1 promoter. This figure depicts the regulatory interactions at the human Snail1 promoter. The central line represents the base-paired sequence, with -750 to -1 bp shown. The relative locations of interactions with various transcription factors are then spatially compared using blocks to represent each regulator’s binding site. Each block, with the base pairs involved denoted at the top, shows where that particular protein binds the Snail1 promoter.
Experiments conducted to elucidate the relationship between p53, a tumor suppressor protein, and Snail1 have shown that p53 acts via miR-34a, -34b, and -34c to repress Snail1 at a 3’ untranslated region (UTR). Consequently, when p53 is repressed, the repression of Snail1 is lifted, and the expression of Snail1 rises [32].
Translational regulation
Two instances of phosphorylation are crucial to Snail1's post-transcriptional regulation. GSK-3β phosphorylates Snail1 at two consensus motifs in serine-rich regions. The first phosphorylation, at motif 2 (S107, S111, S115, S119), results in Snail1's being exported to the cytoplasm. The second instance of phosphorylation (S96, S100, S104) leads to its ubiquitination by β-Trcp, which recognizes the destruction motif D95SGxxS100 and ubiquitinates Lys98, 137, and 146. Consequential proteasomal degradation follows [33],[34]. In conditions that prevent GSK-β from phosphorylating Snail1, the F-box E3 ubiquitin ligase FBXL14 appears to cause proteasomal degradation by ubiquitinating the same lysine residues as β-Trcp [35]. P21-activated kinase 1 (PAK1) also phosphorylates Snail1 at S246[36]. Phosphorylation determines Snail1’s subcellular location, as GSK-3β -mediated phosphorylation induces Snail1's export to the cytoplasm through exportins such as chromosome region maintenance 1 (CRM1) [33],[37]. By contrast, PAK1 phosphorylation promotes Snail1’s presence in the nucleus and, therefore, increases its activity [36]. In the cytoplasm, Snail1 is quickly degraded; it has a half-life of only twenty-five minutes [33]. To protect from this degradation, Snail1 has nuclear localization signals (NLS): one monopartite from amino acids 151-152 and one bipartite overlapping the SNAG domain between amino acids 8 and 16 [38]. These signals are responsible for the nuclear transport of Snail1, which in turn is required for proper expression. β-catenin, Lef-1, and IκB employ similar systems [38] (Figure 3, Table 1).

Snail1 stability and localization. This figure shows the effects of GSK-3β and PAK1-mediated phosphorylation on Snail1 stability and subcellular localization. The outer circle represents the cell membrane, and the inner circle represents the nucleus. Nuclear Snail1 is phosphorylated by GSK-3β at motif 2 and is consequently exported from the nucleus. If Snail1 remains in the cytoplasm, it is ultimately ubiquitinated and degraded. By contrast, phosphorylation by PAK1 favors the nuclear localization of Snail1, which increases its stability.
TNFα, NF-κB, FGF, Wnt, and microRNA signals also influence the regulation of GSK-3β-mediated phosphorylation. The TNFα/NF-κB pathway induces CSN2, which protects Snail1 from degradation by interfering with the binding of GSK-3β and β-Trcp. Thus, Snail1 is neither phosphorylated nor ubiquitylated [39]. FGF operates through the PI3K/Akt pathway to downregulate GSK-3β, and receptor tyrosine kinase induces EGF suppression of GSK-3β [34],[40]. Wnt can also suppress GSK-3β and, thus, the phosphorylation of Snail1 [41]. Additionally, miR-148a causes the phosphorylation of AKT and GSK-3β, which results in less Snail1 localized in the nucleus. This, in turn, inhibited EMT in hepatocellular carcinoma [42].
Phosphorylation of upstream targets also influences the regulation of Snail1. For example, RANKL, in association with IκB, activates the NF-κB p65 subunit, and Akt influences the nuclear localization of NF-κB through its phosphorylation of IKKα and IκB in turn [43],[44]. TGF-β1 induces the phosphorylation of SMAD2 and SMAD3, which is necessary for their binding to Snail1 and the consequential upregulation of Snail1’s activities [45]. However, the cooperation of Ras signals is required for this pathway, since TGF-β1-mediated induction of Snail1 ceases with the silencing of Ras [46].
Other mechanisms of regulation contribute to the expression levels of Snail1, too. The small C-terminal domain phosphatase (SCP) induces dephosphorylation of both GSK-3β and the affected Snail1 motifs, thereby stabilizing Snail1 [47]. Additionally, histone deacetylase inhibitors promote the acetylation, likely of lysines, and increase Snail 1’s nuclear localization by inhibiting ubiquitination [48].
Snail 1’s targets
The variety of targets regulated by Snail1, detailed below, show that Snail 1’s EMT program is driven by multiple mechanisms (Table 2). While it directly represses epithelial markers like E-cadherin and claudins, Snail1 also upregulates markers of the mesenchymal phenotype, including vimentin and fibronectin. Frequently, the expression levels of Snail 1’s targets serve as prognostic indicators. For example, decreased E-cadherin expression correlates with lower patient survival rates while overexpression of MMPs associates with invasiveness. In addition to replacing epithelial with mesenchymal markers, Snail1 upregulates co-repressors, as in the case of ZEB-1, to complete its EMT program.
E-cadherin
E-cadherin is a transmembrane glycoprotein responsible for calcium-dependent cell-to-cell adhesion [49]. E-cadherin is a type I cadherin encoded by the gene CDH1, which is located on human chromosome 16q22.1 [50]. The founding member of the cadherin superfamily, E-cadherin plays a pivotal role in cadherin-catenin-cytoskeleton complexes, and it grants anti-invasive and anti-migratory properties to epithelial cells [51]. E-cadherin expression naturally decreases during gastrulation in order to properly form the mesoderm, and its expression increases once more for kidney organogenesis [52],[53]. The CDH1 promoter contains multiple E-boxes, and Snail1, Slug, ZEB1, ZEB2, and Twist, among others, have been shown to directly repress E-cadherin [54]. Total E-cadherin knockout in mice resulted in immediate death at implantation [55]. Decreases in E-cadherin expression correlate with epithelial-mesenchymal transition, metastasis, and lower patient survival rates [10].
Four Snail1 complexes have been identified as mechanisms of E-cadherin repression. (1) Snail1 interacts with G9a, which concurrently recruits DNA methyltransferases (DNMTs) to the E-cadherin promoter. Snail1's zinc fingers are thought to interact with the G9a ankyrin repeats, SET domain, or both. The complex has been shown to increase H3K9me2 and decrease H3K9 acetylation [56]. (2) The Snail1-Ajuba-PRMT5 complex promotes the methylation of H4R3. This, too, operates at the E-cadherin promoter [57]. The demethylation of H3K4 by Co-REST, CtBP, and HDAC complexes also factors into the last two mechanisms [58]. (3) Snail1 works in conjunction with Sin3A and HDAC1/2 to deacetylate H3 and H4, which suppress E-cadherin [59]. (4) In perhaps the most elucidated case, the Snail1 SNAG domain interacts with the LSD1 AO domain to form a Snail1-LSD1-CoREST complex. Snail1 residues Pro2, Arg3, Ser4, Phe5, Arg8, and Lys9 have been shown to be particularly crucial to this union, since mutants could not interact with LSD1. Likewise, LSD1 requires functional Asp375 and Glu379, Glu553, Glu555 and Glu556 to cooperate with Snail1. LSD1 inhibitors, histone H3, and SNAG peptides also hamper the activity of the complex. The formation of the Snail1-LSD1-CoREST complex results in the demethylation of H3K4me2 and consequential suppression of E-cadherin, while also increasing the stability of each of the components of the complex [60]. In a proposed second step to this mechanism, Snail1 recruits Suv39H1 to the E-cadherin promoter. Similar to prior cases, the Snail1 SNAG domain interacts with the Suv39H1 SET domain to suppress E-cadherin. Knockdown of Suv39H1 restored E-cadherin expression by inhibiting H3K9me3 [61].
RKIP
Raf kinase inhibitor protein (RKIP), a member of the phosphatidylethanolamine-binding protein (PEBP) group, suppresses metastasis by inhibiting the Raf-MEK-ERK and NF-κB pathways [62] [65]. In prostate, breast, and colorectal cancers, among others, RKIP expression is downregulated [64],[66]. Furthermore, elevated RKIP expression is a positive prognostic indicator for survival [66],[67]. Expression levels of RKIP correlate with those of E-cadherin, another Snail1 target, as they are both repressed by means of the E-boxes in their promoters [68].
PTEN
Phosphatase and tensin homolog deleted in chromosome 10 (PTEN) dephosphorylates phosphoinositide-3,4,5-triphosphate (PIP3) and, thus, inhibits the PI3K pathway [69]. In this way, PTEN functions as a tumor suppressor. Snail1 binds to the PTEN promoter, which contains two E-boxes, and represses PTEN [70]. The specificity of this interaction is emphasized by the fact that neither Slug nor ZEB1 expression significantly alters PTEN levels [70]. Snail1's association with the PTEN promoter inhibits the binding of p53, which activates PTEN during apoptosis, and it consequently increases resistance to gamma radiation-induced apoptosis [70],[71]. A positive feedback loop has been established around this interaction as well, since the repression of PTEN increases the expression of Akt [72]. Akt, operating through NF-κB, increases the expression of Snail1 [44]. Through this pathway, Snail1 may contribute to raising its own expression levels [70].
Occludin
Occludin, an integral membrane protein crucial to the integrity of tight junctions, was first identified in 1993. The transmembrane protein has four hydrophobic domains within its 522 amino acid sequence and a molecular weight of 65 kDa [73],[74]. Though it is considered similar to connexins in gap junctions, occludin is found exclusively at tight junctions in epithelial and endothelial cells [73]. Snail1 functions as a transcriptional repressor of occludin, just as it does E-cadherin in adherens junctions. By binding to the E-box in the occludin promoter sequence, Snail1 can completely repress the promoter activity [75]. Immunoblot analysis and immunocytochemistry confirm the considerable reduction of occludin expression in the presence of Snail1 [13]. This repression, along with that of E-cadherin and claudins, is critical to the loss of cell-to-cell adhesion observed in EMT.
Claudins
The claudin family contains more than twenty members, all of which are integral proteins spanning the membrane four times. Family members range from 20-27 kDa, but they all share PDZ binding motifs, which allow them to interact with ZO-1, ZO-2, and MUPP-1, among others [76]. Claudins are components of tight junctions, and claudin-1 binds with occludin [76],[77]. The expression of claudins is frequently low or nonexistent in breast cancer cell lines, and it shares an inverse relationship with Snail1 expression levels in invasive breast tumors [77].
Specifically, claudin-1, -3, -4, and -7 are all susceptible to repression by Snail1. The promoter sequence of each of these proteins contains multiple E-box binding motifs: claudin-1 has two E-boxes, claudin-3 has six, claudin-4 has 8, and claudin-7 has eight. As such, Snail1 can completely inhibit their transcription [75]. The destruction of tight junctions that accompanies the repression of claudins and occludin leads to epithelial cells' loss of apical polarity and increases proliferation [78]. This mechanism helps drive Snail1-induced EMT.
Mucin-1
Mucin-1, a transmembrane glycoprotein encoded by MUC1, is an epithelial marker expressed at the apical surface of epithelial cells in the reproductive tract, digestive tract, lungs, kidney, liver, eyes, and other tissues [79] [81]. Additionally, it is expressed in hematopoietic and T cells [80]. Mucin-1's functions include lubrication and protection from pathogens, and its association with β-catenin has implicated Mucin-1 in cell signaling [80]. O-linked glycosylation affects the protein significantly, as the core protein ranges from 120-225 kDa and the glycosylated form can reach up to 500 kDa [82]. In epithelial tumors, Mucin-1 is upregulated, and disparities in splice variants and glycosylation become apparent [79],[80]. Splice variants differ greatly-the protein can vary from 4-7 kb [82]. Perhaps most importantly, Mucin-1 also loses its apical restriction in malignant cases [80].
The 2872 bp promoter facilitates much of Mucin-1's regulation, and it notably includes five sites for YY1 binding [79]. Snail1 interacts with the two E-boxes that begin -84 bp from the start of transcription. Like E-cadherin, Mucin-1 is an epithelial marker repressed by Snail1 during the induction of EMT [83].
ZEB-1
ZEB-1, like Snail1, is a zinc-finger transcription factor that assists in the induction of EMT. Using E-boxes and co-repressors such as CtBP and BRG1, ZEB-1 represses E-cadherin and Mucin-1 [83],[84]. However, ZEB-1 is at least ten times less potent a repressor of both E-cadherin and Mucin-1 than Snail1 [83]. Interference with the interaction between ZEB-1 and BRG1 results in the upregulation of E-cadherin and simultaneous downregulation of vimentin, so an abundance of functional ZEB-1 is associated with a mesenchymal phenotype [84]. In contrast to the lethal effects of Snail1 knockout, ZEB-1 knockout does not prevent development to term and, thus, is not as critical for gastrulation [83].
The presence of Snail1 increases both RNA and protein levels of ZEB-1 during EMT. Snail1 expression in MDCK clones causes a 2.5-fold increase in ZEB-1 promoter activity compared to control cells. The abilities of Snail1 and ZEB-1 to repress E-cadherin are additive, and the two transcription factors work together to achieve a complete EMT [83].
Vimentin
Vimentin is 57 kDa intermediate filament generally restricted to mesenchymal cells [85]. Vimentin regulation is a complex interplay of epigenetic and post-translational modifications in addition to transcriptional regulation. Of note, the human vimentin promoter contains an NF-κB binding site as well as a TGF-β1 response element [86],[87]. Akt1 protects vimentin from caspase proteolysis via phosphorylation of Ser39 [88]. During EMT, epithelial cells, which normally express keratin intermediate filaments, begin to express vimentin. Overexpression of vimentin is evident in breast and prostate cancers, among many other types, and overexpression generally correlates with invasiveness, migration, and poor prognosis [89] [91]. Snail1 upregulates vimentin during EMT [54].
Fibronectin
Fibronectin is a glycoprotein involved in cell adhesion, differentiation, and migration [92],[93]. A dimer with two 250 kDa components, fibronectin is greatly affected by splicing, and at least twenty variants of the human form have been identified [94]. Fibronectin interacts with many integrins in addition to heparin, collagen, and fibrin [95] [99]. Inactivation of fibronectin is lethal in mice [100]. Snail1 upregulates fibronectin, a mesenchymal marker indicative of EMT [54].
Cytokeratin 18
Cytokeratins exist in two types, and each cytokeratin works with a complementary partner to form keratin filaments [101]. Cytokeratin 18 is the first type, acidic, and interacts with the basic cytokeratin 8 [101]. The cytokeratin 18 protein is encoded by the CK18 gene, which is located on chromosome 12q13. Cytokeratin 18 is an intermediate filament protein involved in cell structure, cell signaling, and the cell cycle [101] [104]. Cytokeratin 18 serves as an epithelial marker, and it localizes in epithelial organs, such as the kidney, liver, gastrointestinal tract, and mammary glands [105]. Snail1 represses cytokeratin 18 during the induction of EMT [83]. Unlike other targets, though, cytokeratin 18 expression is not completely subdued by Snail1's presence [75].
MMP 2/9
Matrix metalloproteinases (MMP) cleave extracellular matrix substrates and, thereby, alter cell-matrix adhesions [106]. MMP-2 and -9 are a subcategory within the MMP group because they specifically act on gelatin, collagen, elastin, and fibronectin [107] [111]. The genes that encode MMP-2 and -9 both contain fibronectin type II domains and are consequently three exons longer than the other MMP genes [107]. MMP-2 is a 72 kDa protein while MMP-9 is 92 kDa, and the main difference between them is the MMP-9's 54 amino acid hinge region [107],[112]. Additionally, MMP-2 localizes in the nucleus and MMP-9 in the cytoplasm [113]. Overexpression of MMP-2 and MMP-9 is frequently associated with invasive, metastatic tumors [114] [117].
Snail1's presence increases the mRNA levels of both MMP-2 and -9 [118]. One suggested interaction includes the upregulation of MMP-2 and -9 by Snail1 to trigger EMT and, then, the coordinated effort of Snail1 and Slug to sustain EMT by continually stimulating MMP-9 [113].
LEF-1
Lymphoid enhancer-binding factor 1 (LEF-1) is a T-cell factor commonly detected in tumors [119],[120]. The transcription factor represses E-cadherin by forming complexes with β-catenin, which, like Snail1, is degraded as a result of GSK-3β-mediated phosphorylation [11],[121] [123]. LEF-1 interacts with Snail1 via Wnt, PI3K and TGF-β1 pathways, and both Snail1 and LEF-1 are necessary for a complete EMT [124]. LEF-1 is considered a mesenchymal marker, and Snail1 induces its expression and continues to upregulate it [82],[125].
Snailexpression in cancer
Snail1 is expressed in many types of cancer. Snail1 overexpression usually correlates with increased migration, invasion, and metastasis. An inverse relationship with E-cadherin is expected, and Snail1 consequently corresponds with poor differentiation as well. Frequently, more advanced malignancies and poor prognosis also accompany elevated Snail1 expression (Table 3).
Breast carcinoma
Invasive breast carcinomas, including infiltrating ductal (IDC) and infiltrating lobular carcinomas (ILC), spread to surrounding breast tissues, lymph nodes and the pleural cavity. Assigned histological grades, with three being the highest, correlate with prognosis [126]. Breast carcinomas can give rise to malignant pleural effusions, and typical survival rates at that point are a matter of months [127].
Snail1 is not present in normal breast epithelium, nor is it present in ILCs (n = 21). Of 17 patients, Snail1 was expressed in 47% of IDCs, and its expression correlated with lymph node metastases and high histologic grades [128]. E-cadherin and Snail1 expression levels are inversely related, and high expression levels of Snail1 correlate with shorter effusion-free, disease-free, and overall survival rates (n = 16) [129]. As such, Snail1 has prognostic significance as a marker of IDC malignancy [128].
Hepatocellular carcinoma
Snail1 mRNA and protein levels are inversely correlated with E-cadherin in hepatocellular carcinoma (HCC) [130]. Snail1 overexpression, which in one study included 23% of cases (n = 47), is associated with portal vein invasion, metastasis, and poor differentiation. Furthermore, Snail1 expression correlates with a poor prognosis in recurrence-free survival and, thus, is considered a potential risk factor for early recurrence [131],[132].
Ovarian carcinoma
Overall, Snail1 expression is lower in ovarian carcinoma than in breast carcinoma, though its expression is still associated with distant metastases [129]. Expression is higher among primary tumors and metastases than effusions, and effusions show complete cytoplasmic localization of Snail1 [133]. Snail1 represses E-cadherin and upregulates MMPs, and E-cadherin expression correlates with disease-free survival while MMP-2 is considered a marker of poor prognosis [129].
Gastric carcinoma
E-cadherin expression is drastically reduced in gastric carcinoma, and Snail1 expression levels once again share an inverse relationship with E-cadherin expression levels [129]. Snail1 expression levels are more comparable to breast than ovarian carcinomas, and Snail1 expression is still higher in diffuse rather than intestinal varieties of gastric carcinomas [129],[134]. Elevated Snail1 expression increases cells' capacities for migration and invasion. Overexpression correlates with tumor size, depth of invasion, and lymph node metastasis. Shortened survival rates are also directly related to Snail1 overexpression, and Snail1 is considered a predictor of poor prognosis [135].
Oral squamous carcinoma
Oral squamous carcinoma is another case of E-cadherin/Snail1 expression inversion, and the higher the Snail1 expression, the more invasive the cancer. E-cadherin positive cells maintain their cuboidal shape while E-cadherin negative cells turn spindle-shaped. This is a typical sign of EMT, and it shows Snail1's repression of E-cadherin [136].
Pancreatic carcinoma
Pancreatic carcinoma tissues show significantly reduced E-cadherin levels and relatively high Snail1 expression [129]. In one study, 78% (n = 36) of ductal adenocarcinoma tissues expressed Snail1, and Snail1 expression is higher in undifferentiated cell lines than in differentiated ones [137].
Colorectal carcinoma
Colorectal cancer (CRC) begins in gland cells that line the colon and rectum, and it is one of the most commonly newly diagnosed cancers and a leading cause of cancer-related deaths [138]. Snail1 expression is again inversely correlated to E-cadherin expression in CRC, and the expression level of Snail1 is quite high in CRC (78%, n = 59) [130],[139]. Interestingly, the mean age of the Snail1-positive group was nine years older than the Snail1-negative group in one study, with a standard deviation of 12.7 years (58.9 years vs. 49.8 years, n = 59) [139]. In another study, Snail1 expression was detected by Western blot in all tested CRC lines, and its expression increased both migratory and invasive properties. Additionally, Snail1 expression led to a stem-cell like phenotype and spindle shape, as usually accompanies the loss of E-cadherin [140]. Snail1 expression also increased with the stage of the tumors, with 15/23 stage III expressing Snail1 and 6/6 of stage IV. The significantly higher rate of metastasis associated with Snail1 expression suggests that Snail1's presence indicates a high risk of distant metastases [139],[140].
Bladder carcinoma
Though the expression level of Snail1 is lower in bladder carcinoma than in other types of cancer, its presence still has a significant impact on the cancer's progression. In one study, only 16% of the 120 tested tissues expressed Snail1, indicating that Slug and Twist, whose expression levels were 63% and 44% respectively, play larger roles. However, Snail1 expression increased in node-positive compared to node-negative tumors, and Snail1's presence lowered the three-year progression free survival rate to only 15% [141]. Since Snail1 expression is closely linked with tumor recurrence, its elevation is considered a significant prognostic factor [141],[142].
Melanoma
In melanoma, there is increased Snail1 mRNA and low E-cadherin in the presence of Snail1 expression. By contrast, no Snail1 mRNA was detected in primary melanocytes [143]. Snail1 expression confers both invasive and immunosuppressive properties in melanoma [144].
Synovial sarcoma
Saito et al. reported that Snail1 mRNA was found in all cases tested of synovial sarcoma (n = 20) and E-cadherin mRNA was detected by RT-PCR in 14/20 cases. This does not show the same strong inverse correlation that has come to be expected of Snail1 and E-cadherin. In this case, mutations of the CDH1 gene, which encodes E-cadherin, seem to be more influential than the presence of Snail1 [145].
Prostate cancer
Prostate cancer is the second most commonly diagnosed cancer in men worldwide, with estimates of over 900,000 new cases per year [146]. A Gleason grade, which describes the two most important histopathological patterns of that patient's cancer, accompanies a diagnosis. The grade ranges from 2-10 with a higher score meaning less differentiated [147]. Significant losses of E-cadherin and syndecan 1, two proteins involved in cellular adhesion, have been observed in malignant prostate cancer [148],[149]. Both promoters contain E-boxes, so Snail1 can directly bind and repress them [150],[151]. The presence of E-boxes may explain the inverse correlation between E-cadherin/syndecan 1 and Snail1 expression levels. Poblete et al. found that high Snail1 expression correlated with a high Gleason grade and increased malignancy. Furthermore, in more malignant cell lines, like PC3, Snail1 had exclusively nuclear localization. By contrast, Snail1 had both cytoplasmic and nuclear localization in less malignant cell lines [152].
Cervical carcinoma
Cervical cancer is one of the most common malignancies in women worldwide [138]. Chen et al. found Snail1 expressed in 94% of samples (n = 70), and the elevated expression of Snail1 correlated with late FIGO stage, lymph node metastasis, and poor differentiation [153].
Snailand cancer stem cells
Snail1-induced EMT causes a stem-like phenotype, a property closely related to metastasis and resistance. Cancer stem cells (CSCs), or tumor-initiating cells, are subpopulations within tumors that possess self-renewing capabilities [154]. In breast tissue, for example, populations with a CD44high/CD24low phenotype have a higher tumor-initiating capacity than do their CD44low/CD24high counterparts within the same tumors [155]. CSCs are also associated with chemoresistance, relapse, and metastasis [156].
Mani et al. reported that EMT could induce stem-like properties in non-cancerous mammary epithelial cells [14]. The CD44high/CD24low phenotype correlates with both breast CSCs and normal mammary stem cells, and both Snail1- and Twist-induced EMTs stimulated this same phenotype in nontumorigenic human mammary epithelial cells (HMLEs). These EMTs also increased the HMLEs' mammosphere-forming ability thirty-fold, and the CD44high/CD24low cells are able to produce more CD44high/CD24low cells in addition to CD44low/CD24high cells. Furthermore, these CD44high/CD24low cells exhibited a decrease of E-cadherin expression along with elevated fibronectin, vimentin, Snail1, and Twist, as measured by RT-PCR [14]. Thus, EMT promotes self-renewal capabilities and the stem-like phenotype.
Given that Snail1 induced EMT and a stem-like phenotype in human colorectal cancer cells (as mentioned in "Colorectal Carcinoma," above), Zhou et al. examined human pancreatic cancer cells and reached similar conclusions [15]. Epithelial BxPC-3 cells were compared with more morphologically diverse Panc-1 cells, and the comparison identified Panc-1 cells, which had higher Snail1 expression and were more poorly differentiated than BxPC-3 cells, as CSChigh with a larger ALDHhigh population [15]. Stem cells' pluripotent capabilities are maintained in part by the polycomb complex protein BMI-1 (Bmi-1), homeobox protein Nanog, sex-determining region Y-box 2 (Sox2), and octamer-binding transcription factor 4 (Oct4) [157][159]. Snail1 silencing resulted in a decrease in ALDH, Sox-2, Oct-4, and invasive properties. Following Snail1 knockdown, E-cadherin expression increased as vimentin and ZEB1 expressions both decreased. Without Snail1, the Panc-1 cells underwent MET and consequently lost their stem-like phenotype [15].
In a similar study of non-small cell lung cancer, Wang et al. compared ciplatin-resistant A549 cells with their A549 counterparts [16]. A549/CDDP cells showed increased expression levels of Nanog, Oct4, and Bmi-1, as detected by Western blot. RT-PCR also showed increased CD44 and Sox2. Migratory and invasive capacities were increased in A549/CDDP cells, as well. Interestingly, only Snail1 expression was elevated in A549/CDDP cells-Slug, Twist, and ZEB1 were not influential factors in this comparison. Snail1 knockdown again caused a decline in migration, invasiveness, Bmi-1 expression, Oct-4 expression, and mammosphere-forming ability. E-cadherin increased as vimentin decreased, and the cells became more responsive to cisplatin [16]. Since β-catenin had effects on the system comparable to active Snail1, an antagonist of the PI3K/Akt pathway was introduced, and this resulted in a decrease in β-catenin, Snail1, Nanog, migration, invasiveness, and mammosphere-forming ability [16]. Thus, the Akt pathway plays a crucial role in stem-like phenotype in lung cancer cells.
Poor differentiation, sphere-forming capacity, self-renewal, and typical markers such as ALDH and CD44, among other properties, characterize the stem-like phenotype [15]. Clearly, Snail1 overexpression is associated with all of these properties. After Snail1 induces EMT, cells adopt a mesenchymal morphology, become more invasive, increase migratory capacity, and express a stem-like phenotype. Knockdown of Snail1 causes the reverse process, mesenchymal-epithelial transition (MET), which prompts cells to become less invasive, migratory, and stem-like, as well as more sensitized to drugs. Thus, Snail1-induced EMT is a critical link between resistance, metastasis, and stem-like characteristics.
Regulation of EMT, in part, by Snail1
Snail1 drives EMT primarily through the direct repression of E-cadherin [53]. Other targets that contribute to Snail1's EMT program were detailed above (See Section "Snail1's Targets", Table2). However, other transcription factors, notably, TGF-β, RANKL, Notch1, and Cox-2, Notch1 are crucial to the EMT phenotype as well.
Zhu et al. have examined the relationship between the expression of the Response Gene to Complement-32 (RGC-32) and TGF-β-mediated EMT [160]. RGC-32 is over-expressed in many cancers and correlates with the lower level of expression of E-cadherin in pancreatic cancer. Stimulation of cells with TGF-β was associated with the upregulation of RGC-32 and EMT. Noteworthy, the findings that RGC-32 mediated TGF-beta-induced EMT and cell migration was corroborated with the use of RGC-32 siRNA. The authors extrapolated that RGC-32 regulates Snail1 expression and EMT.
Snail1 is a target of NF-κB activity and its expression and role in EMT are well recognized. Since NF-κB is activated by many signals, clearly, such signals will also regulate Snail1 among other target gene products. Tsubaki et al. have reported that various solid tumors express the Receptor Activator of Nuclear Factor-κB (RANK) and it is activated by RANK-ligand resulting in the promotion of tumor cell growth, migration, metastasis, and anchorage independence in breast cancer cells [42]. In addition, they reported that RANKL induces EMT by activating NF-κB and enhances the expression of Snail1, Twist, vimentin, and N-cadherin and decreases the expression of E-cadherin. Inhibitors of NF-κB are shown to inhibit RANKL-mediated EMT, cell migration, and invasion.
Huang et al. investigated the expression level of Notch1 in lung adenocarcinoma and its relationship to metastasis [161]. They found that lung tumors express low levels of Notch1 and were associated with advanced clinical stage and lymph node metastasis. In contrast, patients with positive Notch1 expression had the prolonged progression of overall survival. Thus, Notch1 expression regulates negatively the EMT phenotype. Dysregulation of the Notch signaling pathway plays an important role in the pathogenesis of many cancers. Notch1 is one receptor of the Notch signaling pathway. Notch1 is involved in the regulation of tumor cell growth, proliferation, apoptosis, metastasis, and chemoradioresistance. Notch1 protects Snail1 from degradation by preventing GSK-3β-mediated phosphorylation via LOXL2 oxidation, as detailed above [18].
The relationship between the expression of cyclooxegnase-2 (Cox-2) and the downregulation of E-cadherin and its relationship to the EMT phenotype was reported by Fujii et al. [162]. These investigators examined Head and Neck Squamous Cell Carcinoma (HNSCC) cells and treated the cells with Cox-2 inhibitors (Celecoxib, NS-398 and SC-791) and examined EMT-associated gene products by quantitative real-time PCR and Western blot. The findings demonstrated that the inhibitors upregulated E-cadherin and inhibited its transcriptional repressors such as Snail1. The investigators suggested that the administration of Cox-2 inhibitors may suppress EMT and metastasis via re-expression of E-cadherin.
Snailregulates chemo and immune resistance
Reducing Snail1 expression has proven Snail1's involvement in tumor resistance to many chemotherapeutic drugs and immunotherapies. In melanoma, Snail1 knockdown, as a result of siRNA treatment, stops both tumor metastasis and immunosuppression. Tumor-specific T cell responses also intensify as a result of this knockdown [144]. Similarly, shRNA treatment induces apoptosis in adriamycin-resistant melanoma cells, and Snail1 reduction leads to cisplatin sensitization in lung adenocarcinoma, head and neck squamous, and ovarian cancers [13],[163][165]. Additionally, Snail1 has been implicated in resistance to radiation and paclitaxel in ovarian cancer cell lines as well as protection against 5-fluorouracil and gemcitabine in Panc-1 cells [166],[167].
Snail1 also factors into resistance because of its involvement in survival pathways. Snail1's activation of MAPK and PI3K survival pathways leads to resistance to serum depletion and TNF-α[168]. The repression of NF-κB and therefore Snail1, its downstream target, sensitizes tumor cells to cisplatin and TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Treatments with nitric oxide, the proteasome inhibitor NPI-0052, and rituximab all achieve this repression and consequential resistance reversal. These treatments have proven effective in prostate cancers and B-Non-Hodgkin’s Lymphoma, respectively [168][171].
Akalay et al. reported that the overexpression of Snail1 in breast cancer cell lines resulted in resistance to CTL-mediated killing and was associated with the EMT phenotype. The resistant cells exhibited amodulation of the formation of the immunologic synapse with CTLs along with the induction of autophagy in the target cells. The findings also showed that the inhibition of autophagy by targeting Beclin-1 sensitized the EMT cells to CTL killing. Hence, tumor cells’ resistance to CTL is mediated by EMT-induced activation of autophagy-dependent mechanisms [172],[173].
Chemical inhibitors targeting Snail1
Few chemical inhibitors target Snail1 directly. However, Snail1-induced EMT has been successfully abrogated by a select few chemical inhibitors. LSD and HDAC inhibitors, as well as drugs targeting Snail1/p53 and Snail1/E-cadherin interactions, have shown efficacy (Figure 4, Table 4). Their interactions are detailed below.

K-Ras-induced Snail1 represses p53, a tumor suppressor encoded by the TP53 gene, by binding directly and inducing exocytosis [174]. Lee et al. have developed two chemical inhibitors, GN25 and GN29, which prevent this binding and thereby protect p53 and its downstream targets, like p21, from Snail1 [175]. In K-Ras-mutated A549, HCT116, and MKN45 cell lines, both inhibitors were shown to be effective, though GN25 was more so. GN25 and GN29 also inhibited proliferation with more success than did Nutlin-3, which interferes with p53/MDM2 binding. In vivo studies indicated that the presence of GN25 reduced tumor progression as well as increased tumor regression. While this mechanism did not have cytotoxic effects on normal cells in this study, it does have some limitations. GN25 only activated wild-type p53 and was not effective in normal fibroblasts and Panc-1 cells. Additionally, this mechanism is effective exclusively in K-Ras-activated cancer cells, not N-Ras/Myc-transformed cells [175].
Harney et al. reported that Co(III)-Ebox, a Co(III) Schiff base complex, interferes with Snail1/E-cadherin binding and thereby inhibits Snail’s repression of the E-cadherin promoter in breast cancer cells [176]. Both the zinc finger region and ability to bind to E-box sequences are critical to this mechanism. With the introduction of Co(III)-Ebox, an increase in E-cadherin gene activity was observed. A 15 nM dose of Co(III)-Ebox achieved maximum results. While Co(III)-Ebox decreased DNA binding, it did not have an effect on Snail1 protein levels in this study [176].
Javaid et al. showed that LSD1, LSD2, and HDAC inhibitors are also effective in countering Snail1-induced EMT [177]. In breast epithelial cells, the LSD1/LSD2 inhibitor Tranylcypromine (TCP) and the HDAC class I and II inhibitor Trichostatin A (TSA) individually decreased Snail1’s effects on epithelial and mesenchymal markers. TSA almost completely reversed EMT markers’ expressions, indicating that HDAC inhibitors can obstruct EMT maintenance in addition to induction. Treatment with both TCP and TSA simultaneously inhibited Snail1-induced EMT, as well as TGF-β-induced EMT. The LSD1 inhibitor Pargyline and the HDAC1, HDAC2, HDAC3, and HDAC6 inhibitor LBH589 were also successful in inhibiting Snail1-induced EMT [177].
Furthermore, Shah et al. found that the HDAC inhibitor entinostat (ENT) reverses Snail1-induced EMT in breast cancer cells [178]. Treating MDA-MB-231 and Hs578T cells with ENT caused an increase in E-cadherin transcription with a concomitant reduction of N-cadherin mRNA. ChIP showed increased E-cadherin promoter activity as well as a reduction in the association of Twist and Snail1. ENT reduced the percentage of CD44high/CD24low cells in time and dose dependent manners, and Western blot showed downregulation of Twist and Snail1. Consequently, N-cadherin was reduced, cytokeratin 18 was upregulated, and vimentin was downregulated. Phosphorylation of vimentin increased, and remodeling resulted in a more rounded cell shape. As such, cell morphology became increasingly epithelial and cell migration decreased. ENT thus reverses EMT in triple-negative breast cancer cells, limiting invasive and metastatic potential [178].
Many chemical inhibitors have been developed to target gene products upstream of Snail1. MEK is an attractive target for selective inhibition because of its allosteric binding site, which allows for noncompetitive inhibition, and because all tumors dependent on MAPK signaling are potentially vulnerable to MEK inhibitors [179]. For example, trametinib, a MEK inhibitor, showed higher progression-free and overall survival at six months in phase III trials and was approved by the FDA in May 2013. Selumetinib, which is in phase II trials, has also shown increased PFS and OS [180]. Since PI3K and mTOR have similar catalytic sites, ATP-competitive compounds that target both have been developed in an attempt to increase efficacy. Pre-clinical studies show that dual PI3K/mTOR inhibitors reduce proliferation and induce apoptosis [181].
Ongoing clinical trials targeting Snail1
Very few ongoing clinical trials relate to Snail1’s role in cancer [182]. In one study, “Polyethylene Glycol 3350 in preventing cancer in patients at risk of colorectal cancer“ (NCT00828984), Snail1’s presence will be quantified by immunohistochemistry and RT-PCR. However, Snail1’s role is secondary to EGFR, the true target. The phase II study, which is being conducted by the National Cancer Institute, is listed as recruiting and was last verified in October 2013 [182].
The use of Snail1 as a search term generates a list including collections of tissue samples to study resistance (NCT00880503, NCT00026663) as well as stem cell transplants (NCT01239368, NCT00923845, NCT00074490), but none of these mentions Snail1 specifically in their research descriptions [182]. A phase I HDAC inhibitor study, “A phase I study of belinostat in combination with cisplatin and etoposide in adults with small cell lung carcinoma and other advanced cancers“ (NCT00926640), also appears in this list, though it does not cite Snail1 as a target either. The NCI is conducting this study, which was listed as recruiting in its most recent update on March 14, 2014 [182].
Conclusions and future directions
Snail1, the founding member of the Snail superfamily, is a zinc-finger transcriptional repressor critical to many biological processes. The repression of epithelial markers like E-cadherin, claudins, and mucin-1, in addition to the upregulation of vimentin, fibronectin, and MMPs, facilitates the loss of cell adhesion. Thus, Snail1 confers migratory and invasive properties on epithelial cells. This progression of changing from epithelial cells to a mesenchymal phenotype, known as EMT, is crucial to processes such as gastrulation. Snail1 has also been implicated in cell differentiation and survival.
Snail1 is widely expressed in various cancers, and overexpression is frequently associated with migration, invasion and metastasis. Also correlated with recurrence and a lack of differentiation, Snail1 serves as a poor prognostic indicator in hepatocellular carcinomas, gastric carcinomas, and bladder carcinomas, among others. Therefore, combatting Snail1’s presence could prove pivotal in improving cancer prognoses.
To that end, the development of chemical inhibitors for both Snail1 and targets further upstream has begun [183][187]. PI3K, MEK, and mTOR inhibitors are making great strides, and combinations of these prove even more effective. However, many more Snail1-targeting therapies are possible. There are few Snail1-specific chemical inhibitors, and even fewer in clinical trials. Snail1 is ineffective when its nuclear localization is compromised. As such, more can be done to facilitate the phosphorylation and consequential degradation of Snail1 by GSK-3β and proteasomes, respectively. MicroRNA and epigenetic modifications are continually expanding areas of research.
Snail1’s roles in metastasis, recurrence, and resistance make it a novel and pleiotropic target in cancer, and improving our understanding of Snail1 could thus provide new ways of approaching the treatment of metastatic cancer.
Authors’ contributions
SK was responsible for reviewing the literature, summarizing data and preparing a draft of the manuscript. BB conceptualized and developed an outline for the manuscript as well as edited the manuscript for publication. Both authors read and approved the final manuscript.
Abbreviations
- Akt:
-
Protein kinase B
- ALDH:
-
Aldehyde dehydrogenase
- β-Trcp:
-
Beta-transducin repeat-containing protein
- CDDP:
-
Cisplatin
- Co-REST:
-
REST corepressor 1
- CSN2:
-
COP9 signalosome 2
- CtBP:
-
C-terminal binding protein
- EGF:
-
Epidermal growth factor
- Egr-1:
-
Early growth response factor-1
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinase
- FGF:
-
Fibroblast growth factor
- FIGO:
-
International Federation of Gynecology and Obstetrics
- GSK-3β:
-
Glycogen synthase kinase-3 beta
- HB-EGF:
-
Heparin-binding EGF-like growth factor
- HDAC:
-
Histone deacetylase
- HGF:
-
Hepatocyte growth factor
- HIF-1α:
-
Hypoxia-inducible factor 1-alpha
- HMGA2:
-
High-mobility group A2
- HRE2:
-
Hypoxia response element-2
- IKKα:
-
IκB kinase alpha
- ILK:
-
Integrin-linked kinase
- IL-6:
-
interleukin 6
- JAK:
-
Janus kinase
- LEF-1:
-
Lymphoid enhancer-binding factor 1
- LOXL2:
-
Lysyl oxidase-like 2
- LSD1:
-
Lysine-specific demethylase 1
- MAPK:
-
Mitogen-activated protein kinase
- MDM2:
-
Mouse double minute 2 homolog
- MEK:
-
MAPK kinase
- MTA3:
-
Metastasis-associated protein 3
- MUPP-1:
-
Multi-PDZ domain protein 1
- NF-κB:
-
Nuclear factor kappa-B
- NuRD:
-
Nucleosome remodeling deacetylase
- Oct-4:
-
Octamer-binding transcription factor 4
- PAK1:
-
p21-activated kinase 1
- PARP-1:
-
Poly(ADP-ribose) polymerase-1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PRMT5:
-
Protein arginine methyltransferase 5
- RANKL:
-
Receptor activator of nuclear factor kappa-B ligand
- STAT3:
-
Signal transducer and activator of transcription 3
- Sox-2:
-
Sex determining region Y-box 2
- TGF-β1:
-
Transforming growth factor beta 1
- TNFα:
-
Tumor necrosis factor alpha
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- YY1:
-
Yin Yang 1
- ZEB1/2:
-
Zinc finger E-box-binding homeobox 1/2
- ZO-1/2:
-
Zonula occludens protein 1/2
References
Nieto MA: The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002, 3: 155-166.
Boulay J, Dennefeld C, Alberga A: The Drosophila developmental gene snail encodes a protein with nucleic acid binding fingers. Nature. 1987, 330: 395-398.
Manzanares M, Locascio A, Nieto MA: The increasing complexity of the snail gene superfamily in metazoan evolution. Trends Genet. 2001, 17: 178-181.
Grau Y, Carteret C, Simpson P: Mutations and chromosomal rearrangements affecting the expression of snail, a gene involved in embryonic patterning in Drosophila melanogaster. Genetics. 1984, 108: 347-360.
Nusslein-Volhard C, Weischaus E, Kluding H: Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. I. Zygotic loci on the second chromosome. Wilheim Roux’s Arch Dev Biol. 1984, 193: 267-282.
Twigg S, Wilkie AOM: Characterization of the human snail (SNAI1) gene and exclusion as a major disease gene in craniosynostosis. Hum Genet. 1999, 105: 320-326.
Paznekas W, Okajima K, Schertzer M, Wood S, Jabs E: Genomic organization, expression, and chromosome location of the human snail gene (SNAI1) and a related processed pseudogene (SNAI1P). Genomics. 1999, 62: 42-49.
Barrallo-Gimeno A, Nieto MA: Evolutionary history of the snail/scratch superfamily. Trends Genet. 2009, 25: 248-252.
Human Snail1: sequence retrieved from and alignments run through NIH BLAST http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi.,http://www.uniprot.org/uniprot/O95863
Kalluri R, Weinberg R: The basics of epithelial-mesenchymal transition. J Clin Invest. 2009, 119: 1420-1428.
Carver EA, Jiang R, Gridley T: The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001, 21: 8184-8188.
Barrallo-Gimeno A, Nieto MA: The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005, 132: 3151-3161.
Kajita M, McClinic K, Wade P: Aberrant expression of the transcription factors Snail and Slug alters the response to genotoxic stress. Mol Cell Biol. 2004, 24: 7559-7566.
Mani S, Guo W, Liao MJ, Eaton E, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA: The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008, 133: 704-715.
Zhou W, Lv R, Qi W, Wu D, Xu Y, Liu W, Mou Y, Wang L: Snail contributes to the maintenance of stem cell-like phenotype cells in human pancreatic cancer. PLoS One. 2014, 9: e87409-
Wang H, Zhang G, Zhang H, Zhang F, Zhou BP, Ning F, Wang HS, Cai SH, Du J: Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur J Pharmacol. 2014, 723: 156-166.
Majmundar AJ, Wong WJ, Simon MC: Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010, 40: 294-309.
Peinado H, Del Carmen Iglesias-de la Cruz M, Olmeda D, Csiszar K, Fong KS, Vega S, Nieto MA, Cano A, Portillo F: A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J. 2005, 24: 3446-3458.
Zhu GH, Huang C, Feng ZZ, Lv XH, Qiu ZJ: Hypoxia-induced snail expression through transcriptional regulation by HIF-1alpha in pancreatic cancer cells. Dig Dis Sci. 2013, 58: 3503-3515.
Barbera MJ, Puig I, Dominguez D, Julien-Grille S, Guaita-Esteruelas S, Peiro S, Baulida J, Franci C, Dedhar S, Larue L, Garcia de Herreros A: Regulation of snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene. 2004, 23: 7345-7354.
Brandl M, Seidler B, Haller F, Adamski J, Schmid RM, Saur D, Schneider G: IKKalpha controls canonical TGFBeta-SMAD signaling to regulate genes expressing snail and slug during EMT in Panc1 cells. J Cell Sci. 2010, 123: 4231-4239.
Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A: HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem. 2008, 283: 33437-33446.
McPhee T, McDonald P, Oloumi A, Dedhar S: Integrin-linked kinase regulates E-Cadherin expression through PARP-1. Dev Dyn. 2008, 237: 2737-2747.
Yadav A, Kumar B, Datta J, Teknos T, Kumar P: IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011, 9: 1658-1667.
Zhang XH, Liang X, Wang TS, Liang XH, Zuo RJ, Deng WB, Zhang ZR, Qin FN, Zhao ZA, Yang ZM: Heparin-binding epidermal growth factor-like growth factor (HB-EGF) induction on Snail expression during mouse decidualization. Mol Cell Endocrinol. 2013, 381: 272-279.
Li X, Deng W, Lobo-Ruppert S, Ruppert J: Gli1 acts through Snail and E-Cadherin to promote nuclear signaling by Beta-catenin. Oncogene. 2007, 26: 4489-4498.
Fujita N, Jaye D, Kajita M, Geigerman C, Moreno C, Wade P: MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003, 113: 207-219.
Dhasarathy A, Kajita M, Wade P: The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. 2007, 21: 2907-2918.
Grotegut S, von Schweinitz D, Christofori G, Lehembre F: Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25: 3534-3545.
Palmer M, Majumder P, Cooper J, Yoon H, Wade P, Boss J: Yin Yang 1 regulates the expression of Snail through a distal enhancer. Mol Cancer Res. 2009, 7: 221-229.
Peiro S, Escriva M, Puig I, Barbera MJ, Dave N, Herranz N, Larriba MJ, Takkunen M, Franci C, Munoz A, Virtanen I, Baulida J, Garcia de herreros A: Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res. 2006, 34: 2077-2084.
Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI: A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesencymal transition. J Cell Biol. 2011, 195: 417-433.
Zhou BP, Deng J, Xia W, Xu J, Li Y, Gunduz M, Hung MC: Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004, 6: 931-940.
Katoh M, Katoh M: Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006, 5: 1059-1064.
Vinas-Castells R, Beltran M, Valls G, Gomez I, Garcia JM, Montserrat-Sentis B, Baulida J, Bonilla F, Garcia de herreros A, Diaz VM: The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J Biol Chem. 2010, 285: 3794-3805.
Yang Z, Rayala S, Nguyen D, Vadlmudi R, Chen S, Kumar R: Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchhyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005, 65: 3179-3184.
Dominguez D, Montserrat-Sentis B, Virgos-Soler A, Guaita S, Grueso J, Porta M, Puig I, Baulida J, Franci C, Garcia de Herreros A: Phosphorylation regulates the subcellular location and activity of the snail transcriptional repressor. Mol Cell Biol. 2003, 23: 5078-5089.
Ko H, Kim H, Kim N, Lee S, Kim K, Hong S, Yook J: Nuclear localization signals of the E-Cadherin transcriptional repressor Snail. Cells Tissues Organs. 2007, 185: 66-72.
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP: Stabilization of snail by NFkappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009, 15: 416-428.
Wu Y, Zhou BP: Snail: more than EMT. Cell Adhes Migrat. 2010, 4: 199-203.
Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ: Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005, 280: 11740-11748.
Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM: MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling.Oncogene 2013, Epub ahead of print.,
Tsubaki M, Komai M, Fujimoto SI, Itoh T, Imano M, Sakamoto K, Shimaoka H, Takeda T, Ogawa N, Mashimo K, Fujiwara D, Mukai J, Sakaguchi K, Satou T, Nishida S: Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J Exp Clin Cancer Res. 2013, 32: 62-
Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L: Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007, 26: 7445-7456.
Cheng JC, Chang HM, Leung P: TGF-Beta1 inhibits trophoblast cell invasion by inducing snail-mediated down-regulation of ve-cadherin. J Biol Chem. 2013, 288: 33181-33192.
Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M: Role of Ras signaling in the induction of snail by transforming growth factor-beta. J Biol Chem. 2009, 284: 245-253.
Wu Y, Evers BM, Zhou BP: Small C-terminal domain phosphatase enhances snail activity through dephosphorylation. J Biol Chem. 2009, 284: 640-648.
Jiang GM, Wang HS, Zhang F, Zhang KS, Liu ZC, Fang R, Wang H, Cai SH, Du J: Histone deacetylase inhibitor induction of epithelial-mesenchymal transitions via up-regulation of Snail facilitates cancer progression. Biochim Biophys Acta. 1833, 2013: 663-671.
Takeichi M: Functional correlation between cell adhesive properties and some cell surface proteins. J Cell Biol. 1977, 75: 464-474.
Berx G, Staes K, van Hengel J, Molemans F, Bussemakers M, von Bokhoven A, van Roy F: Cloning and characterization of the human invasion suppressor gene E-cadherin (CDH1). Genomics. 1995, 26: 281-289.
Van Roy F, Berx G: The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008, 65: 3756-3788.
Takeichi M, Matsunami H, Inoue T, Kimura Y, Suzuki S, Tanaka T: Roles of cadherins in patterning of the developing brain. Dev Neurosci. 1997, 19: 86-87.
Vestweber D, Kemler R: Identification of a putative cell adhesion domain of uvomorulin. EMBO J. 1985, 4: 3393-3398.
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA: The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000, 2: 76-83.
Larue L, Ohsugi M, Hirchenhain J, Kemler R: E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A. 1994, 91: 8263-8267.
Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou P, Evers B, Zhou B: G9a interacts with snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Investig. 2012, 122: 1469-1486.
Hou Z, Peng H, Ayyanathan K, Yan KP, Langer EM, Longmore GD, Rauscher FJ: The LIM protein AJUBA recruits protein arginine methyltransferase 5 to mediate SNAIL-dependent transcriptional repression. Mol Cell Biol. 2008, 28: 3198-3207.
Shi Y, Whetstine JR: Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007, 25: 1-14.
Peinado H, Ballestar E, Esteller M, Cano A: Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004, 24: 306-319.
Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP: The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29: 1803-1816.
Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP: Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013, 32: 1351-1362.
Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, Fee F, Katsanakis KD, Rose DW, Mischak H, Sedivy JM, Kolch W: Suppression of Raf-1 kinase activity and MAP kinase signaling by RKIP. Nature. 1999, 401: 173-177.
Yeung K, Rose DW, Dhillon AS, Yaros D, Gusafsson M, Chatterjee D, McFerran B, Wyche J, Kolch W, Sedivy JM: Raf kinase inhibitor protein interacts with NF-kappaB-inducing kinase and TAK1 and inhibits NF-kappaB activation. Mol Cell Biol. 2001, 21: 7201-7217.
Chatterjee D, Bai Y, Wang Z, Beach S, Mott S, Roy R, Braastad C, Sun Y, Mukhopadhyay A, Aggarwal BB, Darnowski J, Pantazis P, Wyche J, Fu Z, Kitagwa Y, Keller ET, Sedivy JM, Yeung KC: RKIP sensitizes prostate and breast cancer cells to drug-induced apoptosis. J Biol Chem. 2004, 279: 17515-17523.
Park S, Yeung ML, Beach S, Shields JM, Yeung KC: RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005, 24: 3535-3540.
Al-Mulla F, Hagan S, Behbehani AI, Bitar MS, George SS, Going JJ, Garcia JJ, Scott L, Fyfe N, Murray GI, Kolch W: Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J Clin Oncol. 2006, 24: 5672-5679.
Fu Z, Kitagawa Y, Shen R, Shah R, Mehra R, Rhodes D, Keller PJ, Mizokami A, Dunn R, Chinnaiyan AM, Yao Z, Keller ET: Metastasis suppressor gene Raf kinase inhibitor protein (RKIP) is a novel prognostic marker in prostate cancer. Prostate. 2005, 66: 248-256.
Beach S, Tang H, Park S, Dhillon AS, Keller ET, Kolch W, Yeung KC: Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008, 27: 2243-2248.
Vazquez F, Devreotes P: Regulation of PTEN Function as a PIP3 Gatekeeper through Membrane. Cell Cycle. 2006, 5: 1523-1527.
Escriva M, Peiro S, Herranz H, Villagrasa P, Dave N, Montserrat-Sentis B, Murray SA, Franci C, Gridley T, Virtanen I, Garcia de herreros A: Repression of PTEN Phosphatase by Snail1 Transcriptional Factor during Gamma Radiation-Induced Apoptosis. Mol Cell Biol. 2008, 28: 1528-1540.
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW: Regulation of PTEN transcription by p53. Mol Cell. 2001, 8: 317-325.
Yamada KM, Araki M: Tumor suppressor PTEN: modulator of cell signalling, growth, migration and apoptosis. J Cell Sci. 2002, 114: 2375-2382.
Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S, Tsukita S: Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993, 123: 1777-1788.
Ando-Akatsuka Y, Saitou M, Hirase T, Kishi M, Sakakibara A, Itoh M, Yonemura S, Furuse M, Tsukita S: Interspecies diversity of the occludin sequence: cDNA cloning of human, mouse, dog, and rat-kangaroo homologues. J Cell Biol. 1996, 133: 43-47.
Ikenouchi J, Matsuda M, Furuse M, Tsukita S: Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003, 116: 1959-1967.
Findley M, Koval M: Regulation and roles for claudin-family tight junction proteins. IUBMB Life. 2009, 61: 431-437.
Martinez-Estrada O, Culleres A, Vilaro S: The transcription factors Slug and Snail act as repressors of Claudin-1 expression in epithelial cells. Biochem J. 2006, 394: 449-457.
Martin T, Jiang W: Loss of tight junction barrier function and its role in cancer metastasis. BBA Biomembranes. 2009, 1788: 872-891.
Zaretsky J, Barnea I, Aylon Y, Gorivodsky M, Wreschner D, Keydar I: MUC1 gene overexpressed in breast cancer: structure and transcriptional activity of the MUC1 promoter and role of estrogen receptor alpha (ERalpha) in regulation of the MUC1 gene expression. Mol Cancer. 2006, 5: 57-
Brayman M, Thathiah A, Carson D: MUC1: a multifunctional cell surface component of reproductive tissue epithelia. Reprod Biol Endocrinol. 2004, 2: 4-
Hollingsworth M, Swanson B: Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004, 4: 45-60.
Gendler S, Spicer A: Epithelial mucin genes. Annu Rev Physiol. 1995, 57: 607-634.
Guaita S, Puig I, Franci C, Garrido M, Dominguez D, Batlle E, Sancho E, Dedhar S, De Herreros AG, Baulida J: Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem. 2002, 277: 39209-39216.
Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A: ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010, 29: 3490-3500.
Satelli A, Li S: Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011, 68: 3033-3046.
Lilienbaum A, Paulin D: Activation of the human vimentin gene by the Tax human T-cell leukemia virus. I. Mechanisms of regulation by the NF-kappa B transcription factor. J Biol Chem. 1993, 268: 2180-2188.
Wu Y, Zhang X, Salmon M, Lin X, Zehner ZE: TGFbeta1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP-1 and Sp1 family members. Biochim Biophys Acta. 2007, 1773: 427-439.
Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, Nguyen T, Ding Z, Belousov R, Bill K, Luo X, Lazar A, Dicker A, Mills GB, Hung MC, Lev D: Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene. 2011, 30: 457-470.
Gilles C, Polette M, Mestdagt M, Nawrocki-Raby B, Ruggeri P, Birembaut P, Foidart JM: Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 2003, 63: 2658-2664.
Lang SH, Hyde C, Reid IN, Hitchcock IS, Hart CA, Bryden AA, Villette JM, Stower MJ, Maitland NJ: Enhanced expression of vimentin in motile prostate cell lines and in poorly differentiated and metastatic prostate carcinoma. Prostate. 2002, 52: 253-263.
Zhao Y, Yan Q, Long X, Chen X, Wang Y: Vimentin affects the mobility and invasiveness of prostate cancer cells. Cell Biochem Funct. 2008, 26: 571-577.
Hynes RO, Yamada KM: Fibronectins: multifunctional modular glycoproteins. J Cell Biol. 1982, 95: 369-377.
Mosher DF: Fibronectin. 1989, Academic Press, Inc., San Diego
Pankov R, Yamada K: Fibronectin at a glance. J Cell Sci. 2002, 115: 3861-3863.
Benecky MJ, Kolvenback CG, Amrani DL, Mosesson MN: Evidence that binding to the carboxyl-terminal heparin-binding domain (HepII) dominates the interaction between plasma fibronectin and heparin. Biochem. 1988, 27: 7565-7571.
Ingham KC, Brew SA, Atha DH: Interaction of heparin with fibronectin and isolated fibronectin domains. Biochem J. 1990, 272: 605-611.
Mostafavi-Pour Z, Askari JA, Whittard JD, Humphries MJ: Identification of a novel heparin-binding site in the alternatively spliced IIICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 2001, 20: 63-73.
Liao YF, Gotwals PJ, Koteliansky VE, Sheppard D, Van De Water L: The EIIIA segment of fibronectin is a ligand for integrins α9β1 andα 4β1 providing a novel mechanism for regulating cell adhesion by alternative splicing. J Biol Chem. 2002, 277: 14467-14474.
Erat MC, Sladek B, Campbell ID, Vakonakis I: Structural analysis of collagen type I interactions with human fibronectin reveals a cooperative binding mode. J Biol Chem. 2013, 288: 17441-17450.
George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO: Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993, 119: 1079-1091.
Moll R, Franke WW, Schiller DL, Geiger B, Krepler R: The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982, 31: 11-24.
Fuchs E, Cleveland DW: A structural scaffolding of intermediate filaments in health and disease. Science. 1998, 279: 514-519.
Coulombe PA, Omary MB: ‘Hard‘ and ‘soft‘ principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol. 2002, 14: 110-122.
Galarneau L, Loranger A, Gilbert S, Marceau N: Keratins modulate hepatic cell adhesion, size and G1/S transition. Exp Cell Res. 2007, 313: 179-194.
Oshima RG, Baribault H, Caulín C: Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev. 1996, 15: 445-471.
Lin MH, Liu SY, Su HJ, Liu YC: Functional role of matrix metalloproteinase 28 in the oral squamous cell carcinoma. Oral Oncol. 2006, 42: 907-913.
Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA: Matrix Metalloproteinases: a review. Crit Rev Oral Biol Med. 1993, 4: 197-250.
Senior RM, Griffin GL, Fliszar CJ, Shapiro SD, Goldberg GI, Welgus HG: Human 92- and 72- kilodalton type IV collagenases are elastases. J Biol Chem. 1991, 266: 7870-7875.
Seltzer JL, Adams SA, Grant GA, Eisen AZ: Purification and properties of a gelatin-specific neutral protease from human skin. J Biol Chem. 1981, 256: 4662-4668.
Seltzer JL, Eisen AZ, Bauer EA, Morris NP, Glanville RW, Burgeson RE: Cleavage of type VII collagen by interstitial collagenase and type IV collagenase (Gelatinase) derived from human skin. J Biol Chem. 1989, 264: 3822-3826.
Gadher SJ, Schmid TM, Heck LW, Woolley DE: Cleavage of collagen type X by human synovial collagenase and neutrophil elastase. Matrix. 1989, 9: 109-115.
Huhtala P, Tuuttila A, Chow LT, Lohi J, Keski-Oja J, Tryggvason K: Complete structure of the human gene for 92-kDa type IV collagenase. Divergent regulation of expression for the 92- and 72-kilodalton enzyme genes in HT-1080 cells. J Biol Chem. 1991, 266: 16485-16490.
Qiao B, Johnson N, Gao J: Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-β1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int J Oncol. 2010, 37: 663-668.
Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S: Metastatic potential correlates with enzymic degradation of basement membrane collagen. Nature. 1980, 284: 67-68.
Garbisa S, Pozzati R, Muschel RJ, Saffiotti U, Ballin M, Goldfarb RH, Khoury G, Liotta LA: Secretion of type IV collagenolytic protease and metastatic phenotype: induction by transfection with C-Ha-ras but not C-Ha-ras plus Ad2-Ela. Cancer Res. 1987, 47: 1523-1528.
Nakajima M, Welch DR, Belloni PN, Nicholson GL: Degradation of basement membrane type IV collagen and lung subendothelial matrix by rat mammary adenocarcinoma cell clones of differing metastatic potentials. Cancer Res. 1987, 47: 4869-4876.
Bernhard EJ, Muschel RJ, Hughes EN: Mr 92,000 gelatinase release correlates with the metastatic phenotype in transformed rat embryo cells. Cancer Res. 1990, 50: 3872-3877.
Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T, Ohba Y, Nishihara H, Shirato H, Tsuda M, Tanaka S: Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2013, 0: 1-15.
Porfiri E, Rubinfeld B, Albert I, Hovanes K, Waterman M, Polakis P: Induction of a β-catenin-LEF-1 complex by wnt-1 and transforming mutants of β-catenin. Oncogene. 1997, 15: 2833-2839.
Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P: Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997, 275: 1790-1792.
Jamora C, DasGupta R, Kocieniewski P, Fuchs E: Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003, 422: 317-322.
Kim K, Lu Z, Hay ED: Direct evidence for a role of betacatenin/LEF-1 signalling pathway in the induction of EMT. Cell Biol Int. 2002, 26: 463-476.
Waterman ML: Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. 2004, 23: 41-52.
Medici D, Hay E, Goodenough D: Cooperation between Snail and LEF-1 transcription factors is essential for TGF-β1-induced epithelial-mesenchymal transition. Mol Biol Cell. 2006, 17: 1871-1879.
De Craene B, van Roy F, Berx G: Unraveling signaling cascades for the Snail family of transcription factors. Cell Signal. 2005, 17: 535-547.
Elston CW, Ellis IO: Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience with long-term follow-up. Histopathology. 1991, 19: 403-410.
Dieterich M, Goodman SN, Rojas-Corona RR, Emralino AB, Jimenez-Joseph D, Sherman ME: Multivariate analysis of prognostic features in malignant pleural effusions from breast cancer patients. Acta Cytol. 1994, 38: 945-952.
Blanco MJ, Moreno-Bueno G, Sarrio D, Locascio A, Cano A, Palacios J, Nieto MA: Correlation of Snail expression with histological grade and lymph node status in breast carcinomas. Oncogene. 2002, 21: 3241-3246.
Elloul S, Bukholt Elstrand M, Nesland JM, Trope CG, Kvalheim G, Goldberg I, Reich R, Davidson B: Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005, 103: 1631-1643.
Jiao W, Miyazaki K, Kitajima Y: Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br J Cancer. 2002, 86: 98-101.
Miyoshi A, Kitajima Y, Miyazaki K: Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer. 2005, 92: 252-258.
Woo HY, Min AL, Choi JY, Bae SH, Yoon SK, Jung CK: Clinicopathologic significance of the expression of Snail in hepatocellular carcinoma. Korean J Hepatol. 2011, 17: 12-18.
Elloul S, Silins I, Trope CG, Benshushan A, Davidson B, Reich R: Expression of E-cadherin transcriptional regulators in ovarian carcinoma. Virchows Arch. 2006, 449: 520-528.
Rosiavitz E, Becker I, Specht K, Fricke E, Luber B, Busch R, Hofler H, Becker KF: Differential expression of the epithelial-mesenchymal transition regulators Snail, SIP1, and Twist in gastric cancer. Am J Pathol. 2002, 161: 1881-1891.
Shin NR, Jeong EH, Choi CI, Moon HJ, Kwon CH, Chu IS, Kim GH, Jeon TY, Kim DH, Lee JH, Park do Y: Overexpression of Snail is associated with lymph node metastasis and poor prognosis in patients with gastric cancer. BMC Cancer. 2012, 12: 521-
Yokoyama K, Kamata N, Hayashi E, Hoteiya T, Ueda N, Fujimoto R, Nagayama M: Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001, 37: 65-71.
Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG: Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007, 13: 4769-4776.
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 2009, 59: 225-249.
Roy H, Smyrk T, Koetsier J, Victor T, Wali R: The transcriptional repressor SNAIL is overexpressed in human colon cancer. Dig Dis Sci. 2005, 50: 42-46.
Fan F, Samuel S, Evans KW, Lu J, Xia L, Zhou Y, Sceusi E, Tozzi F, Ye XC, Mani SA, Ellis LM: Overexpression of Snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1: 5-16.
Yu Q, Zhang K, Wang X, Liu X, Zhang Z: Expression of transcription factors snail, slug, and twist in human bladder carcinoma. J Exp Clin Cancer Res. 2010, 29: 119-
Bruyere F, Namdarian B, Corcoran NM, Pedersen J, Ockrim J, Voelzke BB, Mete U, Costello AJ, Hovens CM: Snail expression is an independent predictor of tumor recurrence in superficial bladder cancers. Urol Oncol. 2010, 28: 591-596.
Poser I, Dominguez D, Garcia de Herreros A, Varnai A, Buettner R, Bosserhoff AK: Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J Biol Chem. 2001, 276: 24661-24666.
Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y: Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009, 15: 195-206.
Saito T, Oda Y, Tsuneyoshi M: E-cadherin gene mutations frequently occur in synovial sarcoma as a determinant of histological features. Am J Pathol. 2001, 159: 2117-2124.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D: Global cancer statistics. CA Cancer J Clin. 2011, 61: 69-90.
Delahunt B, Miller RJ, Srigley JR, Evans AJ, Samaratunga H: Gleason grading: past, present and future. Histopathology. 2012, 60: 75-86.
Pecina-Slaus N: Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3: 17-18.
Edwards IJ: Proteoglycans in prostate cancer. Nat Rev Urol. 2012, 21: 196-206.
Smith B, Odero-Marah V: The role of Snail in prostate cancer. Cell Adh Migr. 2012, 6: 433-441.
Nackaerts K, Verbeken E, Deneffe G, Vanderschueren B, Demedts M, David G: Heparan sulfate proteoglycan expression in human lung-cancer cells. Int J Cancer. 1997, 74: 335-345.
Poblete C, Fulla J, Gallardo M, Munoz V, Castellon EA, Gallegos I, Contreras HR: Increased SNAIL expression and low syndecan levels are associated with high Gleason grade in prostate cancer. Int J Oncol. 2014, 44: 647-654.
Chen Z, Li S, Huang K, Zhang Q, Wang J, Li X, Hu T, Wang S, Yang R, Jia Y, Sun H, Tang F, Zhou H, Shen J, Ma D, Wang S: The nuclear protein expression levels of SNAI1 and ZEB1 are involved in the progression and lymph node metastasis of cervical cancer via the epithelial-mesenchymal transition pathway. Hum Pathol. 2013, 44: 2097-2105.
Reya T, Morrison SJ, Clarke MF, Weissman IL: Stem cells, cancer, and cancer stem cells. Nature. 2001, 414: 105-111.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003, 100: 3983-3988.
Jones RJ, Matsui WH, Smith BD: Cancer stem cells: are we missing the target?. J Natl Cancer Inst. 2004, 96: 583-585.
Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006, 126: 663-676.
Moon JH, Heo JS, Kim JS, Jun EK, Lee JH, Kim A, Kim J, Kim J, Whang KY, Kang YK, Yeo S, Lim HJ, Han DW, Kim DW, Oh S, Yoon BS, Schöler HR, You S: Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res. 2011, 21: 1305-1315.
Moon JH, Yun W, Kim J, Hyeon S, Kang PJ, Park G, Kim A, Oh S, Whang KY, Kim DW, Yoon BS, You S: Reprogramming of mouse fibroblasts into induced pluripotent stem cells with Nanog. Biochem Biophys Res Commun. 2013, 431: 444-449.
Zhu L, Qin H, Li PY, Xu SN, Pang HF, Zhao HZ, Li DM, Zhao Q: Response gene to complement-32 enhances metastatic phenotype by mediating transforming growth factor beta-induced epithelial-mesenchymal transition in human pancreatic cancer cell line BxPC-3. J Exp Clin Cancer Res. 2012, 31: 29-
Huang J, Song H, Liu B, Yu B, Wang R, Chen L: Expression of Notch-1 and its clinical significance in different histological subtypes of human lung adenocarcinoma. J Exp Clin Cancer Res. 2013, 32: 84-
Fujii R, Imanishi Y, Shibata K, Sakai N, Sakamoto K, Shigetomi S, Habu N, Otsuka K, Sato Y, Watanabe Y, Ozawa H, Tomita T, Kameyama K, Fujii M, Ogawa K: Restoration of E-cadherin expression by selective Cox-2 inhibition and the clinical relevance of the epithelial-to-mesenchymal transition in head and neck squamous cell carcinoma. J Exp Clin Cancer Res. 2014, 33: 40-
Zhuo W, Wang Y, Zhuo X, Zhang Y, Ao X, Chen Z: Knockdown of Snail, a novel zinc finger transcription factor, via RNA interference increases A549 cell sensitivity to cisplatin via JNK/mitochondrial pathway. Lung Cancer. 2008, 62: 8-14.
Hsu DS, Lan HY, Huang CH, Tai SK, Chang SY, Tsai TL, Chang CC, Tzeng CH, Wu KJ, Kao JY, Yang MH: Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clin Cancer Res. 2010, 16: 4561-4571.
Haslehurst AM, Koti M, Dharsee M, Nuin P, Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, Davey S, Squire J, Park PC, Feilotter H: EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer. 2012, 12: 91-
Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, Bapat SA: Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009, 27: 2059-2068.
Yin T, Wang C, Liu T, Zhao G, Zha Y, Yang M: Expression of Snail in pancreatic cancer promotes metastasis and chemoresistance. J Surg Res. 2007, 141: 196-203.
Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA: Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18: 1131-1141.
Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B: Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res. 2009, 69: 8376-8385.
Jazirehi AR, Huerta-Yepez S, Cheng G, Bonavida B: Rituximab (chimeric anti-CD20 monoclonal antibody) inhibits the constitutive nuclear factor-{kappa}B signaling pathway in non-Hodgkin's lymphoma B-cell lines: role in sensitization to chemotherapeutic drug-induced apoptosis. Cancer Res. 2005, 65: 264-276.
Vega MI, Baritaki S, Huerta-Yepez S, Martinez-Paniagua MA, Bonavida B: A potential mechanism of rituximab-induced inhibition of tumorgrowth through its sensitization to tumor necrosis factor-related apoptosis-inducing ligand-expressing host cytotoxic cells. Leuk Lymphoma. 2011, 52: 108-121.
Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, Chouaib S: EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy. 2013, 9: 1104-1106.
Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AK, Sabbah M, Tan TZ, Keira JH, Hung NT, Thiery JP, Mami-Chouaib F, Chouaib S: Epithelial-to mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73: 2418-2427.
Lee SH, Lee SJ, Chung JY, Jung YS, Choi SY, Hwang SH, Choi D, Ha NC, Park BJ: p53, secreted by K-Ras-Snail pathway, is endocytosed by K-Ras-mutated cells; implication of target-specific drug delivery and early diagnostic marker. Oncogene. 2009, 28: 2005-2014.
Lee SH, Shen GN, Jung YS, Lee SJ, Chung JY, Kim HS, Xu Y, Choi Y, Lee JW, Ha NC, Song GY, Park BJ: Antitumor effect of novel small chemical inhibitors of Snail-p53 binding in K-Ras-mutated cancer cells. Oncogene. 2010, 29: 4576-4587.
Harney A, Meade T, LaBonne C: Targeted inactivation of snail family EMT regulatory factors by a Co(III)-Ebox conjugate. PLoS One. 2012, 7: e32318-
Javaid S, Zhang J, Anderssen E, Black JC, Wittner BS, Tajima K, Ting DT, Smolen GA, Zubrowski M, Desai R, Maheswaran S, Ramaswamy S, Whetstine JR, Haber DA: Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep. 2013, 5: 1679-1689.
Shah P, Gau Y, Sabnis G: Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. 2014, 143: 99-111.
Hatzivassiliou G, Haling JF, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, Luoh SM, Anderson DJ, Ludlam MJ, Wiesmann C, Ultsch M, Friedman LS, Malek S, Belvin M: Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013, 501: 232-236.
Miller C, Oliver K, Farley J: MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol Oncol. 2014, 133: 128-137.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Chiarini F, Evangelisti C, Cocco L, Martelli AM: Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012, 3: 1068-1111.
NIH Database.. .,http://clinicaltrials.gov
Mimasu S, Sengoku T, Fukuzawa S, Umehara T, Yokoyama S: Crystal structure of histone demethylase LSD1 and tranylcypromine at 2.25Å. Biochem Biophys Res Commun. 2008, 366: 15-22.
Pubchem Database.. [],http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=444732&loc=ec_rcs
Pubchem Database.. [],http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=4688&loc=ec_rcs
Pubchem Database.. [],http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=6918837
Pubchem Database.. [],http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=4261
Acknowledgments
The authors acknowledge the collaborators and co-authors of publications related to Snail1 and include Drs. Kam Yeung (University of Toledo, Ohio), Devasis Chatterjee (Brown University) and Stavroula Baritaki (UCLA). The authors acknowledge the Jonsson Comprehensive Cancer Center at UCLA and various donors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kaufhold, S., Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Res 33, 62 (2014). https://doi.org/10.1186/s13046-014-0062-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-014-0062-0