
Abstract
E-cadherin tumor suppressor genes are particularly active area of research in development and tumorigenesis. The calcium-dependent interactions among E-cadherin molecules are critical for the formation and maintenance of adherent junctions in areas of epithelial cell-cell contact. Loss of E-cadherin-mediated-adhesion characterises the transition from benign lesions to invasive, metastatic cancer. Nevertheless, there is evidence that E-cadherins may also play a role in the wnt signal transduction pathway, together with other key molecules involved in it, such as beta-catenins and adenomatous poliposis coli gene products.
The structure and function of E-cadherin, gene and protein, in normal as well as in tumor cells are reviewed in this paper.
Similar content being viewed by others


Introduction
The control of cellular adhesion and motility is one of the crucial mechanisms responsible for tumor initiation and progression. The genes involved are also contributors to malignancy along with genes responsible for cell proliferation and survival. I propose to summarize the current knowledge on one very important tumor suppressor gene, viz. E-cadherin [1, 2].
E-cadherin is one of the most important molecules in cell-cell adhesion in epithelial tissues. It is localized on the surfaces of epithelial cells in regions of cell-cell contact known as adherens junctions [3]. As a member of a large family of genes coding for calcium-dependent cell adhesion molecules (CAMs), the cadherin glycoproteins are expressed by a variety of tissues, mediating adhesion through homotypic binding. Classical cadherins – E- and N-cadherins being the best characterized – play important roles in the formation of tissues during gastrulation, neurulation and organogenesis [4].
E-cadherin has probably been studied in most detail. It is essential for the formation and maintenance of epithelia, was first identified in chicken, and was originally called L-CAM [5]. The mouse counterpart of this protein, uvomorulin [6], has 80% identity in both nucleotide and amino acid sequences to the human counterpart [7].
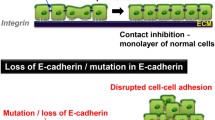
Besides its role in normal cells, this highly conserved gene can play a major role in malignant cell transformation, and especially in tumor development and progression. The suppression of E-cadherin expression is regarded as one of the main molecular events responsible for dysfunction in cell-cell adhesion. Most tumors have abnormal cellular architecture, and loss of tissue integrity can lead to local invasion. Thus, loss of function of E-cadherin tumor suppressor protein correlates with increased invasiveness and metastasis of tumors [8], resulting in it being referred to as the "suppressor of invasion" gene.
E-cadherin gene and protein structure
The human epithelial (E)-cadherin gene CDH1 maps to chromosome 16q22.1. Berx et al. [1] isolated the full-length gene by using recombinant lambda phage, cosmid and P1 phage clones. The gene they cloned encompasses 16 exons and spans a region of ~100 kb. The exons range from 115 to 2245 bp. Further analysis of the gene showed 15 introns ranging from 120 bp (intron 4) to 65 kb (intron 2). The intron-exon boundaries are highly conserved in comparison with other "classical cadherins", and in intron 1 a 5' high-density CpG island was identified that may have a role in transcription regulation [1]. This island covers the region from exon 1 to exon 2 of the human E-cadherin gene, while other exons lacked such features (fig. 1), including the biggest (exon 16 of 2245 bp).

Genomic organization of the human E-cadherin gene. Positions of exons are shown in color boxes with the base pair number of each exon. The connecting lines are introns. The region from exon 1 to exon 2, a sequence of about 1500 bp, is a high-density CpG island.
The chromosomal location of CDH1 on 16q22.1 was later confirmed by fluorescent in situ hybridization (FISH) analysis. It is interesting that the human P-cadherin gene was recently located only 32 kb upstream from E-cadherin [9], and also the M-cadherin gene was positioned on chromosome 16q24.1-qter [10], which further suggests clustering of cadherin genes originating probably from gene duplication, while possible co-evolution might be explained by gene conversion [1]. All classical cadherin genes analyzed so far have 16 exons separated by 15 introns.
CDH1 encodes a 120 kDa glycoprotein with a large extracellular domain, a single transmembrane segment and a short cytoplasmic domain, which interacts with the actin cytoskeleton through linker molecules, alpha- beta- and gamma-catenins [11]. On the cytoplasmic side of the membrane, a bundle of actin filaments is linked to the E-cadherin molecules via a protein complex. Alpha-catenin and either beta- or gamma-catenins are included in this complex. Beta- and gamma-catenins share significant homology and bind to a specific domain at the E-cadherin C-terminus. Alpha-catenin links the bound beta- or gamma-catenin to the actin cytoskeleton. (fig. 2).

Shematic illustration of E-cadherin in adherens junction. E-cadherin homodimer on the cytoplasmic membranes of adjacent cells is shown. The juxtamembrane region with the interacting molecules is also shown. CM – cytoplasmic membrane; AJ – adherens junction; ED – extracellular domain; ID – intracellular domain; AC – actin cytoskeleton; 1-beta-catenin; 2-alpha-catenin; 3-p120.
Alpha-catenin has structural similarities with vinculin, one of the key components of fibroblast membrane attachment sites of microfilaments, beta-catenin shows homology to Armadillo of Drosophila melanogaster and gamma-catenin is identical to plakoglobin, a protein found in desmosomes [12].
The C-terminal cytoplasmic domain of ~150 residues is highly conserved in sequence, and has been shown experimentally to regulate the cell-cell binding function of the extracellular domain of E-cadherin, possibly through interaction with the cytoskeleton. The juxtamembrane region of the cadherin cytoplasmic tail has been identified as a functionally active region supporting cadherin clustering and adhesive strength; one of the interacting proteins involved in clustering and cell adhesion is p120ctn [13].
The structure of the extracellular domain of classical E-cadherin contains five tandem repeats of a 100-residue-amino-acid-motif, and the biggest part of N-terminal of these repeats contains the sites with adhesive activity. This part of the molecule also has binding sites for calcium ions situated in the pockets between the repeats. The amino acid sequences that form the Ca2+ binding pockets are highly conserved between different members of the cadherin family and between different species. Cell-cell adhesion is mediated through homotypic interactions of E-cadherin extracellular domains in a process of lateral dimerization. Parallel dimers are able to interdigitate with dimers from neighbouring cells forming the points of adhesion. Those findings introduce a cadherin-cadherin interface at the cellular surface. Until recently cadherins were thought to be involved only in homophilic interactions; however, E-cadherin has now been shown to be a ligand for two integrins, alphaEbeta7 and alpha2beta1. The first interaction might serve to retain intraepithelial lymphocytes in mucosal tissue, while the second may contribute to the organization of epithelial multilayers [14].
Cadherins have been identified in a large variety of species, including mammals, Xenopus, Drosophila, Caenorhabditis elegans. Many new cadherin family members have been isolated and the cadherin group of genes has grown very fast in the last decade. Proteins of several isolated molecules show a great deal of homology with the "classical" cadherins [15]. Other cadherin like molecules share with the classical cadherins putative Ca2+ binding motifs in repeated extracellular domains but diverge considerably in various regions, particularly in the cytoplasmic domains. Even more deviations were observed for new cadherins, such as K-cadherin [16] and LI-cadherin [17], while the ret protooncogene product also shows some similarity to cadherin [18, 19]. Although the extracellular domain has several similar repeats with putative Ca2+ binding motifs, this transmembrane tyrosine kinase is considered to be unrelated to the cadherins. Since the ret gene lacks matching of the splice sites to the CDH1 gene, this might be the explanation. It is possible that this kinase has acquired its cadherin-like motifs by convergent evolution [1].
Role of E-cadherin in normal cells, wnt signalling, adherens junctions
Expression of E-cadherin in embryonic development is very early, at the two-cell stage [12, 20]. Epithelial differentiation and polarization (processes fundamental to cell differentiation) occur early in ontogeny in the morula stage, when the embryo compacts and each cell polarizes along its apicobasal axis to generate an epithelial-like phenotype. E-cadherin plays an important role in the adhesion of the blastomeres, and early embryo's ability to compact [21]. E-cadherin is expressed in the membrane even before compaction of the morula occurs, is distributed in a non-polar manner, and does not exhibit adhesive function [22, 23]. The mechanism that renders E-cadherin functional is unknown, but it does include phosphorylation of the protein [24]. Controlled epithelial-mesenchymal conversion is the most important exhibit of E-cadherin's function in development [25]. Loss of epithelial adhesion and polarity causing mesenchymal cell morphology occurs during mesoderm formation. Rietmacher and co-workers [12] introduced a targeted mutation in mouse embryonal stem cells and generated a mouse without E-cadherin sequences essential for Ca2+ binding and for adhesive function. Heterozygous mutant animals were normal and fertile. In vitro, they were able to form normal blastocysts with normal blastocoels that consequently expanded. On the other hand, the homozygous E-cadherin -/- embryos showed severe abnormalities before implantation. This included failure to maintain a polarized and compacted state and also failure to form a trophectoderm epithelium; they distort at the blastocyst stage, making the mutation lethal. The initial compaction that was observed in -/- embrios is probably due to the presence of E-cadherin proteins from maternal sources [24].
Investigation of zebrafish E-cadherin expression during early embryogenesis confirmed observed expression in blastomeres, but also led to the detection of a protein expressed in the anterior mesoderm during gastrulation and developing epithelial structures [26]. In the developing nervous system, CDH1 was detected at the pharyngula stage in the midbrain-hindbrain boundary and was preceded by wnt 1 expression [26].
As far as normal adult epithelial tissue structure and integrity is concerned, E-cadherin is also involved in its maintenance and homeostasis. As already mentioned, its function lies primarily in the formation of adherens junctions.
Cadherin mediated adhesion is a dynamic process that is regulated by several signal transduction pathways. There is also evidence that cadherins are not only targets for signaling pathways that regulate adhesion, but may themselves send signals that regulate basic cellular processes, such as migration, proliferation, apoptosis and cell differentiation [4, 27, 28].
The image of individual adhesion molecule performing its function, or linear downstream signalling cascade is somewhat abandoned scheme. Instead of separating and dividing into distinct fields, the cellular mechanisms of signalling and adhesion are nowadays thought to be closely connected mechanisms where components have double (or more) functions and interconnect in a signalling-structural network. The clearest example is recently discovered interaction of E-cadherin with epithelial growth factor receptor (EGFR) [29].
The recently discovered wnt/wingless pathway, of mouse and Drosophila respectively is one of the most interesting kind of signal transduction, in which key components have multiple functions in adhesion and signalling. Information on Wnt signalling can be found on the wnt gene site [30] and at the connection map at Science STKE Web site [31]. In vertebrate cells, it is named after Wnt proteins, a family of highly conserved secreted signaling molecules that regulate cell-to-cell interactions during embryogenesis. Insights into the mechanisms of Wnt action have emerged from several systems: genetics in Drosophila and Caenorhabditis elegans; biochemistry in cell culture; and ectopic gene expression in Xenopus embryos. Many Wnt genes in the mouse have been mutated, leading to very specific developmental defects. As currently understood, Wnt proteins bind to receptors of the Frizzled family on the cell surface. Through several cytoplasmic relay components, the signal is transduced to beta-catenin, which then enters the nucleus and forms a complex with TCF to activate transcription of Wnt target genes [28].
It has been well documented that wnt genes, together with other components of wnt signalling pathway, are implicated in cancer [32]. Another tantalizing compartment of the wnt signalling pathway lies in downstream transcriptional activation. In response to WNT signalling, cytoplasmic beta-catenin is stabilized, accumulates in the cytoplasm and enters the nucleus, where it finds a partner, a member of the DNA binding protein family LEF/TCF (T cell factor-lymphoid enhancer factor). Together they activate new gene expression programs. One of the target genes for β-catenin/TCF encodes c-MYC protein [2], explaining why constitutive activation of the wnt pathway can lead to cancer.
Role of E-cadherin in malignant cells
Progressive accumulation of somatic mutations in a number of different genes characterizes the process of tumorigenesis. Many genes involved in the process of tumorigenesis are components of one of a great many signal transduction pathways through which signals traffic via molecular networks. It is now apparent that epithelial malignancy can in certain aspects be explained by alterations in the adhesive properties of neoplastic cells.
Epithelial-mesenchymal conversion is also observed in malignant tumors of epithelial origin. This process is similar to developmental events but with the important difference that it is uncontrolled. Malignant carcinoma cells are characterized in general by poor intercellular adhesion, loss of the differentiated epithelial morphology and increased cellular motility. Downregulation or a complete shutdown of E-cadherin expression, mutation of the E-cadherin gene, or other mechanisms that interfere with the integrity of the adherens junctions, are observed in carcinoma cells. In human tumors, the loss of E-cadherin-mediated cell adhesion correlates with the loss of the epithelial morphology and with the acquisition of metastatic potential by the carcinoma cells [12]. Thus, a tumor invasion/suppressor role has been assigned to this gene. Additional data also support this role. Loss of heterozygosity on 16q is detected frequently in metastasizing malignancies derived from the liver, prostate, and breast [33]. Mutations in CDH1 have been described in a number of human cancers including breast, stomach, endometrium, ovary and thyroid [34, 35]. Transgenic mouse model with loss of E-cadherin expression developed invasive carcinoma from well-differentiated adenomas [36] and finally germ-line mutations have recently been reported in early onset, diffuse-type stomach cancers [37].
Many immunohistochemical studies have examined changes in expression of the E-cadherin gene in human malignancies. In almost all non-colonic tumors examined, the patterns of changes in the expression of this gene have been similar to that seen in colorectal cancer, i. e. loss of protein expression is positively correlated to loss of tumor differentiation. In oesophageal, pulmonary, squamous head and neck tumors, pancreatic and cervical tumors, loss of expression has been correlated with a high grade and an advanced stage of the disorder, with poor prognosis [2]. It has been reported that inactivating mutations of E-cadherin gene are highly frequent in infiltrating lobular breast carcinomas [38] and diffuse gastric carcinomas [34, 35]. These mutations mostly occur in combination with loss of heterozygosity of the wild-type allele.
Another interesting paper [39] on E-cadherin expression demonstrates its reappearance in metastatic cells. Significant increase in re-expression of E-cadherin, together with alpha and beta-catenin, was observed in metastatic deposit of primary lobular breast cancers. This protein product was missing in the primary tumors of the same origin. Lobular breast cancers harbour E-cadherin mutations as well as losses of the gene locus, even in situ disease. That suggests that the mutated protein has a role even before invasion process started. The findings reveal another dimension of adhesion molecules in tumor metastasis: reestablishment of cellular contact may prevent apoptosis. I would like to cite M. Iiyas [40] "adhesion molecule expression is the phoenix in tumor metastasis".
And finally, let us not forget yet another level of CDH1 expression regulation, i. e. E-cadherin promotor hypermethylation. This process has been known to be an inactivating event in some tumors and is currently extensively researched [41].
Mutation reports
Most interesting genetic mutations and their consequences are listed in this section. Becker et al. [42] suggested that E-cadherin mutations contribute to the histopathologic appearance of stomach cancer because 13 of 26 diffuse gastric carcinomas, which had reduced homophilic cell-to-cell interactions, had abnormal gene transcripts that were not seen in non-cancerous tissue from same patients. Berx et al. [34] reported of 69 somatic mutations of the CDH1 gene. In addition to a few missense mutations, those were mainly splice site and truncation mutations caused by insertions, deletions, and nonsense mutations. There was a major difference in mutation type between diffuse gastric and infiltrative lobular breast cancers. In diffuse gastric tumors, the predominant defects were exon skippings, which caused in-frame deletions. In contrast, most mutations found in infiltrating lobular breast cancers were out-of-frame mutations, which yield secreted truncated E-cadherin products. In most cases these mutations occurred in combination with loss of heterozygosity.
Guilford et al. [37] reported germline mutations in the CDH1 gene in 3 familial gastric cancer kindreds of Maori origin from New Zealand. Furthermore, Richards et al. [43] analyzed 8 UK gastric cancer kindreds and identified novel germline CDH1 mutations (a nonsense and a splice site mutation) in 2 families. Both mutations were predicted to truncate the E-cadherin protein in the signal peptide domain. Gayther et al. [44] also described germline CDH1 mutations in familial gastric cancer. In a family with a strong history of diffuse gastric carcinoma, Chun et al. [45] found the 1558insC germline mutation in the CDH1 gene. The gastric cancer was of the early onset, histologically diffuse type. In another family with early onset diffuse gastric cancer, Guilford et al. [37] found that the 30-year-old proband was heterozygous for A-to-T transition at nucleotide 2095, which resulted in a nonsense mutation. The mutation was predicted to result in an E-cadherin peptide lacking both the transmembrane and cytoplasmic domains. Same authors [37] described a family in which multiple members with gastric cancer were heterozygous for the insertion of an additional C residue in a run of 5 cytosines at positions 2382 to 2386. The resulting frameshift led to an E-cadherin molecule lacking about half of its cytoplasmic domain. In another diffuse gastric cancer family, a heterozygous G-to-T transversion at nucleotide 70 in exon 2 of the CDH1 gene, was also identified. The mutation converted a glutamic acid (glu24) to a TAG stop codon in the signal peptide of the E-cadherin precursor protein.
Richards et al. [43] identified a splice acceptor site mutation, an A-to-G transition at position -2 from nucleotide 49 at the start of exon 2 of the CDH1 gene and also a germline G-to-A transition at nucleotide 59 in exon 2. The mutation, a trp20-to-ter substitution was predicted to truncate the E-cadherin gene product in the signal peptide domain, which is cleaved from the N terminus of the mature protein.
In a family segregating diffuse gastric cancer, Gayther et al. [44] found a 2095C-T transition in the CDH1 gene, resulting in a truncating mutation, arg598 to ter.
Same authors [44] found a 1-bp insertion in the CDH1 gene in the proband of a family with familial diffuse gastric cancer. Insertion of a G after nucleotide 1711 created a frameshift that would truncate the protein at codon 587. Another 1-bp insertion (C) at nucleotide 1588 in exon 11 was also identified in a family segregating diffuse gastric cancer.
Mutations were also found in other types of carcinoma. In an endometrial carcinoma, Risinger et al. [46] identified a GCA-to-ACA transition in codon 617 predicting a substitution of thr for ala in the E-cadherin molecule. Somatic loss of heterozygosity was identified in the tumor tissue. They also identified a CTG-to-GTG transversion resulting in a leu711-to-val amino acid substitution in E-cadherin. The wild type allele was not lost.
In an ovarian carcinoma, same authors [46] identified an AGC-to-GGC transition in codon 838 resulting in substitution of glycine for serine. The tumor tissue also showed somatic loss of heterozygosity. In an infiltrative lobular breast carcinoma, Berx et al. [1] found a GAA (glu)-to-TAA (stop) nonsense mutation in the CDH1 gene. Tumor-specific loss of heterozygosity of chromosomal region 16q22.1 was demonstrated in this case. Here I have assessed only selected examples of CDH1 mutations in carcinoma tissue. The number of mutations is growing each day and can be reached at the following web site [47].
I should also mention the type of genetic changes that my group has encountered so far. Pećina-Šlaus et al. [48] indicated yet another type of genomic instability of CDH1 gene encountered in tumor tissue. When searching for allelic loss i. e. Loss of Heterozygosity (LOH) of the CDH1 gene in clear cell renal cell carcinoma, we came across R eplication ER ror (RER) positive samples, an instability problem [49] that characterizes tumor development and progression. Replication/repair machinery seems to be targeted in 10% of our clear cell renal cell carcinoma sample.
Conclusions
Reduced expression of E-cadherin is regarded as one of the main molecular events involved in dysfunction of the cell-cell adhesion system, triggering cancer invasion and metastasis. Therefore, E-cadherin is an important tumor suppressor gene. Research on E-cadherin has elucidated insights into both embryogenesis and oncogenesis. One of the most crucial exhibit of E-cadherin's function in development is the controlled epithelial-mesenchymal conversion.
The involvement of E-cadherin in wnt signaling, indicates that same molecule may have different functions and that E-cadherin can regulate cellular response generated by external signals the cell receives. In this way it can regulate migration, proliferation, apoptosis and cell differentiation.
The method of blocking E-cadherin downregulation in tumors is one of the important future approaches in gene therapy. To target this molecule is the logical path to prevent metastazing potential of almost any epithelial tumor. Nevertheless, it will not be an easy enterprise since its downregulation is caused by many different mechanisms, ranging from mutations and gross deletions to repression of gene transcription, as well as signal transduction stimulation of E-cadherin adhesion complex formation.
References
Berx G, Staes K, van Hengel J, Molemans F, Bussemakers MJ, van Bokhoven A, van Roy F: Cloning and characterization of the human invasion suppressor gene E-cadherin (CDH1). Genomics. 1995, 26: 281-289. 10.1016/0888-7543(95)80212-5.
Guilford P: E-cadherin downregulation in cancer: fuel on the fire?. Mol Med Today. 1999, 5: 172-177. 10.1016/S1357-4310(99)01461-6.
Gumbiner BM: Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell. 1996, 84: 345-357.
Barth AI, Nathke IS, Nelson WJ: Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997, 9: 683-690. 10.1016/S0955-0674(97)80122-6.
Gallin WJ, Sorkin BC, Edelman GM, Cunningham BA: Sequence analysis of a cDNA clone encoding the liver cell adhesion molecule, L-CAM. Proc Natl Acad Sci USA. 1987, 84: 2808-2812.
Ringwald M, Baribault H, Schmidt C, Kemler R: The structure of the gene coding for the mouse cell adhesion molecule uvomorulin. Nucleic Acids Res. 1991, 19: 6533-6539.
Mansouri A, Spurr N, Goodfellow PN, Kemler R: Molecular cloning and chromosomal localization of the gene encoding the human cell adhesion molecule ovomorulin. Differentation. 1998, 38: 67-71.
Vleminckx K, Vakaet L, Mareel M, Fiers W, van Roy F: Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion supressor role. Cell. 1991, 66: 107-119.
Bussemakers MJG, van Bokhoven A, Völler M, Smit FP, Schalken JA: The genes for the calcium-dependent cell adhesion molecules P- and E-cadherin are tandemly arranged in the human genome. Biochem Biophys Res Commun. 1994, 203: 291-1294.
Kaupmann K, Becker-Follmann J, Scherer G, Jockusch H, Starzinski-Powitz A: The gene for the cell adhesion molecule M-cadherin maps to mouse chromosome 8 and human chromosome 16q24.1-qter and is near the E-cadherin (uvomorulin) locus in both species. Genomics. 1992, 14: 488-490.
Humphries MJ, Newham P: The structure of cell-adhesion molecules. Trends Cell Biol. 1998, 8: 78-83. 10.1016/S0962-8924(97)01188-4.
Rietmacher D, Brinkmann V, Birchmeier CA: Targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc Natl Acad Sci USA. 1995, 92: 855-859.
Yap AS, Niessen CM, Gumbiner BM: The juxtamembrane region of the chaderin cytoplasmic tail supports lateral clustering, adhesive strenghtening, and interaction with p120ctn. J Cell Biol. 1998, 141: 779-789. 10.1083/jcb.141.3.779.
Huber AH, Weis WI: The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001, 105: 391-402.
Takeichi M: Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol. 1993, 5: 806-11.
Xiang YY, Tanaka M, Suzuki M, Igarashi H, Kiyokawa E, Naito Y, Othaware Y, Shen Q, Sugimura H, Kino I: Isolation of complementary DNA encoding K-cadherin, a novel rat cadherin preferentially expressed in fetal kidney and kidney carcinoma. Cancer Res. 1994, 54: 3034-3041.
Berndorff D, Gessner R, Kreft B, Schnoy N, Lajouspetter AM., Loch N, Reutter W, Hortsch M, Tauber R: Liver-intestine cadherin: Molecular cloning and characterization of a novel Ca2+-dependent cell adhesion molecule expressed in liver and intestine. J Cell Biol. 1994, 125: 1353-1396.
Schneider R: The human protooncogene ret: A communicative cadherin?. Trends Biochem Sci. 1992, 17: 468-469. 10.1016/0968-0004(92)90490-Z.
Iwamoto T, Taniguchi M, Asai N, Ohkusu K, Nakashima I, Takahashi M: cDNA cloning of mouse ret proto-oncogene and its sequence similarity to the cadherin superfamily. Oncogene. 1993, 8: 1087-1091.
Larue L, Ohsugi M, Hirchenhain J, Kemler R: E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci USA. 1994, 91: 8263-8267.
Fleming T, Hay M, Javed Q: Epithelial differentiation and intercellular junction formation in the mouse early embryo. Development Suppl. 1992, 17: 105-113.
Hyafil F, Morello D, Babinet C, Jacob F: A cell surface glycoprotein involved in the compaction of embryonal carcinoma cells and cleavage stage embryos. Cell. 1980, 21: 927-934.
Vestweber D, Kemler R: Rabbit antiserum against a purified surface glycoprotein decompacts mouse preimplantation embryos and reacts with specific adult tissues. Exp Cell Res. 1984, 152: 169-178.
Sefton M, Johnson M., Clayton L: Synthesis and phosphorylation of uvomorulin during mouse early development. Development (Cambridge, UK). 1992, 115: 313-318.
Thiery JP: Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002, 2: 442-454. 10.1038/nrc822.
Babb SG, Barnett J, Doedens AL, Cobb N, Liu Q, Sorkin BC, Yelick PC, Raymond PA, Marrs JA: Zebrafish E-cadherin: Expression during early embryogenesis and regulation during brain development. Dev Dyn. 2001, 221: 231-237. 10.1002/dvdy.1132.
Hulsken J, Birchmeier W, Behrens J: E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994, 127: 2061-2069.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW: Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997, 275: 1787-1790. 10.1126/science.275.5307.1787.
Pece S, Gutkind JS: Signaling from E-cadherin to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem. 2000, 275: 41227-41233. 10.1074/jbc.M006578200.
The Wnt gene Homepage. [http://www.stanford.edu/~rnusse/wntwindow.html]
Signal Transduction Knowledge Environment. [http://www.stke.org]
Peifer M, Polakis P: Wnt signaling in Oncogenesis and Embriogenesis – a look outside the nucleus. Science. 2000, 287: 1606-1609. 10.1126/science.287.5458.1606.
Efstathiou JA, Liu D, Wheeler JMD, Kim HC, Beck NE, Ilyas M., Karayiannakis AJ, Mortensen NJMcC, Kmiot W, Playford RJ, Pignatelli M, Bodmer WF: Mutated epithelial cadherin is associated with increased tumorigenicity and los of adhesion and of responsiveness to the motogenic trefoil factor 2 in colon carcinoma cells. PNASOnline. 1999, 96: 2316-2321. 10.1073/pnas.96.5.2316.
Berx G, Becker KF, Hofler H, van Roy F: Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998, 12: 226-237. 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D.
Machado JC, Soares P, Carneiro F, Rocha A, Beck S, Blin N, Berx G, Sobrinho-Simoes M: E-cadherin gene mutations provide a genetic basis for the phenotypis divergence of mixed gastric carcinomas. Lab Invest. 1999, 79: 459-465.
Christofori G, Semb H: The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem Sci. 1999, 24: 73-76. 10.1016/S0968-0004(98)01343-7.
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harrawira P, Taite H, Scoular R, Millers A, Reeve A: E-cadherin germline mutations in familial gastric cancer. Nature. 1998, 392: 402-405. 10.1038/32918.
Huiping C, Sigurgeirsdottir JR, Jonasson JG, Eiriksdottir G, Johannsdottir JT, Egilsson V, Ingvarsson S: Chromosome alterations and E-cadherin gene mutations in human lobular breast cancer. Br J Cancer. 1999, 81: 1103-1110. 10.1038/sj.bjc.6690815.
Bukholm IK, Nesland JM, Børessen-Dale AL: Re-expression of E-cadherin, α-catenin and β-catenin, but not of γ-catenin, in metastatic tissue from breast cancer patiens. J Pathol. 2000, 190: 15-19. 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.3.CO;2-C.
Ilyas M: Adhesion molecule expression in breast cancer: the phoenix in tumor metastasis?. J Pathol. 2000, 190: 3-5. 10.1002/(SICI)1096-9896(200001)190:1<3::AID-PATH490>3.0.CO;2-5.
Tycko B: Epigenetic gene silencing in cancer. J Clin Invest. 2000, 105: 401-407.
Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, Hoefler H: E-Cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994, 54: 3845-3852.
Richards FM, McKee SA, Rajpar MH, Cole TRP, Evans DGR, Jankowski JA, McKeown C, Sanders DSA, Maher ER: Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999, 8: 607-610. 10.1093/hmg/8.4.607.
Gayther SA, Gorringe KL, Ramus SJ, Huntsman D, Roviello F, Grehan N, Machado JC, Pinto E, Seruca R, Halling K, MacLeod P: Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999, 8: 607-610. 10.1093/hmg/8.4.607.
Chun YS, Lindor NM, Smyrk TC, Petersen BT, Burgart LJ, Guilford PJ, Donohue JH: Germline E-cadherin gene mutations: is prophylactic total gastrectomy indicated?. Cancer. 2001, 92: 181-187. 10.1002/1097-0142(20010701)92:1<181::AID-CNCR1307>3.0.CO;2-J.
Risinger JI, Berchuck A, Kohler MF, Boyd J: Mutations of the E-cadherin gene in human gynecologic cancers. Nature Genet. 1994, 7: 98-102.
Online Mendelian Inheritance in Man. [http://www.ncbi.nlm.nih.gov:80/entrez/dispomim.cgi?id=192090]
Pećina-Šlaus N, Pavelić K, Pavelić J: Loss of heterozygosity and protein expression of APC gene in renal cell carcinomas. J Mol Med. 1999, 77: 446-453. 10.1007/s001090050375.
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M: Ubiquitous somatic mutations in simple repaeted sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993, 363: 558-561. 10.1038/363558a0.
Acknowledgements
I would like to thank Prof. Dr. Sc. Denys Wheatley for critically reading the manuscript and Soros Foundation for support. This work was supported by grant 0108215 from Ministry of Science and Technology, Republic of Croatia.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Pećina-Šlaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int 3, 17 (2003). https://doi.org/10.1186/1475-2867-3-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-3-17