
Abstract
Background
Gastric cancer (GC) is a major cancer burden throughout the world with a high mortality rate. The performance of current predictive and prognostic factors is still limited. Integrated analysis is required for accurate cancer progression predictive biomarker and prognostic biomarkers that help to guide therapy.
Methods
An AI-assisted bioinformatics method that combines transcriptomic data and microRNA regulations were used to identify a key miRNA-mediated network module in GC progression. To reveal the module’s function, we performed the gene expression analysis in 20 clinical samples by qRT-PCR, prognosis analysis by multi-variable Cox regression model, progression prediction by support vector machine, and in vitro studies to elaborate the roles in GC cells migration and invasion.
Results
A robust microRNA regulated network module was identified to characterize GC progression, which consisted of seven miR-200/183 family members, five mRNAs and two long non-coding RNAs H19 and CLLU1. Their expression patterns and expression correlation patterns were consistent in public dataset and our cohort. Our findings suggest a two-fold biological potential of the module: GC patients with high-risk score exhibited a poor prognosis (p-value < 0.05) and the model achieved AUCs of 0.90 to predict GC progression in our cohort. In vitro cellular analyses shown that the module could influence the invasion and migration of GC cells.
Conclusions
Our strategy which combines AI-assisted bioinformatics method with experimental and clinical validation suggested that the miR-200/183 family-mediated network module as a “pluripotent module”, which could be potential marker for GC progression.
Similar content being viewed by others

Background
Gastric cancer (GC) remains a major global health problem and is the third leading cause of cancer-associated death worldwide [1]. Although recent advances in techniques have improved the prognosis of patients with GC, many patients are still diagnosed in advanced stages [2], and the mortality rate remains high because of the heterogeneity and complicated regulatory relations at the molecular level [3,4,5,6]. Thus, novel insights into the mechanisms underlying GC progression will be crucial.
Studies are increasingly characterizing the regulatory effects of non-coding RNAs in the initiation and development of GC, as well as drug resistance [7,8,9,10,11,12]. MicroRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have received substantial attention. However, RNA molecules do not function in isolation and can be grouped into “competitive endogenous RNA networks” on the basis of the crosstalk between lncRNAs and mRNAs competing for shared miRNA response elements [13]. This lncRNA-miRNA-mRNA crosstalk, which is involved in various human cancers, may enable effective approaches to studying cancer pathogenesis and progression [14]. In GC, several of these regulatory axes have been determined to play roles in tumorigenesis and cancer progression; examples include LINC01234/miR-204-5p/CBFB [15], HOTAIR/miR-331-3p/HER2 [16], BC032469/miR-1207-5p/hTERT [17], and DLX6-AS1/miR-204-5p/OCT1 axis [18]. However, how lncRNA-miRNA-mRNA interactions control the regulatory mechanism of GC progression and the roles of these interactions have not been fully elucidated.
The bioinformatics methods that are with the help of miRNA-mediated regulated network (miRNet) enable study of the effects of RNA interactions in cancer at system level and global view, and may acid in the development of new therapeutic strategies and discovery of biomarkers. In past decades, several bioinformatics strategies have been proposed to identify module biomarkers or key modules for tumorigenesis and development, on the basis of miRNet. Cui et al. have integrated topological analysis and a random walk with restart algorithm to identify a prognostic signature for GC [19]. He et al. have identified a module using a clique-percolation method with CFinder software, to divide patients into groups according to survival outcomes [20]. Recently, Wang et al. have proposed the network-based matrix factorization framework NSOJNMF for miRNA-mediated regulated co-modules associated with the occurrence and development of cancer [21]. Most of the above methods take full advantage of network structures. Together with advances in “-omics” data, machine learning and AI techniques are powerful tools that can assess module biomarker discovery by integrating multimodal data.
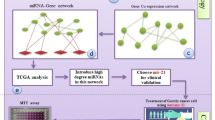
In this study, we identified a miRNet module to characterize GC progression by an AI-assisted bioinformatics method (Fig. 1) based on our previous designed scoring systems (RNs), which integrates various types of high-throughput data including transcriptomic, interactomic and network topological feature data [22]. Subsequently, we explored the prognostic and predictive roles of the module and validated the module in clinical samples and cell lines. Our findings suggested that miR-200/183 family-mediated network modules may have potential as biomarker for GC progression.

Overview of our method for identification of the key miRNA-mediated network module in GC progression
Methods
Derivation of the GC dataset
A data set containing the miRNA, mRNA and lncRNA expression profiles of 257 patients with TNM stage information from The Cancer Genome Atlas (TCGA) was used for identification of the miRNA-mediated network module [23]. The RNA counts were used for further analysis. The differentially expressed genes (DEGs) between early stage (stage I or II, 129 samples) GC groups (ESGC) and late stage (stage III or IV,128 samples) GC groups (LSGC) were identified with the R package DESeq2 [24], with the filter adjusted p-value < 0.05. The detail clinical information of the patients was listed in Additional file 1: Table S1. The association between gene expression and the clinicopathological features of GC patients was evaluated using the chi-square test.
Construction and analysis of a GC progression-specific miRNA mediated (GCP-miR) network
A GCP-miR network was constructed in four steps: (1) the correlation between miRNAs and lncRNAs or mRNAs was determined with Spearman correlation tests, and pairs with p-value < 0.05 were retained and subjected to further steps. (2) The miRNA-mRNA interactions were selected by integrating the miRNA-mRNA pairs (Spearman’s correlation coefficient (SCC) < − 0.3) and the miRNA regulations predicted by miRDB (score > 50) [25, 26]. (3) To obtain more miRNA-lncRNA links, we retained the links meeting one of the following two criteria: (a) miRNA-lncRNA pairs with SCC less than − 0.3; and (b) miRNA-lncRNA pairs with SCC less than zero and were also predicted by starBase [27] or DIANA-LncBase [28]. (4) LncRNA-miRNA and mRNA-miRNA interactions that shared the same miRNAs were regarded as links in the GCP-miR network.
The R package igraph was used to calculate the topological parameter degree (D), betweenness (B) and closeness (C) for each node.
Gene prioritization based on RNs score
The topological features from molecular networks alone are not sufficient to identify disease-associated genes without biological information. To overcome this limitation, we used RNs score (Eq. (1)) which integrated gene expression data via the SVM-RFE algorithm and topological characteristics of network nodes [22]. In our previous work, the RNs score was designed for protein–protein interaction networks. To further validate and extend the application of it, we applied the score to prioritize both coding and non-coding genes in the miRNA-mediated network.
where K is the degree of a node in the network, L is the shortest path length of the node with the remaining nodes in the network, and Rs is the SVM-RFE score ranking the genes by expression level.
Survival model construction
Using the selected genes fitted in a multivariable Cox regression model, we determined a risk score formula based on gene expression. Subsequently, each patient had a risk score, and the patients were divided into low-risk and high-risk groups according to a cutoff mean risk score. The Kaplan–Meier method was used to estimate the survival time and the log rank test was used to compare the survival difference between the low-risk and high-risk groups. A p-value < 0.05 was considered statistically significant.
Functional enrichment analysis
Functional enrichment analysis and visualization were performed using R package clusterProfiler [29, 30]. Gene Ontology (GO) terms with adjusted p-values < 0.01 were considered significantly enriched, whereas Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with p-values < 0.05 were retained as significantly enriched pathways.
Predictive model construction
To explore the predictive significance of gene combinations, we constructed an SVM predictive model on TCGA datasets and then evaluated its performance in our cohort with R package mlr3 [31]. The receiver operator characteristic (ROC) curve was plotted and the area under the curve (AUC) was calculated with R package ROCR [32].
Sample collection and characterization
GC tissue samples and paired non-tumorous adjacent (NT) tissues (located 5 cm from the tumor margin) were obtained from patients with tissue pathology confirmation of GC at the First Affiliated Hospital of Soochow University (Suzhou, China) between March 2017 and August 2018. No patients had received radiotherapy or chemotherapy before surgery, and none of them had cardiac, liver or renal dysfunction. In the GC group, TNM-staging was determined according to the pathological staging criteria (version 8) of the American Joint Committee on Cancer. A total of 20 patients were finally enrolled in the analysis. The clinical characteristics of all patients were summarized in Additional file 1: Table S1.
Cell culture and transfection
The cell lines GES-1, AGS, MKN-45, MKN-28 and HGC-27 were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in RPMI-1640 (Biological Industries, Beit Haemek, Israel) with 10% fetal bovine serum (Biological Industries) and 1% penicillin–streptomycin-amphotericin B (NCM Biotech, Suzhou, China, #C100C8) under 5% CO2 at 37 °C. Cells were transfected with CLLU1 siRNA, control siRNA (RiboBio, Guangzhou, China), has-miR-429 and has-miR-183-5p mimics or control mimics (Genepharma, Shanghai, China) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The sequences of mimics were provided in Additional file 1: Table S3.
Transwell migration and invasion assays
Transwell migration and invasion assays were performed with Transwell plates (8.0 mm pore size, PET membrane, Falcon, USA). The lower chamber was filled with 400 µL RPMI 1640 containing 20% fetal bovine serum. Subsequently, 5 × 104 cells in 400 µL serum-free medium were added to the upper chamber. After 24 h incubation at 37 °C, non-migrating cells were removed from the upper of membrane surface with a cotton swab. The filters were then fixed with 4% methanol for 15 min at room temperature, and stained with Crystal Violet for 10 min. Next, the membranes were washed with phosphate-buffered saline and allowed to dry, and an optical microscope (Olympus, Tokyo, Japan) was used to visualize the stained cells in five random fields on each membrane. Cells penetrating the membrane were counted at a magnification of 100× and the mean number was determined. For Transwell invasion assays, the membrane in the upper chamber was pre-coated with 50 µL Matrigel (Corning, Corning, NY, USA). All assays were performed in triplicate, and the experiment was repeated three times.
Wound healing assay
Wound healing assays were performed to examine the migration ability of cells. Briefly, MKN-45 and HGC-27 cells were transfected with CLLU1 siRNA, or control siRNA for 48 h, then seeded in 12-well plates. When the cells reached 90–95% confluence, a single scratch wound was made across the plate surface with a 200-μL pipette tip. The scratch wounds were photographed over a 48-h period using an inverted microscope (Olympus), and the wound width of was quantified with imaging software. Each assay was performed in triplicate.
RNA extraction and qRT-PCR analysis
Total RNA was extracted from tissue samples with TRIzol reagent (TaKaRa) according to the manufacturer’s instructions. For mRNA and lncRNA expression, 1 μg total RNA was reversed transcribed into cDNA with PrimeScript RT Master Mix (Takara). The qRT-PCR was performed in a CFX96 TouchTM real-time PCR system (Bio-Rad, Hercules, CA, USA) with SYBR Green Master Mix (Vazyme, Nanjing, China). For miRNA expression analysis, 1 µg total RNA was used for first-strand DNA synthesis with a miRNA 1st Strand cDNA Synthesis Kit (Vazyme), and qRT-PCR was performed with miRNA universal SYBR qPCR Master Mix (Vazyme). Relative gene expression was calculated using the 2−ΔΔCt method, with β-actin and small nuclear RNA U6 used as endogenous controls for mRNA/lncRNA and miRNA. The primer sequences for qRT-PCR are provided in Additional file 1: Table S2. The Wilcoxon rank sum test was used to test the difference between the GC and NT groups, as well as the ESGC and LSGC groups.
Results
GCP-miR network construction
Expression data for lncRNAs, mRNAs and miRNAs were collected from 129 patients with ESGC and 128 patients with LSGC in TCGA. First, the significant DEGs between ESGC and LSGC were identified. A total of 1165 mRNAs (649 up-regulated and 516 down-regulated), 15 lncRNAs (11 up-regulated and four down-regulated) and 59 miRNAs (33 up-regulated and 26 down-regulated) were found to be differentially expressed between two groups and the miRNA-lncRNA and miRNA-mRNA pairs with significant negative correlations were used in subsequent analyses.
We then established the GCP-miR network by integrating the above pairs and the results from miRNA target prediction tools as described in Methods. The final GCP-miR network consisted of three types of nodes (22 miRNAs, 126 mRNAs, and 7 lncRNAs), and two type of links (295 miRNA-mRNA and 46 miRNA-lncRNA links; Fig. 2a).

GCP-miR network and its biological function. a Construction and visualization of the GCP-miR network. Diamonds, rectangles, and ellipses indicate mRNAs, miRNAs and lncRNAs, respectively. Pink represents high expression, and blue represents low expression in LSGC compared with ESGC. b GO biological process enrichment analysis of genes in the GCP-miR network (adjusted p-value < 0.01). Each node represents a GO term and each edge represents the overlap between two terms. c KEGG-based enrichment analysis of genes in the GCP-miR network (p-value < 0.05). KEGG terms were sorted by gene ratio
Functional enrichment analysis was performed on genes in the GCP-miR network to explore their biological functions. As shown in Fig. 2b, c, the GO biological process terms and KEGG pathways were highly enriched in cancer development and progression associated pathways, such as circadian entrainment [33], the apelin signaling pathway [34, 35], and the cGMP-PKG signaling pathway [36]. Notably, beyond the cancer-associated pathways, several nervous system-associated terms were enriched, such as glutamatergic synapse. These results indicated that the GCP-miR network is involved in GC progression.
The miR-200/183 family miRNAs are key in GC progression
To identify key genes associated with GC progression, we calculated the RNs score for each gene in the GCP-miR network, which fully accounts for the network topological structure and gene expression levels in cancer samples. The top 10 genes with highly RNs in the network are listed in Table 1, including eight down-regulated miRNAs and two up-regulated lncRNAs in LSGC samples. Because miR-203a-3p had the highest RNs score, and has been reported to predict metastases and poor prognosis in human GC clinical samples [37]. We selected the remaining nine genes for further analysis. Notably, the remaining seven miRNAs were grouped into two families: miR-200 and miR-183 (Table 1) All seven miRNAs were significantly up-regulated in TNM stage I than in other stages (Fig. 3a). Previous studies have demonstrated that down-regulation of miR-200 family members promotes GC progression in vitro in GC cell lines [38,39,40,41] and characterize sub-types of GC with poor-prognosis [42]. Moreover, miR-182-5p and miR-183-5p are involved in GC cell proliferation and have been significantly negatively correlated with ETM scores in lung cancer [43], but their expression patterns have not been consistent in across prior studies [44,45,46]. Beyond miRNAs, two lncRNAs H19 and CLLU1 had high RNs score. H19 affects GC cell proliferation and contributes to GC progression [47, 48]. Although no evidence has indicated a role of CLLU1 in GC progression, it has been reported to be associated with hepatocellular carcinoma prognosis [49]. Its roles in GC cells will are explored below.

The miR-200/183 family-mediated module is key module in GC progression. a Expression of miR-200 and miR-183 families in GC tissues with four TNM stages. b The miR-200/183 family-mediated module for GC progression. Pink and blue denote up and down regulation, respectively, in LSGC samples. c Expression of nine mRNAs in GC tissues with four TNM stages. d Kaplan–Meier analysis was used to estimate the survival of high-risk vs. low-risk patients with GC according to the seven miRNA signatures from the miR-200 and miR-183 families in the training set. e Kaplan–Meier analysis was used to estimate the survival of high-risk vs. low-risk patients with GC according to the seven miRNA signatures from the miR-200 and miR-183 families in the validation cohort. (*p-value < 0.05, **p-value < 0.01, ***p-value < 0.001)
The miR-200/183 family-mediated module is key in GC progression
Because of the important roles of the seven miRNAs and two lncRNAs with high RNs score in GC progression, we selected them and mRNAs, which were interacted with members from both miR-200 and miR-183 families in GCP-miR network, as the key module for GC progression. As shown in Fig. 3b, nine mRNAs were added in the module, which were also significantly up-regulated in LSGC (Fig. 3c) and involved in the adipogenesis, TGF-beta signaling, nuclear receptor and EMT in colorectal cancer pathways. The results indicate that miR-200/183 family-mediated module might contribute the GC progression by regulating their target genes. Moreover, in the module, the gene expression showed significantly negative correlations of lncRNAs and mRNAs with miR-200/183 family members, and significantly positive correlations between miRNAs in GC samples (Additional file 1: Fig. S2).
Then we examined the relationship between miR-200/183 family-mediated module’s members and clinical characteristics of the patients in TCGA dataset. The results (Table 2) showed that the expression level of all the miRNAs were significantly correlated with the age of patients (p < 0.05), six of the seven miRNAs were significantly correlated with the TNM stage (p < 0.05), and five miRNAs were significantly correlated with histological grade. For lncRNAs, CLLU1 expression levels were correlated with histological type (p = 0.041) and grade (p = 0.044). Moreover, the TNM stages and histological grade had also significantly relation with most of the mRNAs in the module (Table 3).
Finally, we investigate the prognostic values of the module in the TCGA dataset and our validation cohort. For TCGA datasets, patients were randomly allocated to the training (n = 180) or testing (n = 77) cohorts using a 7:3 ratio. A risk score formula based on the expression level of miRNAs in the training cohort was created as follows by multi-variable Cox regression model: Risk score = (0.0316 × miR-200b-3p) + (0.0716 × miR-141-3p) + (− 0.0193 × miR-200c-3p) + (− 0.1105 × miR-200a-3p) + (0.1529 × miR-429) + (− 0.5501 × miR-182-5p) + (0.3274 × miR-183-5p). The HR and 95% confidence interval for each miRNA were listed in Additional file 1: Table S4. Then the score for each patient was calculated, and the patients were assigned to high-risk score or low-risk score groups according to the median value of risk score (− 2.4568) in the training cohort. The Kaplan–Meier curves showed the high-risk group had significantly shorter overall survival than the low-risk group in the training group (p-value = 0.039; Fig. 3d), the testing group (p-value = 0.042; Additional file 1: Fig. S3a), and the whole group (p-value = 0.005; Additional file 1: Fig. S3b). For our validation cohort, the results were consistent with those of the TCGA, that is, low-risk score groups exhibited better survival than the high-risk groups (p-value = 0.034; Fig. 3e).
Validation of the miR-200/183 family-mediated module in GC progression
To ascertain the role of the miR-200/183 family-mediated module in human GC progression, we further validated the module by using newly collected samples from patients with GC. The expression levels of seven miRNAs, the two lncRNAs H19 and CLLU1, and five randomly selected mRNAs (LDB3, NOVA1, NPTX1, NR3C1 and ZEB2) in 20 pairs of GC and NT tissues were measured. The miR-200 and miR-183 families members were significantly lower in cancer tissues than NT tissues, and also were significantly lower in LSGC than ESGC. In contrast, their potential targets, two lncRNAs and five mRNAs, were significantly up-regulated in the GC and LSGC with respect to NT tissues and ESGC, respectively (Fig. 4a, p-value < 0.05), thus highlighting the specificity of these candidate biomarkers for GC progression.

Validation of the miR-200/183 family-mediated module in clinical GC samples. a Expression levels of genes in the miR-200/183 family-mediated module (*p-value < 0.05, **p-value < 0.01, ***p-value < 0.001). N: NT samples; C: GC samples; E: ESGC samples; L: LSGC samples. b Validation of the miR-200/183 family-mediated module in GC progression. Green links denote significantly negative correlations with p-values less than 0.05 and SCC less than -0.5. c ROC curve of six gene signatures to stratify the ESGC and LSGC samples
We then performed correlation analysis to validate the association among the genes in the module in our cohort (Additional file 1: Fig. S4). The interactions between the miRNAs and their targets with p-value < 0.5 and SCC < − 0.5 were marked as green lines in the module (Fig. 4b). In agreement with the results based on TCGA datasets, lncRNAs H19 and CLLU1 showed a significant negative correlation with the miR-200 and miR-183 families. Similarly, most mRNAs and miRNAs also showed negative correlations in our cohort. The miR-200 family members displayed significant positive correlations with miR-183 family.
Finally, to evaluate the predictive ability of the module in the classification of ESGC and LSGC, we constructed SVM models according to the expression levels of genes in the module. We first constructed the predictive models with SVM for all combinations of 14 genes in the module with the dataset from TCGA, then evaluated the predictive ability of the combinations to stratify ESGC and LSGC in our independently collected GC samples. Finally, the combination of six genes miR-182-5p, miR183-5p, LDB3, NOVA1, NPTX1 and NR3C1 achieved the highest AUC (0.90, Fig. 4c) among all combinations, thus indicating their ability to predict GC progression.
The miR-200/183 family-mediated module influence the invasion and migration of GC cells
To further validate the biological functions of the miR-200/183 family-mediated module in GC, we performed functional analysis for the RNAs that involved in it. As shown in Table 1, most of the miRNAs in the module, such as miR-200a-3p and miR-141-30, have been reported to affects the invasion and migration of GC cells in multiple studies. Therefore, we selected miR-429 and miR-183-5p as representatives of the miR-200 and miR-183 families and explore their function in GC cells, as well as their predicted targets as shown in Fig. 5a. MKN-45 and HGC-27 cells were transfected with miR-429 mimics, miR-183-5p mimics, or control mimics (Fig. 5b). Transwell invasion and migration assays were then performed to examine the migratory and invasive ability in vitro. As shown in Fig. 5c, d, the invasion and migration of GC cells was more suppressed in the miR-429 and miR-183-5p mimics groups than in the controls (p-value < 0.01). These results indicated that the expression of miR-429 and miR-183-5p efficiently weakened the metastatic potential of GC cells.

Evaluation the effects of miR-200/183 family-mediated module effects on invasion and migration of GC cells. a miR-429 and miR-183-5p regulated sub-module. b Expression of miR-429, miR-183-5p and their targets in miR-429 or miR-183-5p mimics-transfected MKN-45 cells and HGC-27 cells. c, d Transwell migration and invasion assays showed that miR-429 mimics and miR-183-5p inhibited the migratory and invasive capacity of MKN-45 and HGC-27 cells. The data represent means ± SD. *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001
We next validated the interactions among the miRNAs and their targets in Fig. 5a by evaluating the expression of the targets in GC cells that were transfected with miR-429 mimics and or miR-183-5p mimics. We observed that both miRNA mimics significantly decreased the expression of their targets (Fig. 5b). The expression levels of miR-429 targets, including NR3C1, ZEB2, CLLU1 and H19, were significantly lower than those in the NC groups. Similarly, the expression of miR-183-5p targets NR3C1, ZEB2 and CLLU1 in the corresponding mimic group was also significantly lower than that in the NC groups. These expression patterns confirmed the potential interactions among the miRNAs and their targets in GC cells.
Finally, we explored the functions of targets of miR-429 and miR-183-5p in GC cells. Because several studies have reported that high expression of their targets such as ZEB2 [50, 51] and H19 [48, 52] could promote the GC progression, we performed functional analysis of another lncRNA CLLU1 in the module. CLLU1 showed elevated expression in MKN-45 and HGC-27 cell lines compared with normal gastric cells (Fig. 6a). The expression levels of eight other genes with high RNs scores were also measured in GC cell lines (Additional file 1: Fig. S1). We transfected siRNAs to knock down CLLU1 in MKN-45 and HGC-27 (Fig. 6a). Transwell and wound healing assays were then performed to examine the in vitro migratory and invasive ability of CLLU1. As shown in Fig. 6b, c, the invasion and migration of GC cells was greatly suppressed in the knock down group (p-value < 0.01). These results indicated that inhibition of CLLU1 efficiently weakened the metastatic potential of GC cells. Taken together, these results reveal that the miR-429 and miR-183-5p regulated sub-module contribute to the invasion and migration of GC cells.

Knockdown of CLLU1 inhibits the invasion and migration of GC cell lines. a Expression of CLLU1 in GC cell lines and its expression after knockdown by siRNA. b Transwell migration and invasion assays indicating that knockdown of CLLU1 inhibits the migratory and invasive ability of GC cell lines. c Wound healing assay indicating that knockdown of CLLU1 impairs the migratory ability of GC cell lines. The data represent means ± SD. *p-value < 0.05; **p-value < 0.01; ***p-value < 0.001
Discussion
GC is a major cause of global mortality and remains a major health burden in Asian countries including China. It is often diagnosed in advanced stages. Because molecular events in GC progression are promoted by complex genomic interactions, molecules can be grouped into “networks” according to their interactions that contribute to cancer progression; several network-based computational methods have been proposed [53]. The miRNAs interact with different molecules and produce varying outcomes depending on the tumor microenvironment [54,55,56]. In this study, we attempted to delineate the miRNA-mediated molecular mechanism operating in GC progression with an AI-assisted bioinformatics method to integrate transcriptomic, interactomic and network topological feature data. Some major findings are listed as follows.
We first identified the key genes in GC progression by using our previously designed RNs score, which considered both the topological characteristics of genes in the GCP-miR network and the expression profiles of genes in GC samples. Seven miRNAs from the miR-200 and miR-183 families had high RNs scores, thus indicating their important topological roles in the network. These seven miRNAs showed significantly different expression levels during GC progression. Indeed, we validated their significant down-regulation in LSGC in our cohort, and observed robust correlations among them. The results confirmed their important roles in GC progression, thus providing a molecular network perspective corroborating findings from previous reports [38,39,40,41, 44,45,46].
Subsequently, we identified the module for GC progression by selecting the identified miRNAs and their target genes, both of which were regulated by members of the miR-200 and miR-183 families in the GCP-miR network (Fig. 7). The identified module consisted of seven miRNAs, two lncRNAs and five mRNAs. All targets were significantly up-regulated in LSGC, and their expression levels were significantly negatively correlated with the miRNA expression in TCGA datasets and the validation cohort. The combination of miRNAs yielded a highly significant predictive power for patient survival. The model constructed from the six genes in the module could stratify the ESGC and LSGC in independent GC samples. Finally, the contribution of the module to the invasion and migration of GC cells was validated in vitro. Therefore, the miR-200/183 family-mediated module can be potential clinical biomarker for GC.

Schematic diagram of miR-200/183 family-mediated module promoting GC progression
However, there are still many challenges and validation is required for their clinical application. This study has a few limitations. First, the validation cohort was relatively small, which might result in potential performance bias of the model. Second, the follow-up information of the patients in the validation cohort was not sufficient to study the overall survival and to evaluate the prognostic role of the module biomarker. Third, further research is also warranted on the functions of the module in vivo. Large-scale prospective studies are needed to validate the prognosis value of miR-200/183 family-mediated module. In future, we will make efforts to perform large and confirmatory prospective studies to consolidate the findings in present study.
Conclusions
Identifying functional modules in the cancer progression is a challenging task. Our AI-assisted bioinformatics model based on multimodal data revealed a highly modular architecture and indicated that seven miRNAs from the miR-200 and miR-183 families were key regulators in GC progression. The candidate module may serve as an indicator of GC progression and a potential marker to stratify patients with ESGC versus LSGC. Our findings suggest that this module is a “pluripotent module” in gene regulatory network as the two sides of a coin, providing a roadmap to investigate new diagnostic and therapeutic opportunities.
Availability of data and materials
The data and materials in this study are available from the corresponding author on request.
Abbreviations
- AUC:
-
Area under the curve
- DEGs:
-
Differential expressed genes
- ESGC:
-
Early stage gastric cancer
- GC:
-
Gastric cancer
- GO:
-
Gene ontology
- GCP-miR network:
-
GC progression-specific miRNA mediated network
- LSGC:
-
Late stage gastric cancer
- lncRNAs:
-
Long non-coding RNAs
- miRNAs:
-
MicroRNAs
- NT:
-
Non-tumorous adjacent
- S CC :
-
Spearman Correlation Coefficient
- SVM-RFE:
-
Support Vector Machine methods based on a Recursive Feature Elimination
- TCGA:
-
The Cancer Genome Atlas
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Lansdorp-Vogelaar I, Kuipers EJ. Screening for gastric cancer in Western countries. Gut. 2016;65(4):543–4.
Tan P, Yeoh KG. Genetics and molecular pathogenesis of gastric adenocarcinoma. Gastroenterology. 2015;149(5):1153.
Guo JW, Yu WW, Su H, Pang XF. Genomic landscape of gastric cancer: molecular classification and potential targets. Sci China Life Sci. 2017;60(2):126–37.
Ho SWT, Tan P. Dissection of gastric cancer heterogeneity for precision oncology. Cancer Sci. 2019;110(11):3405–14.
Nagaraja AK, Kikuchi O, Bass AJ. Genomics and targeted therapies in gastroesophageal adenocarcinoma. Cancer Discov. 2019;9(12):1656–72.
Wei L, Sun JJ, Zhang NS, Zheng Y, Wang XW, Lv LY, et al. Noncoding RNAs in gastric cancer: implications for drug resistance. Mol Cancer. 2020;19(1):1–17.
Song YX, Sun JX, Zhao JH, Yang YC, Shi JX, Wu ZH, et al. Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat Commun. 2017;8(1):289.
Dong XZ, Zhao ZR, Hu Y, Lu YP, Liu P, Zhang L. LncRNA COL1A1-014 is involved in the progression of gastric cancer via regulating CXCL12-CXCR4 axis. Gastric Cancer. 2020;23(2):260–72.
Yan WY, Qian LJ, Chen JJ, Chen WC, Shen BR. Comparison of prognostic microRNA biomarkers in blood and tissues for gastric cancer. J Cancer. 2016;7(1):95–106.
Lee NK, Lee JH, Ivan C, Ling H, Zhang X, Park CH, et al. MALAT1 promoted invasiveness of gastric adenocarcinoma. BMC Cancer. 2017;17(1):46.
Ueda T, Volinia S, Okumura H, Shimizu M, Taccioli C, Rossi S, et al. Relation between microRNA expression and progression and prognosis of gastric cancer: a microRNA expression analysis. Lancet Oncol. 2010;11(2):136–46.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–8.
Qi X, Lin Y, Chen J, Shen B. Decoding competing endogenous RNA networks for cancer biomarker discovery. Brief Bioinform. 2020;21(2):441–57.
Chen X, Chen Z, Yu S, Nie F, Yan S, Ma P, et al. Long noncoding RNA LINC01234 functions as a competing endogenous RNA to regulate CBFB expression by sponging miR-204-5p in gastric cancer. Clin Cancer Res. 2018;24(8):2002–14.
Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331-3p in gastric cancer. Mol Cancer. 2014;13:92.
Lu MH, Tang B, Zeng S, Hu CJ, Xie R, Wu YY, et al. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016;35(27):3524–34.
Liang Y, Zhang CD, Zhang C, Dai DQ. DLX6-AS1/miR-204-5p/OCT1 positive feedback loop promotes tumor progression and epithelial–mesenchymal transition in gastric cancer. Gastric Cancer. 2020;23(2):212–27.
Cui L, Wang P, Ning D, Shao J, Tan G, Li D, et al. Identification of a novel prognostic signature for gastric cancer based on multiple level integration and global network optimization. Front Cell Dev Biol. 2021;9:631534.
He Q, Tian L, Jiang H, Zhang J, Li Q, Sun Y, et al. Identification of laryngeal cancer prognostic biomarkers using an inflammatory gene-related, competitive endogenous RNA network. Oncotarget. 2017;8(6):9525–34.
Wang Y, Zhou G, Guan T, Wang Y, Xuan C, Ding T, et al. A network-based matrix factorization framework for ceRNA co-modules recognition of cancer genomic data. Brief Bioinform. 2022;23(5).
Yan W, Liu X, Wang Y, Han S, Wang F, Liu X, et al. Identifying drug targets in pancreatic ductal adenocarcinoma through machine learning, analyzing biomolecular networks, and structural modeling. Front Pharmacol. 2020;11:534.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–52.
Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–31.
Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–7.
Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–8.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, et al. clusterProfiler 40: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141.
Lang M, Binder M, Richter J, Schratz P, Pfisterer F, Coors S, et al. mlr3: a modern object-oriented machine learning framework in R. J Open Source Softw. 2019;4(44):1903.
Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21(20):3940–1.
Lévi F, Okyar A, Dulong S, Innominato PF, Clairambault J. Circadian timing in cancer treatments. Annu Rev Pharmacol Toxicol. 2010;50:377–421.
Yang Y, Lv SY, Ye W, Zhang L. Apelin/APJ system and cancer. Clin Chim Acta. 2016;457:112–6.
Masoumi J, Jafarzadeh A, Khorramdelazad H, Abbasloui M, Abdolalizadeh J, Jamali N. Role of Apelin/APJ axis in cancer development and progression. Adv Med Sci. 2020;65(1):202–13.
Xiang T, Yuan C, Guo X, Wang H, Cai Q, Xiang Y, et al. The novel ZEB1-upregulated protein PRTG induced by Helicobacter pylori infection promotes gastric carcinogenesis through the cGMP/PKG signaling pathway. Cell Death Dis. 2021;12(2):150.
Imaoka H, Toiyama Y, Okigami M, Yasuda H, Saigusa S, Ohi M, et al. Circulating microRNA-203 predicts metastases, early recurrence, and poor prognosis in human gastric cancer. Gastric Cancer. 2016;19(3):744–53.
Zuo QF, Zhang R, Li BS, Zhao YL, Zhuang Y, Yu T, et al. MicroRNA-141 inhibits tumor growth and metastasis in gastric cancer by directly targeting transcriptional co-activator with PDZ-binding motif, TAZ. Cell Death Dis. 2015;6(1):e1623.
Tang H, Deng M, Tang Y, Xie X, Guo J, Kong Y, et al. miR-200b and miR-200c as prognostic factors and mediators of gastric cancer cell progression. Clin Cancer Res. 2013;19(20):5602–12.
Jia C, Zhang Y, Xie Y, Ren Y, Zhang H, Zhou Y, et al. miR-200a-3p plays tumor suppressor roles in gastric cancer cells by targeting KLF12. Artif Cells Nanomed Biotechnol. 2019;47(1):3697–703.
Yu L, Wu D, Gao H, Balic JJ, Tsykin A, Han TS, et al. Clinical utility of a STAT3-regulated miRNA-200 family signature with prognostic potential in early gastric cancer. Clin Cancer Res. 2018;24(6):1459–72.
Song F, Yang D, Liu B, Guo Y, Zheng H, Li L, et al. Integrated microRNA network analyses identify a poor-prognosis subtype of gastric cancer characterized by the miR-200 family. Clin Cancer Res. 2014;20(4):878–89.
Kundu ST, Byers LA, Peng DH, Roybal JD, Diao L, Wang J, et al. The miR-200 family and the miR-183~96~182 cluster target Foxf2 to inhibit invasion and metastasis in lung cancers. Oncogene. 2016;35(2):173–86.
Tang X, Zheng D, Hu P, Zeng Z, Li M, Tucker L, et al. Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the β-Catenin/TCF/LEF-1 pathway in gastric cancer cells. Nucleic Acids Res. 2014;42(5):2988–98.
Kong WQ, Bai R, Liu T, Cai CL, Liu M, Li X, et al. MicroRNA-182 targets cAMP-responsive element-binding protein 1 and suppresses cell growth in human gastric adenocarcinoma. FEBS J. 2012;279(7):1252–60.
Li W, Cui X, Qi A, Yan L, Wang T, Li B. miR-183-5p acts as a potential prognostic biomarker in gastric cancer and regulates cell functions by modulating EEF2. Pathol Res Pract. 2019;215(11):152636.
Zhang EB, Han L, Yin DD, Kong R, De W, Chen J. c-Myc-induced, long, noncoding H19 affects cell proliferation and predicts a poor prognosis in patients with gastric cancer. Med Oncol. 2014;31(5):914.
Sun LQ, Li JT, Yan WY, Yao ZD, Wang RQ, Zhou XJ, et al. H19 promotes aerobic glycolysis, proliferation, and immune escape of gastric cancer cells through the microRNA-519d-3p/lactate dehydrogenase A axis. Cancer Sci. 2021;112(6):2245–59.
Yue C, Ren Y, Ge H, Liang C, Xu Y, Li G, et al. Comprehensive analysis of potential prognostic genes for the construction of a competing endogenous RNA regulatory network in hepatocellular carcinoma. Onco Targets Ther. 2019;12:561–76.
Dai YH, Tang YP, Zhu HY, Lv L, Chu Y, Zhou YQ, et al. ZEB2 promotes the metastasis of gastric cancer and modulates epithelial mesenchymal transition of gastric cancer cells. Dig Dis Sci. 2012;57(5):1253–60.
Fardi M, Alivand M, Baradaran B, FarshdoustiHagh M, Solali S. The crucial role of ZEB2: from development to epithelial-to-mesenchymal transition and cancer complexity. J Cell Physiol. 2019;234(9):14783–99.
Liu J, Wang G, Zhao J, Liu X, Zhang K, Gong G, et al. LncRNA H19 promoted the epithelial to mesenchymal transition and metastasis in gastric cancer via activating Wnt/beta-catenin signaling. Dig Dis. 2022;40(4):436–47.
Zeng X, Zhang X, Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief Bioinform. 2016;17(2):193–203.
Esquela-Kerscher A, Slack FJ. Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69.
Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2011;481(7380):190–4.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–22.
Wang Z, Zhao Z, Yang Y, Luo M, Zhang M, Wang X, et al. MiR-99b-5p and miR-203a-3p function as tumor suppressors by targeting IGF-1R in gastric cancer. Sci Rep. 2018;8(1):10119.
Yang F, Bi J, Xue X, Zheng L, Zhi K, Hua J, et al. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012;279(17):3159–65.
Acknowledgements
Not applicable.
Funding
This work was supported by the grants Key Research and Development Program of Jiangsu Province (BE2020656), The Life and Health Special Funds of the Jiangsu Province’s Science and Technology Bureau (BL2014046), The Health Personnel Training Project of Suzhou (GSWS201903), The National Natural Science Foundation of China (32271292, 31872723, 82272439), The Medical and Health Science and Technology Innovation Project of Suzhou (SKY2022010) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
WY: conceptualization, methodology, writing—original draft, writing—review and editing. YC: validation, writing—original draft. GH: methodology, visualization. TS: investigation. XL: software. JL: investigation, resources. LS: resources. FQ: conceptualization, project administration. WC: supervision, funding acquisition, writing—review and editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by Medical Ethics Committee of the First Affiliated Hospital of Soochow University (ethical official number: 2020-126). All patients and volunteers were well informed, and written consent was obtained from the study subjects or the legal surrogates of the patients before enrollment.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Expression of RNAs with high RNs scores in GC cell lines. Figure S2. Correlation plot of expression of genes in the module in TCGA dataset. Each cell contains the corresponding correlation coefficient and p-value, and its color indicates correlation according to the color key. Figure S3. Kaplan–Meier survival curve of patients in high-risk and low-risk groups in test cohort and whole cohort of TCGA. Figure S4. Correlation plot of expression of genes in the module in our newly collected clinical samples. Each cell contains the corresponding correlation coefficient and p-value, and its color indicates correlation according to the color key. Table S1. Demographic information for patients with GC in TCGA dataset and validation cohort. Table S2. Primer sequences used for qRT-PCR. Table S3. Sequences of miRNA mimics. Table S4. Parameters for cox regression model.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, W., Chen, Y., Hu, G. et al. MiR-200/183 family-mediated module biomarker for gastric cancer progression: an AI-assisted bioinformatics method with experimental functional survey. J Transl Med 21, 163 (2023). https://doi.org/10.1186/s12967-023-04010-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04010-z