
Abstract
In the past five years, a series of large-scale genetic studies have revealed novel risk factors for Alzheimer’s disease (AD). Analyses of these risk factors have focused attention upon the role of immune processes in AD, specifically microglial function. In this review, we discuss interpretation of genetic studies. We then focus upon six genes implicated by AD genetics that impact microglial function: TREM2, CD33, CR1, ABCA7, SHIP1, and APOE. We review the literature regarding the biological functions of these six proteins and their putative role in AD pathogenesis. We then present a model for how these factors may interact to modulate microglial function in AD.
Similar content being viewed by others


Introduction
Recent large scale genetic studies have identified a set of single nucleotide polymorphisms (SNP)s that are associated with Alzheimer’s disease (AD) risk [1–7]. Several of the genes underlying these SNPs encode proteins relevant to microglial function and inflammation, including TREM2, CD33, CR1, ABCA7 and SHIP1. Prior to these studies, inflammation was well-recognized to occur in AD with a general consensus that anti-inflammatory agents may be helpful. However, the specific aspects of inflammation that were beneficial or detrimental were unclear (reviewed in [8–13]). These recent genetic studies pinpoint specific proteins and pathways in AD risk. When considered along with the pro-inflammatory role of the prototypical AD genetic risk factor, APOE4, a model emerges showing that AD risk is reduced by processes that suppress inflammatory cytokine signaling and enhance clearance of debris, including amyloid. In this review, we examine these recent genetic studies and their interpretation, integrate the findings into our understanding of inflammation processes in AD and discuss the potential for their translation into AD preventative and/or therapeutic agents.
The advent of well-powered genome wide association studies (GWAS) has been critical to this advance. Simply put, these studies compare SNP frequencies between AD and non-AD populations. SNPs with significantly different minor allele frequencies between these two populations constitute potential AD risk factors. For many years, researchers performed candidate SNP studies to identify those that associate with AD risk. The resulting studies were, in retrospect, typically underpowered and not reproducible. This difficulty was not restricted to AD research but was rather a common issue in genetic association studies (reviewed in [14]). The large-scale nature of these GWAS and the requirement for independent replication has largely mitigated concerns about power and reproducibility [1–5]. Among the positive findings reported recently, only the SNP associated with CD33 has struggled with reproducibility [4, 7]. There are several interpretations of non-confirmatory studies. First, when considering whether new data are inconsistent with prior findings, we suggest that the analysis include the 95 % confidence intervals for the SNP association with AD in both the “positive” and “negative” study. If the confidence intervals overlap, the studies are not necessarily in disagreement, and perhaps a meta-analysis of the overall dataset would be informative. This is especially appropriate if the follow-up study is underpowered relative to the original study or if the original finding exhibited a winner’s curse bias, thus overestimating the SNP’s correlation with AD risk [15]. A second possible interpretation of inconsistent genetic results stems from the recognition that the SNPs used in GWAS high throughput arrays are selected to be reliably assayed and to provide a wide genomic coverage and high minor allele frequency (>5 %). Hence, GWAS SNPs are rarely functional SNPs. More commonly, a positive SNP from a GWAS is co-inherited (in linkage disequilibrium (LD)) with a functional SNP. If the functional SNP is consistently co-inherited with the GWAS SNP across ethnic groups and races, then the GWAS SNP will be an efficient proxy SNP for the functional SNP and show consistent association with AD across cohorts. However, if the GWAS SNP is not consistently co-inherited with the functional SNP in different populations, the GWAS SNP will show variable association with phenotype. Hence, inconsistent follow-up results need to be carefully interpreted and understanding the functional effects associated with GWAS SNPs is critical to integrate genetic findings into our understanding of AD.
Determining the relevance of AD genetics to AD prevention or therapy requires knowledge of the SNP actions. Association studies estimate the magnitude of the polymorphism's effect on AD risk. To understand the extent to which we need to impact the underlying pathway to affect AD risk, we need to quantify the SNP action on gene function. The critical factor here is that many of the recent SNPs implicated by GWAS have modest odds ratios for AD risk even without adjusting for the winner’s curse [16]. Whether the products of implicated genes represent robust drug targets depends upon the molecular impact of the SNP on protein function. For example, if the genetic variant completely inhibits the protein and yet has only a 0.9 odds ratio, the protein may not be a robust drug target because complete inhibition produces only a modest effect on AD risk. Alternatively, if the genetic variant alters protein function by a modest 10 % and has a 0.9 odds ratio, the protein may represent a robust drug target because a drug could be developed that mimics the protective SNP allele to a greater degree and thus could have a greater effect on AD risk. This hypothesis has underlying caveats, including that the dose-dependence of protein function in AD risk does not reach a premature plateau, that the protein is a suitable drug target, etc. However, prima facie, a gene modulated by a SNP that has a modest effect on function and a modest effect on AD risk may represent a robust drug target. A secondary issue to address is the thought process that a genetic-based therapy will apply only to those with the AD-risk allele; this is not necessarily accurate. An ideal intervention will not only mimic the action of a protective allele, it will amplify this effect and therefore may be applicable to individuals regardless of their genotype. That said, the intervention may have more impact on those with the risk allele.
In summary, we present an analysis of the genetics of inflammation and microglia in AD. This analysis focuses upon genes selected by the criteria of (i) the genes contain SNPs implicated in AD risk by compelling genetic studies and (ii) the genes encode proteins that impact microglial activation. These genes include TREM2, CD33, CR1, ABCA7, SHIP1 and APOE. Aspects of AD that are relevant but beyond this focus of inflammation genetics, such as AD neuropathology or amyloid protein precursor metabolism, are reviewed elsewhere [17–20]. Within our analysis of genetics, inflammation, microglia and AD, we will discuss the biology of the relevant protein, the protein’s role in inflammation, and how these proteins may interact to collectively modulate immune function in the AD brain.
Review
TREM2
TREM2 (Triggering Receptor Expressed on Myeloid cells 2) is a type 1 transmembrane receptor protein. In mice, TREM2 is expressed in myeloid cells in the brain and appears increased in microglia in the vicinity of plaques in APP mice [21, 22]. Most but not all human brain studies have suggested that TREM2 expression is within microglia [23–26]. TREM2 expression increases with IL-4 exposure [27], suggesting that TREM2 expression may be increased during alternative activation of microglia.
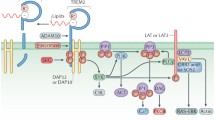
TREM2 ligands include anionic lipids and perhaps other unknown elements from apoptotic neurons [28–31]. TREM2 lacks an extended cytosolic domain, signaling through the immunoreceptor tyrosine-based activating motif (ITAM) of its co-receptor, DAP12 [32, 33]. Activated TREM2 stimulates DAP12 through an intramembrane lysine residue, resulting in phosphorylation of the DAP12 ITAM, and activation of the kinase Syk (Fig. 1, [24, 34]). This leads to activation of PI3K, resulting in actin rearrangement and phagocytic cup formation for target engulfment [35, 36]. TREM2-activated phagocytosis occurs without a commensurate activation of cytokine production [37]. Indeed, TREM2 activation actually decreases cytokine production that occurs in response to Toll-like receptor (TLR) activation [27, 38]. The activation of TREM2, Syk and phagocytosis is balanced by activation of phosphatases, most notably SHP-1, SHP-2, and SHIP1 (encoded by the AD-risk gene INPP5D, see below). Overall, TREM2 stimulation via apoptotic neuronal fragments or TREM2 antibodies appears to result in activation of microglial phagocytosis with minimal changes in cytokine levels.

Several interactions have been reported between the AD risk genes involved in inflammation. TREM2 signals through the ITAM of DAP12 to activate microglial phagocytosis; however, TREM2 expression has also been shown to dampen pro-inflammatory cytokine production activated by TLRs. Activated CD33 recruits SHP-1 and SHP-2 to inhibit Syk signaling; CD33 has also been shown to antagonize CD14/TLR4 signaling. Sialylated apoE, which complexes with Aβ, may serve as a CD33 ligand. ApoE appears to dampen TLR4 and TLR2 signaling and inhibit induction of pro-inflammatory cytokines. SHIP1 antagonizes PI3K action by converting PIP3 to PIP2; SHIP1 has also been shown to bind to and antagonize TREM2 /DAP12 signaling in osteoclasts. SHIP1 also complexes with CD2AP, another AD-implicated protein, to inhibit Syk ubiquitination and degradation. CR1 is a C3b/C4b receptor that promotes phagocytosis; complement components have been shown to complex with Aβ. ABCA7 has been localized to phagocytic cups and linked to Aβ clearance, although its mechanism of action is currently unknown. Proteins encoded by genes associated with AD risk by genetics are shown with solid outlines; proteins that mediate these interactions are shown with dashed outlines
Nonsense, missense, and splice site mutations in TREM2 and its signaling partner DAP12 have been identified as causing Nasu-Hakola disease, a rare, autosomal recessive syndrome marked by early-onset progressive dementia and osteoclast dysfunction resulting in bone cysts [24, 39–42]. In 2012, a genome-wide search for AD risk factors based on the Icelandic population found that a TREM2 missense mutation, R47H (rs75932628), is a significant AD risk factor [43]. This finding was complemented by a large-scale companion study [44]. The odds ratio associated with R47H was initially estimated at 3–4, sparking great interest into TREM2 as a potentially robust therapeutic target [43, 44]. Subsequent studies have confirmed the association but reduced the magnitude of this odds ratio [45–52]. The R47H variant blunts the TREM2-DAP12 response to anionic lipids [29]; therefore, the mutation may reflect a stage of TREM2 function intermediate between full function and the complete loss of function that is associated with the recessive Nasu-Hakola disease.
While the role of TREM2 in phagocytosis may have important implications for Aβ clearance in AD [53], the R47H variant has also been implicated in Parkinson’s disease and frontotemporal dementia (FTD), neither of which centrally involves amyloid [54]. The Q33X nonsense mutation, which appears to lead to loss of TREM2 function, is also associated with FTD risk [55]. Therefore, TREM2-mediated phagocytosis may also target neuronal debris that accumulates with normal synaptic plasticity and with neuronal loss seen in neurodegenerative disorders. Consistent with this possibility, TREM2-transduced macrophages promote clearance of neuronal debris and recovery in an animal model of multiple sclerosis [56].
Based on these findings, many researchers speculate that activators of TREM2 function or expression may represent robust AD preventative and/or therapeutic agents. However, some recent results have called this hypothesis into question. An initial study by Ulrich et al. showed that TREM2 hemizygosity does not affect the prevalence of cortical plaques, soluble Aβ levels, or production of inflammatory cytokines in APPPS1-21 mice [57]. However, TREM2 hemizygosity does affect recruitment of myeloid cells, presumed to be microglia, to plaques [57]. A later report by Wang et al. involving 5xFAD TREM2 knockout and hemizygous mice showed similar results, with greatly reduced microglial clustering around plaques in TREM2-depleted mice [29]. As many researchers had initially hypothesized from the genetic findings, this study found that TREM2-deficient mice develop more plaques and higher soluble and insoluble Aβ levels. However, this effect did not appear to be due to differences in Aβ uptake or metabolism [29]. Instead, TREM2 deletion resulted in decreased microgliosis and microglial survival, at least in part due to decreased response to CSF-1 [29].
In agreement with these two studies, an independent group found that in both 5xFAD and APP-PS1 mouse models, TREM2 knockout mice had negligible immune cells clustering around plaques [58]. Surprisingly, Jay et al. found that TREM2 deletion led to decreased plaque levels in the hippocampus and unchanged plaque levels in the cortex [58]. These amyloid results appear incongruent with the study by Wang et al. and with genetic findings that seem to point to a protective role for TREM2 in neurodegenerative disease [29, 55]. The reason for these discrepancies is unclear, although one variable is that the two groups use different TREM2 knockout strains: Jay et al. use a strain that lacks exons 2–4, which encode the ligand binding domain through the cytosolic domain, while Wang et al. use a strain that lacks exons 3–4, which encode a portion of the TREM2 transmembrane and cytosolic sequence [19] and could produce soluble TREM2.
Even more intriguingly, when Jay et al. examined the “microglial” myeloid cells surrounding plaques in TREM2-positive mice, they found that they had higher CD45 expression than normal microglia, suggesting that they might in fact be bone-marrow derived monocytes infiltrating from the periphery [58]. The authors proposed that the effects of TREM2 in AD are mediated by infiltrating monocytes, rather than microglia: in fact, they report that TREM2 expression in resident microglia in the brain is undetectable by immunohistochemistry. While awaiting further supporting evidence, this model offers insight into therapeutic strategies for AD: pharmacologic agents may not need to cross the blood–brain barrier to be effective but may be able to act on peripheral monocytes that then invade the brain. Overall, studies on TREM2 have focused unparalleled research attention on this inflammation pathway, implicating microglial activation in general and phagocytosis in particular as critical for healthy CNS aging.
CD33
CD33 is a type 1 transmembrane protein and member of the sialic acid-binding immunoglobulin-like lectin (SIGLEC) family of receptors (reviewed in [59]). In the brain, CD33 is expressed largely in microglial cells [60, 61]. CD33 ligands appear to include sialylated cell surface proteins acting in cis as well as other sialyated agents. Many have suggested that CD33 (like other inhibitor SIGLECs) functions to limit immune activation in response to “self” macromolecules that bear a “self-associated molecular pattern” of sialylation (reviewed in [62]). Indeed, amyloid plaques may be “hidden” from microglia because the plaque vicinity is rich in sialylated agents including apoE, apoJ and gangliosides that stimulate CD33 and thereby inhibit microglia [63–65].
Sialic acid binding activates CD33, resulting in phosphorylation of the CD33 immunoreceptor tyrosine-based inhibitory motif (ITIM) domains and activation of the SHP-1 and SHP-2 tyrosine phosphatases [66, 67]. These phosphatases act on multiple substrates, including Syk, to inhibit immune activation [68, 69]. Hence, CD33 activation leads to increased SHP-1 and SHP-2 activity that antagonizes Syk, inhibiting ITAM-signaling proteins, possibly including TREM2/DAP12 (Fig. 1, [70, 71]). Consistent with this possibility, long-term CD33 inhibition (by antibodies or siRNA) induces production of cytokines such as IL1β, TNFα, and IL-8 [72].
A polymorphism in the CD33 proximal promoter was associated with AD risk in several, although not all genome wide studies [4, 6, 7, 73]. Recently, we and others sought to identify the mechanism whereby rs3865444 modulates CD33 to alter AD risk [60, 61, 74–76]. CD33 expression in the brain is modestly increased in AD and decreased with the minor, AD-protective rs3865444A allele [60, 61, 76]. Increased CD33 expression was associated with reduced Aβ phagocytosis [60, 76]. This is consistent with findings that CD33 activation by cell surface sialic acids in cis leads to reduced cellular activation [72, 77], as well as a report suggesting that CD33 negatively regulates CD14, an Aβ receptor [78–80].
Pursuing these molecular actions further, we found that the rs3865444A minor allele was associated with a robust increase in the proportion of CD33 isoforms that lacked exon 2 (D2-CD33) or retained intron 1; the former change results in a translated CD33 protein that lacks its ligand-binding IgV domain, while the latter change results in a frameshift and premature stop codon [61, 74, 75, 81, 82]. Additionally, DNA sequencing established that rs12459419, a SNP within CD33 exon 2, was in near- perfect LD with rs3865444. In vitro minigene splicing studies demonstrated that rs12459419 is a functional SNP that modulates the efficiency of exon 2 splicing [61]. Since D2-CD33 lacks the predicted sialic acid binding domain, D2-CD33 likely encodes a non-functional protein ([81, 82], reviewed in [59]); consistent with this possibility, Aβ42 uptake is inhibited when BV-2 cells were transfected with CD33 but not when cells were transfected with CD33 lacking the IgV domain [60]. Overall, these results provide a genetic basis to support a model wherein TREM2 and CD33 act in opposing directions to modulate tyrosine phosphorylation and, thereby, microglial activation and AD risk.
The utility of CD33 as a target for AD prevention or therapy is an active research area. Of particular interest, the mRNA splicing studies show that each minor allele of the AD-associated SNP results in a 24 % decrease per allele in CD33 mRNA including exon 2 and lacking intron 1 [75]. Hence, an AD odds ratio of ~0.89 correlated with a ~24 % reduction in RNA encoding functional CD33. This suggests that more robust CD33 inhibition may reduce AD risk further. This suggestion is supported further by studies with CD33 deficient mice; these mice develop normally and yet have reduced amyloid accumulation [60, 83], suggesting that CD33 inhibition in humans may be safe and robustly reduce AD risk. Recently, we showed the CD33 antibody Lintuzumab, which has been used safely in human acute myeloid leukemia trials, downregulates cell surface CD33 up to 80 % in PMA-differentiated U937 cells in vitro [75]. Based on linear regression of the allelic dose dependence for CD33 isoforms and AD risk, this 80 % reduction is calculated to reduce the AD odds ratio to 0.68, consistent with the possibility that this level of inhibition could be clinically meaningful.
INPP5D (SHIP1)
The gene INPP5D encodes phosphatidylinositol-3,4,5-trisphosphate-5-phosphatase 1, also known as SHIP1 (SH2-containing inositol 5′ - phosphatase). Since SHIP1 is expressed in macrophage cell lines [84], SHIP1 may be expressed in microglia in the brain. SHIP1 is a phosphatase that hydrolyzes PIP3 to PIP2 on the cytosolic side of plasma membrane, counteracting PI3K induced pathways [85]. The SHIP1 amino-terminal region contains an SH2 domain that binds phosphorylated tyrosine residues on ITIM- or ITAM-containing target proteins [85–90]. SHIP1 has been shown to inhibit monocyte activation and phagocytosis [68, 84, 91–95], in part by transducing inhibitory signaling of FcγRIIB and other ITIM-containing proteins. In dendritic cells, SHIP1 complexes with the AD-relevant protein CD2AP to inhibit the ubiquitination of pro-inflammatory proteins Syk and FcγRIIa [96]. SHIP1 also reduces NF-κB activation, which has been shown to activate BACE1 expression in activated astrocytes [97, 98]. Perhaps most relevant to AD, SHIP1 inhibits TREM2 signaling through DAP12 in osteoclasts, dysfunction of which is another hallmark of Nasu-Hakola disease [99]. Interestingly, SHIP1 does not appear to inhibit the ITIM of CD33 [66, 100, 101].
An INPP5D polymorphism, rs35349669, has recently been associated with AD risk [4]. SHIP1 transcription is initiated at multiple start sites; initiation at an internal site results in production of a protein lacking the SH2 domain [102]. Rs35349669 is near this internal transcription start site, suggesting that this SNP may modulate production of a SHIP1 transcript lacking the SH2 domain. Understanding the actions of the AD-associated SNP rs35349669 may be critical to understanding the role of SHIP1 in AD. Overall, SHIP1 appears to transduce inhibitory signaling of some ITIM-containing proteins and to inhibit signaling of ITAM-containing proteins, such as DAP12. Hence, SHIP1 is anti-inflammatory and anti-phagocytic.
CR1
CR1 (Complement Receptor 1) regulates the complement system, a division of the body’s innate immune response that orchestrates phagocytosis and lysis of cells bearing foreign antigens. CR1 is expressed on some leukocytes in the periphery, as well as the choroid plexus, microglia, and neurons in the brain [103, 104]. CR1 acts as both a positive and negative regulator of the complement pathways by binding to the C3b/C4b peptides, stimulating (i) opsonization and clearance of immune complexes and (ii) destabilization of the C3 and C5 convertases, preventing further complement activation.
The complement pathway has been associated with AD since the 1982 when complement factors were found in amyloid plaques [105]. The complement protein C1q was subsequently shown to bind Aβ, leading to complement activation and inflammation [106]. Addition of C1q to solubilized Aβ1-42 promotes Aβ aggregate formation [107]. Subsequent studies found that complement pathway proteins are upregulated in AD brain [108], and that C3b binding to Aβ leads to inflammation and neuronal lysis (reviewed in [109]). However, some studies have found that the complement cascade can have neuroprotective effects: the complement protein C5a has been shown to activate MAPK, protecting neurons and reducing hippocampal lesions in mouse models [109]. Recent results suggest that CR1 has a mixture of neuroprotective and neurodegenerative effects in AD: antagonizing CR1 prevents Aß phagocytosis by primary rat microglia, but also blocks microglial production of superoxide species and the pro-inflammatory cytokines TNFα and IL-1β. Blockage of CR1 also prevents neuronal death when neurons are treated with microglial conditioned media [110]. These data suggest that CR1 activation may be beneficial to clear Aβ at early disease stages but exacerbate inflammation once amyloid deposits have appeared.
SNPs in CR1 have been associated with AD risk in GWAS since 2009 [1, 4, 6, 7]. The search for functional SNPs that mediate this association with AD has revealed two candidates. First, a rare coding SNP rs4844609 (S1610T) was associated with several indices of AD pathology [111]; however, these initial associations were not replicated in an independent cohort [112]. Second, AD-associated SNPs from GWAS such as rs4844610 were associated with a copy number variant (CNV) that modulates the production of two CR1 isoforms: a larger isoform designated CR1-S (slow migrating on gel electrophoresis) and a smaller isoform designated CR1-F (fast migrating) [113]. The larger CR1-S isoform, which has a 15 % frequency and associates with increased AD risk, contains two copies of low copy repeat 1 and therefore encodes more C3b/C4b binding sites than the smaller CR1-F [113]. This CNV is a better predictor of AD risk than the GWAS-implicated SNP rs4844610, suggesting that CR1 genetics modulate AD risk through the functional CR1 CNV [104]. The action of CR1-S in AD is still unclear: one possibility is that since CR1-S encodes more C3b/C4b binding sites, CR1-S leads to increased complement activation and inflammation and thereby increases AD risk [104, 113]. However, studies have also shown that CR1-S carriers have lower overall CR1 protein expression: therefore, AD risk might be conferred through lower CR1 expression leading to decreased complement activation and impaired clearance of Aβ [104, 114]. Hence, whether CR1 and complement activation are beneficial or deleterious for AD is currently unclear and requires further study.
ABCA7
ABCA7 is a member of the ATP-binding cassette superfamily of transporters that is expressed in the periphery in the spleen, thymus, and bone marrow, as well as in microglia in the brain [115, 116]. ABCA7 was initially thought to modulate lipid homeostasis and was found to transport phospholipids across inner and outer plasma membrane leaflets [117–119]. However, the C. elegans ABCA7 homolog, ced-7, is responsible for apoptotic cell engulfment [120]. In fact, ABCA7 has been localized to the phagocytic cups of activated microglia, although the underlying mechanism is not fully understood [120]. Consistent with the possibility that ABCA7 may modulate lipid transport and thereby contribute to phagocytosis, Rong et al. observed that inflammation modulates membrane phospholipid composition, thus affecting the function of membrane proteins [121]. Moreover, Kim et al. showed ABCA7 deficiency resulted in increased Aβ deposition, suggesting a decrease in phagocytic clearance [122]. Taken together, these findings support the possibility that ABCA7 reduces AD risk by contributing to phagocytic cup formation and Aβ clearance (Fig. 1).
Several ABCA7 SNPs, including rs3764650, have been associated with AD in initial and replicative GWAS [4, 7]. The mechanisms underlying this association are unclear. We recently reported that the AD-protective rs3764650 allele was associated with increased ABCA7 expression [123], leading us to hypothesize that the SNP-associated increase in ABCA7 expression protects from AD. We also saw an increase in ABCA7 expression in AD brains that we attributed to an increase in inflammation in AD, noting that ABCA7 expression is increased as monocytes differentiate into macrophages and, interestingly, by LDL [115]. A recent study of an Icelandic population showed that rare loss-of-function variants in ABCA7 confer an increased risk of AD, with a combined odds ratio of 2.1. In replication populations, this striking association held true with an odds ratio of 1.7. One of these variants, rs200538373, promotes retention of a short intronic sequence after exon 41, resulting in a premature stop codon [124]. Hence, an apparent loss of ABCA7 function increases AD risk while increased ABCA7 expression is associated with reduced AD risk.
APOE
While these recent genetic studies have focused new attention upon genes that are critical to neuroinflammation, APOE polymorphisms have also been suggested to differentially affect inflammation. Of the three common APOE alleles, APOE4 raises AD risk and reduces age of AD onset, while APOE2 lowers AD risk and increases age of onset [125]. In the CNS, APOE is primarily expressed by astrocytes [126], as well as by microglia and ependymal cells and, under certain conditions of neurotoxicity, by neurons [127]. ApoE functions in the transport of cholesterol (reviewed in [128]), and is important for the redistribution of lipids within the CNS, including delivery of cholesterol and phospholipids to neurons.
In addition to its function in lipid transport, apoE is an anti-inflammatory agent. Evidence supporting this perspective includes that apoE deficiency exacerbates neuroinflammation in several rodent injury models including ischemia [129], experimental autoimmune encephalomyelitis [130], traumatic brain injury [131], and induced neuroinflammation [132]. Moreover, apoE deficiency was associated with a reduced clearance of neuronal debris in a model of entorhinal cortex lesion, suggesting that apoE contributes to the clearance of cholesterol-rich neuronal breakdown products [133]. Conversely, an apoE mimetic peptide decreased damage in traumatic brain injury [134], increased axonal regeneration after peripheral nerve injury [135], and decreased lesion volume after focal ischemia [136].
Compared to APOE3, APOE4 is associated with a reduced ability to suppress inflammatory stimuli both in vivo and in vitro (reviewed in [137]). In models comparing APOE-knockout (KO) mice to human APOE-targeted replacement (TR) mice, APOE4 appears to represent a loss of positive function rather than a gain of negative function. For example, in APOE-TR mice treated with intracerebroventricular (ICV) injections of lipopolysaccharide (LPS), levels of activated microglia, astrocytes, invading T-cells and cytokines (IL-1β, and TNFα), and synaptic protein loss were greater in APOE-KO > APOE4-TR > APOE3-TR > APOE2-TR [138]. Similarly, in the periphery, proinflammatory stimuli induced an increase in IL-1β release in APOE-KO > APOE4-TR > APOE3-TR > APOE2-TR [139]. Peripheral LPS injection also induces higher TNFα levels in APOE4-TR compared to APOE3-TR mice [140, 141], while TNFα, IL-6, and IL-1β are greater in APOE-KO mice compared to wild type mice [132]. Although the literature is sparse on APOE isoform-specific effects on Aß-induced neuroinflammation, evidence supports that APOE4 modulates Aß-induced neuroinflammation in vivo. E4FAD mice (APOE-TR mice crossed with 5xFAD mice) [142] exhibit greater microgliosis and astrogliosis around cortical Aβ deposits compared to E3FAD [143]. Multiplex analysis of mRNA levels for neuroinflammatory markers in the cortex of EFAD mice at 6 and 8 months revealed that in 6 month E4FAD mice, select markers related to TLR4 signaling are higher, while IL-4R and related markers are lower compared to E3FAD mice; these age- and APOE-dependent effects suggest an APOE3-specific adaptive response lacking with APOE4 [137].
These in vivo findings that apoE promotes an anti-inflammatory state and that apoE4 is a less effective anti-inflammatory agent than apoE3 are recapitulated in vitro. In response to LPS stimulation, TNFα and IL-6 are upregulated in APOE-KO glial cells compared to wild type [132], while nitric oxide levels are increased in APOE-KO but not APOE3-TR mixed glial cultures [144]. In primary microglia, an apoE-mimetic peptide inhibits LPS-induced JNK activation through interactions with the LDL receptor family members [145, 146]. APOE deletion has also been shown to upregulate TLR4 and TLR2 and enhance TLR signaling [147, 148]. Compared to apoE3, apoE4 increases cytokine production in both LPS-treated peripheral immune cells [132, 133] and LPS- and Aß-treated mixed glial cultures [137, 149, 150]. LPS and oligomeric Aβ-induced TNFα secretion are inhibited by TLR4 antagonists in mixed glial cultures, consistent with APOE isoform specific effects on TLR4 signaling in vivo [137, 149, 150]. APOE4 alleles also dose-dependently increase nitric oxide production in microglial cultures [141].
Overall, the distinction that APOE4 represents a loss of positive function in neuroinflammation rather than a gain of negative function is important with respect to development of AD treatment strategies, suggesting that APOE4 carriers would benefit from strategies designed to correct the structure/function of apoE4 rather than eliminate apoE4.
Proposed Model Integrating Genetics, Neuroinflammation and AD Risk
We propose a testable model wherein each AD genetic risk factor considered here is integrated into a coherent model wherein AD risk is modulated by immune activation. We depict activation as defined by promoting phagocytosis and cytokine production (Fig. 1). We recognize that microglial cytokine production has diverse effects in AD; these effects have been reviewed elsewhere [151].
Hence, we suggest the following:
TREM2
Neuronal debris and perhaps other lipid-rich ligands activate TREM2, which promotes microglial phagocytosis through the ITAM domain of its co-factor, DAP12 and the downstream effector, Syk [28]. We hypothesize that enhanced TREM2 function reduces AD risk, although as discussed above, this hypothesis is not supported by all current data [29, 57, 58].
CD33
Sialic acid-rich areas, such as the vicinity of plaques, stimulate CD33 signaling, leading to activation of the CD33 ITIM, which, in turn activates SHP-1 to inhibit microglial activation, particularly TREM2 signaling via Syk [70, 71]. ApoE is an abundant sialylated protein in the vicinity of plaques and hence could contribute to this action [63–65]. The AD-protective SNP allele reduces the proportion of CD33 encoding functional CD33 and thereby effectively inhibits CD33 to promote microglial activation [60, 61, 74–76].
INPP5D (SHIP1)
The SH2-containing SHIP1 isoform moves from the cytosol to the cell surface to bind the phosphorylated ITAM of DAP12 to inhibit TREM2 signaling [99]. SHIP1 also antagonizes the action of PI3K, an important mediator of phagocytosis [95]. We hypothesize that the risk allele of the AD-associated SNP will increase expression of SHIP1 or a SHIP1 isoform.
CR1
This receptor is stimulated by C3b and C4b, which may bind to Aβ to promote inflammation and/or phagocytosis. The CR1 GWAS signal is most likely due to a CNV which modulates the proportion of the two CR1 isoforms: the shorter CR1-F and longer CR1-S [113]. The CR1-S isoform, which increases AD risk, has more ligand-binding sites but also leads to decreased overall expression; therefore, it is unclear whether CR1 activation is beneficial or deleterious for AD risk [104, 113, 114].
ABCA7
This phagocytic cup protein is critical to phagocytosis of substrates such as apoptotic cells [120]. The AD-risky allele of rs3764650 was associated with modestly decreased ABCA7 expression [7, 123]. More strikingly, rare nonsense SNPs were associated with a more robust AD odds ratio [124]. Overall, decreased ABCA7 is hypothesized to reduce phagocytosis and thereby increase AD risk.
APOE
This protein promotes an anti-inflammatory state, with apoE4 being less effective than apoE3, which is less effective than apoE2. Hence, we propose that the AD risk associated with apoE4 is due, at least in part, to increased pro-inflammatory microglial activation with reduced phagocytosis. Conversely, apoE2 reduces AD risk relative to apoE3 by promoting an anti-inflammatory state, perhaps with increased phagocytosis. These apoE allelic actions could occur in parallel with other apoE allele-dependent mechanisms that also modulate AD, including differential ability to serve as an Aß chaperone (reviewed in [128]).
In aggregate, we propose that microglial activation state reflects a homeostatic balance between proteins like CD33 and SHIP1 that inhibit all microglial activation, proteins like TREM2 that promote phagocytic activity without inflammatory cytokines, and proteins like CR1 that are non-specific immune activators. We would also group apoE4 in the last category, relative to the immunosuppressive activities of apoE3 and, progressively, apoE2 (Fig. 2).

Microglial activation can be neuroprotective and/or neurotoxic; the actions of AD risk proteins modulate these effects. The normal actions of CD33 and SHIP1 (encoded by INPP5D) appear to antagonize both forms of microglial activation, while CR1 action appears to promote both Aβ phagocytosis and the production of neurotoxic pro-inflammatory cytokines such as TNF. TREM2 appears to promote phagocytosis while dampening pro-inflammatory cytokine production. ABCA7 helps to mediate phagocytosis. APOE2 and APOE3 are anti-inflammatory, while APOE4 promotes inflammation and neurotoxicity
Conclusions
Genetics has certainly ignited interest in the role of inflammation as a modulator of AD pathogenesis and risk. Along with this interest has come renewed appreciation that immune activation represents a “double-edged sword” in that activation can be neuroprotective by promoting phagocytosis and clearance of cellular debris and Aβ, but also neurotoxic by promoting pro-inflammatory cytokine production, oxidative stress, and neuronal death. While the actions of some AD-relevant agents, such as TREM2 polarize towards a protective phenotype, other agents, such as CD33, CR1, and SHIP1 (INPP5D) seem to modulate both “types” of inflammation concurrently, suggesting that these two phenotypes may not in fact be distinct or separable [152].
Since the actions of several AD-associated SNPs are still unknown, further research into SNP effects on gene expression and protein function is needed to gain clarity. However, even where there is widespread agreement in the field as to the effect of a genetic variant, results from different variants tell different tales. For example, CD33 inactivation and APOE4 seem to have similar effects on TLR activation and pro-inflammatory cytokine induction [72, 78] but genetic data tell us that CD33 inactivation is AD-protective while APOE4 is of course risky [141, 147]. This suggests that an agent that generally modulates even one facet of microglial activation, such as TLR function or phagocytosis, may not be useful as an AD therapeutic: currently, the field has not yet determined with certainty that any particular microglial function is “good” or “bad” for AD risk. However, we can currently be confident that a genetic risk factor discovered in unbiased, large-scale human studies is protective or risky for AD. This is in part why genetic risk factors offer such great promise as therapeutic targets and research tools, and why it is so critical to understand their effects.
Abbreviations
- SNP:
-
Single nucleotide polymorphism
- AD:
-
Alzheimer’s disease
- GWAS:
-
Genome wide association studies
- LD:
-
Linkage disequilibrium
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
- ITAM:
-
Immunoreceptor tyrosine-based activating motif
- TLR:
-
Toll-like receptor
- FTD:
-
Frontotemporal dementia
- SIGLEC:
-
Sialic acid-binding immunoglobulin-like lectin
- ITIM:
-
Immunoreceptor tyrosine-based inhibitory motif
- SHIP1:
-
SH2-containing inositol 5′ - phosphatase
- CR1:
-
Complement Receptor 1
- CNV:
-
Copy number variant
- KO:
-
Knockout
- TR:
-
Targeted replacement
- ICV:
-
Intracerebroventricular
- LPS:
-
Lipopolysaccharide
References
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–9.
Lambert JC, Zelenika D, Hiltunen M, Chouraki V, Combarros O, Bullido MJ, et al. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging. 2011;32:756 e711–755.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–93.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8.
Jun G, Naj AC, Beecham GW, Wang LS, Buros J, Gallins PJ, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67:1473–84.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421.
Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346.
Wilcock DM. Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer’s disease. Curr Gerontol Geriatr Res. 2012;2012:170276.
Finch CE, Morgan TE. Systemic inflammation, infection, ApoE alleles, and Alzheimer disease: a position paper. Curr Alzheimer Res. 2007;4:185–9.
Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15.
Bamberger ME, Landreth GE. Inflammation, apoptosis, and Alzheimer’s disease. Neuroscientist. 2002;8:276–83.
Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG. Replication validity of genetic association studies. Nat Genet. 2001;29:306–9.
Kraft P. Curses--winner’s and otherwise--in genetic epidemiology. Epidemiology. 2008;19:649–51. discussion 657–648.
Xiao R, Boehnke M. Quantifying and correcting for the winner’s curse in genetic association studies. Genet Epidemiol. 2009;33:453–62.
Zinser EG, Hartmann T, Grimm MO. Amyloid beta-protein and lipid metabolism. Biochim Biophys Acta. 2007;1768:1991–2001.
Xia W. Amyloid metabolism and secretases in Alzheimer’s disease. Curr Neurol Neurosci Rep. 2001;1:422–7.
Thinakaran G. Metabolism of presenilins. J Mol Neurosci. 2001;17:183–92.
Bu G, Cam J, Zerbinatti C. LRP in amyloid-beta production and metabolism. Ann N Y Acad Sci. 2006;1086:35–53.
Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC, et al. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer’s disease. ASN Neuro. 2010;2, e00037.
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56:1438–47.
Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F, et al. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. 2004;20:2617–28.
Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357–61.
Numasawa Y, Yamaura C, Ishihara S, Shintani S, Yamazaki M, Tabunoki H, et al. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol. 2011;18:1179–83.
Satoh JI, Kawana N, Yamamoto Y, Ishida T, Saito Y, Arima K. A survey of TREM2 antibodies reveals neuronal but not microglial staining in formalin-fixed paraffin-embedded postmortem Alzheimer’s brain tissues. Alzheimers Res Ther. 2013;5:30.
Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–4.
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–56.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71.
Cannon JP, O’Driscoll M, Litman GW. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics. 2012;64:39–47.
Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur J Immunol. 2001;31:783–91.
Bouchon A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–5.
Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–22.
Bakker AB, Baker E, Sutherland GR, Phillips JH, Lanier LL. Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Natl Acad Sci U S A. 1999;96:9792–6.
Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol Rev. 2013;256:222–39.
Deschamps C, Echard A, Niedergang F. Phagocytosis and cytokinesis: do cells use common tools to cut and to eat? Highlights on common themes and differences. Traffic. 2013;14:355–64.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57.
Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006;177:2051–5.
Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552–8.
Kondo T, Takahashi K, Kohara N, Takahashi Y, Hayashi S, Takahashi H, et al. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology. 2002;59:1105–7.
Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–62.
Kuroda R, Satoh J, Yamamura T, Anezaki T, Terada T, Yamazaki K, et al. A novel compound heterozygous mutation in the DAP12 gene in a patient with Nasu-Hakola disease. J Neurol Sci. 2007;252:88–91.
Jonsson T, Stefansson H, Ph DS, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2012.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2012.
Ruiz A, Dols-Icardo O, Bullido MJ, Pastor P, Rodriguez-Rodriguez E, Lopez de Munain A, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014;35:444 e441–444.
Guerreiro R, Hardy J. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1569–70.
Bird TD. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1568.
Benitez BA, Cruchaga C, United States-Spain Parkinson’s Disease Research G. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1567–8.
Rajagopalan P, Hibar DP, Thompson PM. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1565–7.
Bertram L, Parrado AR, Tanzi RE. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1565.
Reitz C, Mayeux R, Alzheimer’s Disease Genetics C. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1564–5.
Jonsson T, Stefansson K. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1568–9.
Savage JC, Jay T, Goduni E, Quigley C, Mariani MM, Malm T, et al. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J Neurosci. 2015;35:6532–43.
Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. 2013;8:19.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27.
Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007;4:e124.
Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–95.
Varki A, Angata T. Siglecs--the major subfamily of I-type lectins. Glycobiology. 2006;16:1R–27.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–43.
Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 2013;33:13320–5.
Schwarz F, Fong JJ, Varki A. Human-specific evolutionary changes in the biology of siglecs. Adv Exp Med Biol. 2015;842:1–16.
Salminen A, Kaarniranta K. Siglec receptors and hiding plaques in Alzheimer’s disease. J Mol Med. 2009;87:697–701.
Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, et al. APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol Psychiatry. 2014;19:1243–50.
Xu PT, Schmechel D, Qiu HL, Herbstreith M, Rothrock-Christian T, Eyster M, et al. Sialylated human apolipoprotein E (apoEs) is preferentially associated with neuron-enriched cultures from APOE transgenic mice. Neurobiol Dis. 1999;6:63–75.
Ulyanova T, Blasioli J, Woodford-Thomas TA, Thomas ML. The sialoadhesin CD33 is a myeloid-specific inhibitory receptor. Eur J Immunol. 1999;29:3440–9.
Walter RB, Raden BW, Zeng R, Hausermann P, Bernstein ID, Cooper JA. ITIM-dependent endocytosis of CD33-related Siglecs: role of intracellular domain, tyrosine phosphorylation, and the tyrosine phosphatases, Shp1 and Shp2. J Leukoc Biol. 2008;83:200–11.
Huang ZY, Hunter S, Kim MK, Indik ZK, Schreiber AD. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcgamma receptors FcgammaRIIB and FcgammaRIIA. J Leukoc Biol. 2003;73:823–9.
Reth M, Brummer T. Feedback regulation of lymphocyte signalling. Nat Rev Immunol. 2004;4:269–77.
Linnartz B, Wang Y, Neumann H. Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int J Alzheimers Dis. 2010;2010.
Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 2013;61:37–46.
Lajaunias F, Dayer JM, Chizzolini C. Constitutive repressor activity of CD33 on human monocytes requires sialic acid recognition and phosphoinositide 3-kinase-mediated intracellular signaling. Eur J Immunol. 2005;35:243–51.
Morgan K. The three new pathways leading to Alzheimer’s disease. Neuropathol Appl Neurobiol. 2011;37:353–7.
Raj T, Ryan KJ, Replogle JM, Chibnik LB, Rosenkrantz L, Tang A, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. 2014;23:2729–36.
Malik M, Chiles 3rd J, Xi HS, Medway C, Simpson J, Potluri S, et al. Genetics of CD33 in Alzheimer's disease and acute myeloid leukemia. Hum Mol Genet. 2015;24(12):3557–70.
Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–50.
Gonzalez Y, Herrera MT, Soldevila G, Garcia-Garcia L, Fabian G, Perez-Armendariz EM, et al. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 2012;13:19.
Ishida A, Akita K, Mori Y, Tanida S, Toda M, Inoue M, et al. Negative regulation of toll-like receptor-4 signaling through the binding of glycosylphosphatidylinositol-anchored glycoprotein, CD14, with the sialic acid-binding lectin, CD33. J Biol Chem. 2014;289:25341–50.
Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–89.
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–92.
Hernandez-Caselles T, Martinez-Esparza M, Perez-Oliva AB, Quintanilla-Cecconi AM, Garcia-Alonso A, Alvarez-Lopez DM, et al. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: two isoforms of CD33 are generated by alternative splicing. J Leukoc Biol. 2006;79:46–58.
Perez-Oliva AB, Martinez-Esparza M, Vicente-Fernandez JJ, Corral-San Miguel R, Garcia-Penarrubia P, Hernandez-Caselles T. Epitope mapping, expression and post-translational modifications of two isoforms of CD33 (CD33M and CD33m) on lymphoid and myeloid human cells. Glycobiology. 2011;21:757–70.
Brinkman-Van der Linden EC, Angata T, Reynolds SA, Powell LD, Hedrick SM, Varki A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol Cell Biol. 2003;23:4199–206.
An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, et al. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3 K-independent mechanism. Blood. 2005;105:4685–92.
Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, et al. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1996;93:1689–93.
Mason JM, Beattie BK, Liu Q, Dumont DJ, Barber DL. The SH2 inositol 5-phosphatase Ship1 is recruited in an SH2-dependent manner to the erythropoietin receptor. J Biol Chem. 2000;275:4398–406.
Kavanaugh WM, Pot DA, Chin SM, Deuter-Reinhard M, Jefferson AB, Norris FA, et al. Multiple forms of an inositol polyphosphate 5-phosphatase form signaling complexes with Shc and Grb2. Curr Biol. 1996;6:438–45.
Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev. 1996;10:1084–95.
Barber DL, Mason JM, Fukazawa T, Reedquist KA, Druker BJ, Band H, et al. Erythropoietin and interleukin-3 activate tyrosine phosphorylation of CBL and association with CRK adaptor proteins. Blood. 1997;89:3166–74.
Osborne MA, Zenner G, Lubinus M, Zhang X, Songyang Z, Cantley LC, et al. The inositol 5′-phosphatase SHIP binds to immunoreceptor signaling motifs and responds to high affinity IgE receptor aggregation. J Biol Chem. 1996;271:29271–8.
Cox D, Dale BM, Kashiwada M, Helgason CD, Greenberg S. A regulatory role for Src homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fc gamma receptors and complement receptor 3 (alpha(M)beta(2); CD11b/CD18). J Exp Med. 2001;193:61–71.
Maresco DL, Osborne JM, Cooney D, Coggeshall KM, Anderson CL. The SH2-containing 5′-inositol phosphatase (SHIP) is tyrosine phosphorylated after Fc gamma receptor clustering in monocytes. J Immunol. 1999;162:6458–65.
Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature. 1996;383:263–6.
Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell. 1997;90:293–301.
Kamen LA, Levinsohn J, Swanson JA. Differential association of phosphatidylinositol 3-kinase, SHIP-1, and PTEN with forming phagosomes. Mol Biol Cell. 2007;18:2463–72.
Bao M, Hanabuchi S, Facchinetti V, Du Q, Bover L, Plumas J, et al. CD2AP/SHIP1 complex positively regulates plasmacytoid dendritic cell receptor signaling by inhibiting the E3 ubiquitin ligase Cbl. J Immunol. 2012;189:786–92.
Bourne KZ, Ferrari DC, Lange-Dohna C, Rossner S, Wood TG, Perez-Polo JR. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res. 2007;85:1194–204.
Conde C, Rambout X, Lebrun M, Lecat A, Di Valentin E, Dequiedt F, et al. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-kappaB activation by disturbing the interaction of XIAP with RIP2. PLoS One. 2012;7, e41005.
Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38.
Paul SP, Taylor LS, Stansbury EK, McVicar DW. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood. 2000;96:483–90.
Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, Freeman SD. The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. J Biol Chem. 1999;274:11505–12.
Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–9.
Khera R, Das N. Complement receptor 1: disease associations and therapeutic implications. Mol Immunol. 2009;46:761–72.
Hazrati LN, Van Cauwenberghe C, Brooks PL, Brouwers N, Ghani M, Sato C, et al. Genetic association of CR1 with Alzheimer's disease: a tentative disease mechanism. Neurobiol Aging. 2012;33:2949 e2945–12.
Eikelenboom P, Stam FC. An immunoperoxidase study. Acta Neuropathol. 1982;57:239–42.
Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:10016–20.
Webster S, Rogers J. Relative efficacies of amyloid beta peptide (A beta) binding proteins in A beta aggregation. J Neurosci Res. 1996;46:58–66.
Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol. 1999;154:927–36.
Kolev MV, Ruseva MM, Harris CL, Morgan BP, Donev RM. Implication of complement system and its regulators in Alzheimer’s disease. Curr Neuropharmacol. 2009;7:1–8.
Crehan H, Hardy J, Pocock J. Blockage of CR1 prevents activation of rodent microglia. Neurobiol Dis. 2013;54:139–49.
Keenan BT, Shulman JM, Chibnik LB, Raj T, Tran D, Sabuncu MR, et al. A coding variant in CR1 interacts with APOE-epsilon4 to influence cognitive decline. Hum Mol Genet. 2012;21:2377–88.
Van Cauwenberghe C, Bettens K, Engelborghs S, Vandenbulcke M, Van Dongen J, Vermeulen S, et al. Complement receptor 1 coding variant p.Ser1610Thr in Alzheimer's disease and related endophenotypes. Neurobiol Aging. 2013;34:2235 e2231–2236.
Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert JC, Bettens K, Le Bastard N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–33.
Mahmoudi R, Kisserli A, Novella JL, Donvito B, Drame M, Reveil B, et al. Alzheimer’s disease is associated with low density of the long CR1 isoform. Neurobiol Aging. 2015;36:1766. e1765-1712.
Kaminski WE, Orso E, Diederich W, Klucken J, Drobnik W, Schmitz G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem Biophys Res Commun. 2000;273:532–8.
Kim WS, Guillemin GJ, Glaros EN, Lim CK, Garner B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport. 2006;17:891–6.
Wang N, Lan D, Gerbod-Giannone M, Linsel-Nitschke P, Jehle AW, Chen W, et al. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J Biol Chem. 2003;278:42906–12.
Abe-Dohmae S, Ikeda Y, Matsuo M, Hayashi M, Okuhira K, Ueda K, et al. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem. 2004;279:604–11.
Ikeda Y, Abe-Dohmae S, Munehira Y, Aoki R, Kawamoto S, Furuya A, et al. Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem Biophys Res Commun. 2003;311:313–8.
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol. 2006;174:547–56.
Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–97.
Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK, et al. Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci. 2013;33:4387–94.
Vasquez JB, Fardo DW, Estus S. ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci Lett. 2013;556:58–62.
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47:445–7.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–3.
Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–61.
Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci. 2006;26:4985–94.
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–18.
Sheng H, Laskowitz DT, Mackensen GB, Kudo M, Pearlstein RD, Warner DS. Apolipoprotein E deficiency worsens outcome from global cerebral ischemia in the mouse. Stroke. 1999;30:1118–24.
Karussis D, Michaelson DM, Grigoriadis N, Korezyn AD, Mizrachi-Koll R, Chapman S, et al. Lack of apolipoprotein-E exacerbates experimental allergic encephalomyelitis. Mult Scler. 2003;9:476–80.
Lynch JR, Pineda JA, Morgan D, Zhang L, Warner DS, Benveniste H, et al. Apolipoprotein E affects the central nervous system response to injury and the development of cerebral edema. Ann Neurol. 2002;51:113–7.
Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT. Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol. 2001;114:107–13.
Fagan AM, Murphy BA, Patel SN, Kilbridge JF, Mobley WC, Bu G, et al. Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (−/−) mice but impaired clearance of axonal degeneration products following injury. Exp Neurol. 1998;151:314–25.
Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, et al. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma. 2010;27:1983–95.
Li FQ, Fowler KA, Neil JE, Colton CA, Vitek MP. An apolipoprotein E-mimetic stimulates axonal regeneration and remyelination after peripheral nerve injury. J Pharmacol Exp Ther. 2010;334:106–15.
Tukhovskaya EA, Yukin AY, Khokhlova ON, Murashev AN, Vitek MP. COG1410, a novel apolipoprotein-E mimetic, improves functional and morphological recovery in a rat model of focal brain ischemia. J Neurosci Res. 2009;87:677–82.
Tai LM, Ghura S, Koster KP, Liakaite V, Maienschein-Cline M, Kanabar P, et al. APOE-modulated Abeta-induced neuroinflammation in Alzheimer’s disease: current landscape, novel data, and future perspective. J Neurochem. 2015;133:465–88.
Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW, et al. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia. 2012;60:559–69.
Azevedo OG, Bolick DT, Roche JK, Pinkerton RF, Lima AA, Vitek MP, et al. Apolipoprotein E plays a key role against cryptosporidial infection in transgenic undernourished mice. PLoS One. 2014;9, e89562.
Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–33.
Vitek MP, Brown CM, Colton CA. APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009;30:1350–60.
Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, et al. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–86.
Rodriguez GA, Tai LM, LaDu MJ, Rebeck GW. Human APOE4 increases microglia reactivity at Abeta plaques in a mouse model of Abeta deposition. J Neuroinflammation. 2014;11:111.
Laskowitz DT, Matthew WD, Bennett ER, Schmechel D, Herbstreith MH, Goel S, et al. Endogenous apolipoprotein E suppresses LPS-stimulated microglial nitric oxide production. Neuroreport. 1998;9:615–8.
Pocivavsek A, Burns MP, Rebeck GW. Low-density lipoprotein receptors regulate microglial inflammation through c-Jun N-terminal kinase. Glia. 2009;57:444–53.
Pocivavsek A, Mikhailenko I, Strickland DK, Rebeck GW. Microglial low-density lipoprotein receptor-related protein 1 modulates c-Jun N-terminal kinase activation. J Neuroimmunol. 2009;214:25–32.
Goldklang M, Golovatch P, Zelonina T, Trischler J, Rabinowitz D, Lemaitre V, et al. Activation of the TLR4 signaling pathway and abnormal cholesterol efflux lead to emphysema in ApoE-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1200–8.
Michelsen KS, Doherty TM, Shah PK, Arditi M. TLR signaling: an emerging bridge from innate immunity to atherogenesis. J Immunol. 2004;173:5901–7.
Jofre-Monseny L, Loboda A, Wagner AE, Huebbe P, Boesch-Saadatmandi C, Jozkowicz A, et al. Effects of apoE genotype on macrophage inflammation and heme oxygenase-1 expression. Biochem Biophys Res Commun. 2007;357:319–24.
Tsoi LM, Wong KY, Liu YM, Ho YY. Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-alpha and IL-6 in macrophages. Arch Biochem Biophys. 2007;460:33–40.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405.
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The University of Kentucky Research Foundation holds a patent (US Patent No. 9,066,928) on the use of CD33 inhibitors in Alzheimer’s disease.
Authors’ contributions
Each author contributed part of the overall text. MM and SE integrated the parts into the final version. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Malik, M., Parikh, I., Vasquez, J.B. et al. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegeneration 10, 52 (2015). https://doi.org/10.1186/s13024-015-0048-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-015-0048-1