
Abstract
As a promising alternative platform for cellular immunotherapy, natural killer cells (NK) have recently gained attention as an important type of innate immune regulatory cell. NK cells can rapidly kill multiple adjacent cancer cells through non-MHC-restrictive effects. Although tumors may develop multiple resistance mechanisms to endogenous NK cell attack, in vitro activation, expansion, and genetic modification of NK cells can greatly enhance their anti-tumor activity and give them the ability to overcome drug resistance. Some of these approaches have been translated into clinical applications, and clinical trials of NK cell infusion in patients with hematological malignancies and solid tumors have thus far yielded many encouraging clinical results. CAR-T cells have exhibited great success in treating hematological malignancies, but their drawbacks include high manufacturing costs and potentially fatal toxicity, such as cytokine release syndrome. To overcome these issues, CAR-NK cells were generated through genetic engineering and demonstrated significant clinical responses and lower adverse effects compared with CAR-T cell therapy. In this review, we summarize recent advances in NK cell immunotherapy, focusing on NK cell biology and function, the types of NK cell therapy, and clinical trials and future perspectives on NK cell therapy.
Similar content being viewed by others

Introduction
Globally, cancer is a great threat to human health and one of the leading causes of death [1]. For decades, surgery, chemotherapy, and radiotherapy have been the main methods of treating tumors in patients [2]. Nevertheless, the development of resistance to chemotherapy and/or radiotherapy is associated with a high incidence of cancer recurrence [3,4,5]. Treatment of these cancers can reduce physical strength and impair immune response, which can lead to recurrence as well as metastasis of remaining tumor cells in the body after treatment. Therefore, scholars should urgently uncover novel strategies for eliminating these resistant cancer cells. For many years, immune cells have been demonstrated to be significant targets for cancers. By 1984, immunotherapy was considered the fourth therapy after surgery, chemotherapy, and radiotherapy [6].
Immune cells roughly confer innate and adaptive immunity, which actively avert cancer development through immunosurveillance [7,8,9,10]. Innate immune cells consist of natural killer (NK) cells, dendritic cells (DCs), monocytes, and macrophages [11, 12]. These cells mediate the release of cytokines through an immediate and short-lived immune response. Cytokines then drive the following processes: (i) the direct lysing of cancer cells or the capture of dead cancer cells; (ii) antigen processing performed by phagocytose cancer cells; (iii) the activation of T cells mediated adaptive anti-tumor immune responses; and (iv) the release of cytoplasmic granules containing perforin and granzymes, which directly kill cancer cells and so on [13,14,15,16,17]. T cells and B cells constitute the adaptive immune cells, which are responsible for long-lived, antigen-distinct reactions and effective immune memory [13].
Despite innate and adaptive immune reactions in our body, cancer cells can evade immunosurveillance through several mechanisms [18,19,20,21,22,23,24] (Fig. 1). For instance, they secrete the immunosuppressive cytokines TGF-β and IL-10, which repress the adaptive anti-tumor immune response [25, 26] or polarize tumor-associated macrophages (TAMs) toward an M2 phenotype that has significantly less anti-tumor potential but highly promotes tumor growth and metastasis capacity [27]. Some cancers affect the release of IL-6 [28], IL-10 [29], VEGF [30], or GM-CSF [31] and impair the functions of DC either by inactivating or suppressing maturation. Some tumors induce T regulatory cells to repress tumor-distinct T cell reactions [18]. Some cancers also induce the expression of PD-L1, thereby exhausting T cells through interaction with PD-1 [32]. In normal conditions, a “don’t eat me” signal is expressed by erythroblasts that avoid phagocytosis by macrophages, whereas senescent red blood cells cease to have the ability to express CD47 and are engulfed by macrophages [33,34,35]. Other cancers also express CD47 to avoid phagocytosis by macrophages in the tumor microenvironment (TME) [27, 36, 37]. Furthermore, malignant cells express minimal levels of tumor-associated antigens, dislodge NK cell-activating receptor ligands, or alter the expression of MHC-I as well as costimulatory biomolecules to escape immune reactions [13, 38].

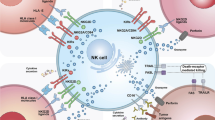
Schematic diagram of tumor-infiltrating immune cells interactions among each other and with cancer cells. Innate immune response cells (macrophages, mast cells and neutrophils) and adaptive immune response cells (lymphocytes) interact with tumor cells through chemokines, adipose cytokines and cytokines
Immunotherapies are developed to trigger either a prospective active or passive anti-tumor reaction against cancers by activating immune responses [39, 40]. To date, several cancer immunotherapies have been used in clinical practice, such as cytokines [10, 41, 42], monoclonal antibodies [43], vaccines [44,45,46], adoptive cell transfer (T [47,48,49,50,51,52], DC [53,54,55], NK [56, 57], and NK-T [58, 59]), and toll-like receptor (TLR) agonists [60,61,62]. In particular, NK cell immunotherapy has remained highly promising for over 30 years. Recently, in the NK cell biology field, remarkable advancements have been made in the comprehension of the function of NK cells as an effective cancer immunotherapy tool. Notably, NK cell therapy has been subjected to clinical phase I/II trials. In this paper, we review recent advancements in NK cell immune therapy, with an emphasis on the biology of NK cells, the function and types of NK cell therapy, and clinical trials as well as future perspectives on NK cell therapy.
Development, classification, and distribution of NK cells
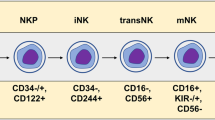
NK cells are unique lymphocyte subpopulations that are larger than T as well as B lymphocytes and contain unique cytoplasmic granules [63]. They were first identified and discovered in 1975 by Herberman et al. [64] and Kiessling et al. [65]. NK cells originate from CD34+ hematopoietic progenitor cells in a continuous process in which common lymphocyte progenitors (CLPs) steadily downregulate CD34 while upregulating CD56. These events prompt the differentiation of NK cells along with their maturation [66,67,68,69]. In humans, a pluripotent progenitor cell close to CLPs has the potential to produce all subpopulations of ILCs. ILC-restricted common ILC precursor (CILCP) arises from CLP and subsequently generates the NK-restricted NKP. NKs are associated with CD122 expression and the loss of CD34 and CD127. Notably, the expression of T-bet and Eomes is required for the further differentiation of functional NK cells [70,71,72]. In mice, CLP cells produce CILCP, which successively generates NK cells as well as helper-like ILCs. Beginning with CILCPs, the development of NK cells comprises at least five phases: NKP → rNKP → CD27+CD11b−NK → CD27+CD11b+NK → CD27−CD11b+NK [73, 74] (Fig. 2).

NK cell development. In mice, common lymphoid progenitor (CLP) produces common ILC precursor, CILCP). CILCP can produce NK cells and helper-like ILCs. There are at least five other stages in NK cell development from CILCP: NK progenitor cells (NKP), refined-NKP (rNKP), CD27+CD11b−NK, CD27+CD11b+NK and CD27−CD11b+ NK. In humans, after CILCP is developed from CLP, NK-restricted NKP will be developed from the latter. NKs is characterized by expressing CD122, losing CD34 and CD127. The expression of T-bet and Eomes is needed for further differentiation into functional NK cells. The expression of CD56 can divide NK cells into two subgroups: CD56dim and CD56bright. The two subsets express activated surface receptors NKp46 and NKp80. CD56+ NK cells can differentiate into CD56− NK cells by expressing CD16, PEN5 and CD57
Since NK cells express many surface markers, unique approaches exist for classifying distinct subsets of NK cells [70, 75, 76]. Based on CD56 expression, they are categorized into two cell subsets, namely CD56 low/dim and CD56bright. In particular, CD56low/dim NK cells are anti-tumor cytotoxic, but both subsets can secrete cytokines [57, 77, 78]. NKp46- and NKp80-activating surface receptors are expressed in both NK cell subpopulations [79,80,81]. CD56bright NK cells specialize into CD56dim NK cells by expressing CD16, and PEN5 [82, 83]. Similarly, distinct subsets categorized based on their expression of CD11b, CD226 (DNAM-1), CD27, and KLRG1 have been studied in mice [84, 85]. However, no equivalence has thus far been established between the subsets of human NK cells and those of mice [86].
NK cells are extensively distributed throughout lymphoid as well as nonlymphoid tissues, consisting of the BM, liver, lungs, lymph nodes (LNs), spleen, liver, and peripheral blood (PB) [87, 88]. Studies have also found NK cells in several other organs, including the uterus, intestines, skin, adipose tissue, bladder, thymus, tonsils, kidneys, pancreas, and brain of mice [88]. In general, the CD56low/dim subpopulation is dominant in human blood, whereas the CD56bright subset is more dominant in lymph nodes [13, 89].
The activation and inhibition of NK cells
Inhibitory and activating signals
Several repressive and activating signal molecules are secreted by NK cells. At steady state, the suppressive receptors prevent the activation of NK cells and their subsequent killing effects. NK cells identify target cells in a non-MHC-restrictive manner, which then recognize the MHC-I molecule expressed on the intended cells. This recognition represses the stimulation of NK cells and prevents them attacking the body [13, 89,90,91,92]. Under stress, target cells diminish MHC-I expression. Consequently, NK cells lose repressive signaling and are activated through the “missing-self recognition” process [13, 89,90,91,92,93,94,95]. Some non-MHC biomolecules, such as Clr-b, CD48, and LLT-1 identified by the repressive receptors NKR-P1B [96], 2B4 [97], and NKR-P1A [98], respectively, also carry out the activation role. Other inhibitory receptors are killer cell immunoglobulin-like receptors (KIRs; e.g., KIR2DL and KIR3DL) [99] and c-type lectin receptors (CD94/NKG2A/B) [100]. Table 1 presents the inhibitory receptors associated with NK cells in a steady state. Accumulating evidence suggests that NK cells express activating receptors with potential recognition of either pathogen-encoded molecules or self-expressed proteins [13]. Under normal conditions, pathogen-coded biomolecules are not secreted by the host, and such recognition is termed “non-self-recognition” [13]. Additionally, disease-infected or transformed cells have been found to upregulate self-produced proteins. This phenomenon has been termed "stress-triggered self-recognition." [13]. Stimulating receptors comprise the cytotoxicity receptors NKp44 [101], NKp46 [79], and NKp30 [102]; the C-type lectin receptors NKG2E/H [103], CD94/NKG2C [104, 105], NKG2F [103], and NKG2D [106]; and the KIRs KIR-3DS and KIR-2DS [99], whereas repressive receptors comprise CD94/NKG2A/B and the KIRs KIR-2DL and KIR-3DL [99, 100]. Table 2 presents the activating receptors associated with NK cells in a steady state. Collectively, NK cells recognize their targets through the identification of numbers of stimulating as well as suppressive signals, whose outcomes depend on the nature of the target cells.
Regulatory cytokines that increase NK cells in the TME
Ongoing research on NK cells relays increasing evidence regarding the critical roles they play in the early regulation of viral infection, in HSC transplantation (HSCT; improved grafting, graft vs. tumor effect, and graft vs. host disease), and in cancer immune surveillance among others [107]. Several regulatory cytokines induce the functions of NK cells as tumor targets [108]. Numerous cytokines (IL-2, IL-21, IL-12, IL-18, and IL-15) and type I interferons can be adopted for in vitro multiplication along with the induction of NK cells prior to adoptive transfer. Notably, individual activating receptors trigger cytokine secretion or insufficient cytotoxicity in naïve NK cells. Exposure to cytokines plays a remarkable role in the preactivation of NK cells. IL-12 enhances signaling from the activating receptors of NK cells [109]. IL-12 combined with IL-15 and IL-18 is especially attractive since it induced a memory-like NK cell population, which grew in immune-incompetent mice inoculated with exogenous IL-2 [110]. Patients transfused with NK cells are frequently given IL-2 to promote in vivo expansion [111]. One study found that repeated administration of IL-2 at low doses was well tolerated; however, no clinical benefit of IL-2 treatment was reported in a corresponding-pairs assessment [112]. The anti-malignant influences of IL-12 and IL-18 as mono-agents are quite minimal [108]. By contrast, IL-21 is potent, particularly in combination with cancer-targeting monoclonal antibodies (mAbs) [113]. The IL-15 cytokine is the one that demonstrates the best promise, with advancements being made in establishing IL-15 signaling mechanisms in NK cells [108]. A phase I clinical trial involving individuals with metastatic cancers found that daily administration of IL-15 induced the proliferation of NK cells and elevated NK cell numbers substantially [114]. Even though no objective response was reported in the trial, some individuals manifested few marker lesions. In another study, IL-15-triggered NK cells were found to induce a clinical reaction in four out of six pediatric individuals with solid refractory cancers [115]. Currently, IL-15 is undergoing a trial with the infusion of NK cells for managing solid cancers as well as hematologic malignancies (NCT01385423 and NCT01875601 clinical trials). The superagonist ALT-803 (IL15N72D:IL15RαSu/IgG1 Fc complex) demonstrates a remarkable biological influence relative to that of native IL-15. This promising inducer of NK cell anti-metastatic roles is undergoing a clinical trial (NCT02099539). Moreover, genetic engineering of the ectopic expression of IL-15 is another potential approach for promoting the role of NK cells. Type I IFN is a proinflammatory cytokine that potentially preactivates NK cells, preparing them for activation by activating receptors [110]. To this end, regulatory cytokines play indispensable roles in the stimulation of NK cells for killing tumors. Numerous cytokines that activate NK cells are presently undergoing clinical or preclinical development.
NK cell-mediated anti-tumor mechanisms
Direct cancer killing
According to previous studies, NK cells have evolved multiple mechanisms for identifying healthy cells from tumor cells. Notably, NK cell killing of tumor cells is non-MHC-I- and non-antibody-dependent [13]. To escape recognition by tumor-invading cytotoxic T cells, the expression of MHC-I on the surface of tumor cells is frequently diminished or lost [13]. However, tumors that have relinquished the self-expression of MHC-I or that harbor “altered-self” stress-inducible proteins cause unregulated ligand expression on cancer cells for NK cell-stimulating receptors [13]. Therefore, because NK cells are stimulated through the initial recognition of certain “stress” or “danger” signals, there is no doubt that tumor cells are ideal NK cell targets [13]. The “self-deletion” model of NK cell recognition of tumor cells was first demonstrated through an analysis of the selective rejection of MHC-I-deficient homologous cancer cells using NK cells [116]. Additionally, the NK cell inhibitory receptors could detect this deficiency in MHC-I expression. Through their activation receptors, NK cells can kill specific MHC-I adequate cancer cells by detecting stress-triggered self-ligands [90]. In a nutshell, direct cytotoxicity mediated by NK cells exerts significant anticancer effects. NK cells kill cancer cells directly through various mechanisms, which are described as follows: (1) NK cells release the killing mediators perforin and granzyme to cause apoptosis of malignant cells, a process that requires direct contact of the NK cell recognition receptor with tumor cells; the CD56 low/dim NK cell subpopulation mainly kills target cells through this mechanism [117]. (2) NK cells trigger apoptosis through the binding of membrane TNF family molecules (FasL, TRIAL, and mTNF) to tumor cell membrane ligands. The process does not require direct contact of the NK cell recognition receptor with the tumor cell, and the CD56 bright NK cell subpopulation kills the malignant cells [57]. (3) NK cells act as a bridge between the anti-tumor antibodies IgG1 and IgG3, whereby Fab specifically recognizes the tumor while the Fc segment binds to the NK cell FcRγ IIIa to trigger antibody-dependent cell-mediated cytotoxicity (ADCC) [118]. (4) NK cells generate numerous cytokines constituting IFN-γ that exert anti-tumor effects through different mechanisms, such as the inhibition of tumor angiogenesis and activation of adaptive immune responses [119] (Fig. 3).

NK cells in tumor immunosurveillance. This figure shows the potential role of NK cells in tumor immunosurveillance. NK cells initially recognize tumor cells through stress or danger signals. Activated NK cells directly kill target tumor cells through at least four mechanisms: cytoplasmic granule release, death receptor-induced apoptosis, effector molecule production, or ADCC. In addition, NK cells interact as regulatory cells with dendritic cells to improve their antigen uptake and presentation and promote the generation of antigen-specific CTL responses. Also, activated NK cells induce CD8+ T cells to become CTLs by producing cytokines such as IFN-γ. Activated NK cells also promote CD4+ T cells to differentiate toward Th1 responses and promote CTL differentiation. Cytokines produced by NK cells may also regulate the production of anti-tumor antibodies by B cells. Abs, antibodies; ADCC, antibody-dependent cellular cytotoxicity; CTL, cytotoxic T lymphocyte; DC, dendritic cell; IFN, interferon; NK, natural killer
Indirect cancer killing
NK cells have immunomodulatory effects, as demonstrated by their prospective effect on the functions of numerous immune cells, consisting of DCs, macrophages, T cells, and B cells [107, 120]. Furthermore, various cytokines, growth factors, and chemokines are produced through the cross-talk of these immune cells [107, 120]. The secretion of IFN-γ by stimulated NK cells triggers the transformation of CD8+ T cells into cytotoxic T lymphocytes (CTLs) and the specialization of CD4+ T cells into Th1 cells. This consequently promotes CTL differentiation [121]. NK cell-originated cytokines may also modulate anti-tumor antibody produced by B cells [122]. Additionally, cancer cells killed by NK cells can deliver cancer antigens to DCs, triggering them to maturation and presentation of antigens [123]. Activated NK cells can offer more antigenic cellular debris to other DCs through the lysis of peripheral DCs that have phagocytosed and then process exogenous antigens [123]. Thus, to promote anti-tumor immunity, activated NK cells regulate DC stimulation and maturation. These DCs potentially promote the production of antigen-distinct CTL responses as they can cross-present cancer-distinct antigens (which originated from NK cell-triggered tumor lysis) and CD8 T cells [124].
NK cell-based therapeutic strategies
Many clinical approaches have been applied to kill cancer cells through NK cell stimulation. Cytokines, autologous as well as allogeneic NK cells, and gene-edited CAR-NK cell immune therapy are presently being pioneered in the field of NK cell treatment (Fig. 4).

NK cell-based therapeutic strategies. A Autologous NK cell transfer:Cytokines IL-2, IL-12,IL-15, IL-18, as well as IL-21 in vitro can stimulate NK cells from patients’ blood and promote in vivo NK-cells proliferation and activation after NK cells are infused into cancer patients. NK cells release perforin, as well as granzyme, to cause apoptosis of tumor cells after they contact and recognize receptor on the tumor cells. B Allogeneic NK cell transfer: NK cells from healthy donors’ peripheral cord blood expand in vitro and are infused into cancer patients. NK cells with donor KIR release perforin, as well as granzyme, to cause apoptosis of tumor cells after they contact and recognize HLA receptor on the tumor cells. If KIR-ligand mismatch, no negative signal exists. C CAR-engineered NK cells: There are four different generations of CARs and they deliver stimulation signals to NK cells. Through genetic engineering modification, CAR can bind to tumor specific antigens are expressed on the surface of NK cells. After transfusion, tumor cells with specific antigens can be specifically recognized and immune responses can be triggered to achieve the purpose of tumor cell clearance
Cytokines
The cytokines IL-18, IL-2, IL-15, IL-21, and IL-12 can attenuate the immunosuppressive microenvironment in tumors by stimulating NK cell. The conflicting character of cytokines either inhibiting or activating NK cell activity disrupts this phenomenon [125]. For instance, the repressive influences of NK cells against malignant cells increase with the injection of IL‐2. However, the use of IL‐2 is minimal because of its adverse toxicity caused by the expansion of modulatory T cell populations [126]. A recent report documented that IL‐2 diphtheria toxic fusion promotes in vivo NK cell growth. Simultaneously, it cleared Treg cells. Moreover, TIM-3, a repressive checkpoint receptor, was elevated in human NK cells using IL-2, IL-21, and IL-15 alone or in combination in vitro. A recombinant fusion protein that comprised diphtheria toxin along with truncated IL-2 (IL-2DT) was designed to deplete Tregs [127]. The results demonstrated enhanced in vivo proliferation of haploid NK cells, leading to acute myeloid leukemia (AML) remission [127].
IL-15 has attracted much attention in NK cell treatment because of its beneficial properties, where regulatory T cells do not amplify but remarkably activate NK cells [128]. The cross-talk of IL-15 and its receptor generates a distinct complex that reduces the affinity of IL-2Rβ to NK cells and inhibits the activation influence of IL-2 on Treg cell expansion [129]. An IL-15 superagonist complex, namely ALT-803, was recently revealed to potentially elevate NK cells and enhance their cytotoxicity. However, in vivo as well as in vitro experiments have documented that constant inoculation of NK cells with IL-15 depletes more NK cells compared with intermittent exposure. However, ALT-803 has still yielded clinical responses in both solid tumors and hematological malignancies [130, 131]. An ALT-803 regimen given to patients in combination with PD-1 mAb reverted, with the refractory disease presenting support for the anti-tumor influence for a novel group of agents in NSCLC [130]. In a phase I first-in-human multi-center trial of ALT-803, it was found to be a safe and well-tolerated agent in study subjects who experienced relapse > 60 days post allo-HCT. It also remarkably elevated NK as well as CD8+ T cells along with function. This immune-activation IL-15 super-agonist should be further studied to promote anti-cancer immunity alone as well as in combination with other immunotherapies [131].
Furthermore, IL-12 can coordinate with IL-15, IL-2, and/or IL-18 to activate NK cells [132]. Prestimulating NK cells with IL-12 and activating them with IL-15 and IL-18 results in memory-like NK cells with extended survival and enhanced function. IL-12 activation requires the stimulation of STAT4, whereas the synergistic effects of IL-15, IL-18, and IL-12 do not [15]. Diminished cytotoxicity of NK cells has been documented in IL-18-deficient mice; however, IL-18 alone cannot sufficiently induce IFN-γ production. IL-12 alone or combined with IL-15 can be used to distinguish CD34+ HSC from NK cells in vitro for treatment purposes [15].
IL-21 and IL-2 synergize, causing the elevation of NKG2A, perforin, CD25, granzyme B, CD69, and CD86, and it is related to the high cytotoxicity of human NK cells [85]. IL-21 also reverses NK depletion to promote cancer regression in mice [133]. IL-21-activated NK cells modulate the amplification of intracellular pathogens such as hepatitis C virus along with Mycobacterium tuberculosis [15]. In a recent study, human NK cells were elevated through IL-21 along with autologous feeder cells to generate CAR-NK cells [134]. Even though research utilizing NK-92 cell lines has documented potent results, optimizing the in vitro elevation of functional NK cells in the donor or patient is essential for the generation of safe and effective CAR-NK cells.
Despite numerous clinical and translational experiments conducted to uncover more potent cytokines to trigger NK cells, research related to NK cells along with cytokines remains immature. The fundamental necessity of understanding the impact of various chemokines on the design process of NK cells warrants further exploration.
Allogeneic and autologous NK cell treatment
Allogeneic or autologous NK cells originate from the peripheral blood [57]. They can also originate from umbilical cord blood or bone marrow as well as stimulated pluripotent or human embryonic stem cells, which are currently being explored as potential origins of NK cells with clinical significance [57]. NK cell progenitors or mature NK cells can be infused with other cells as part of the HSCT or alone following the pre-enrichment process [57].
Inhibitory receptors on donor allogeneic NK cells (e.g., KIR) do not recognize human leukocyte antigen (HLA) class I on recipient cells in case of a class mismatch. Therefore, the donor NK cells are relieved of their repressive receptor-triggered inhibition. In this case, cancer cells lack the suitable class I MHC ligands to engage the repressive KIR, and thus, they are removed by allo-reactive NK cells [13]. Numerous reports have revealed that allogeneic NK cells potentially trigger remission or suppress relapse in individuals with hematological malignancies, including AML and multiple myeloma (MM) [57, 125]. This is due to the in vitro expansion and activation of HSCT or peripatetic NK cell treatment [57, 125]. In a clinical trial of haploidentical NK cells for AML, the authors reported the induction of complete remission in dismal prognosis or elderly individuals and a 100% event-free survival rate at 18 months in a pediatric cohort [135]. Allo-reactive NK cells have the capacity to avert graft-versus-host disease (GVHD) through the elimination of host antigen-presenting cells [136]. Nevertheless, this protective influence has been challenged by a study in which allogeneic HSCT followed by the infusion of donor NK cells stimulated by IL-15 along with CD137L (ligand for co-stimulatory receptor CD137) exacerbated acute GVHD by promoting the underlying T cell allogeneic response [137]. Differences in the origin as well as development of the infused NK cells may explain these contradictory findings. Indeed, inadequate T cell depletion in KIR-mismatched grafts may cause severe GVHD and offset the clinical benefit of allogeneic NK cells [138]. Collectively, further studies are warranted to explore the discrete conditions and types of cancers that would benefit from allogeneic NK cell infusion.
In individuals with hematological cancer undergoing autologous HSCT, the number of blood NK cells recovers early after transplantation. Several NK cells were linked to positive results in these patients, which illustrated the anti-cancer ability of NK cells [139, 140]. Other reports have indicated that autologous NK cell expansion and infusion in individuals with metastatic melanoma, advanced gastrointestinal cancer, or renal cell carcinoma do not translate into a clinical response [141, 142]. Notably, NK cells that persist in the circulation from a secondary infusion cannot kill tumor cells unless they are restimulated in vitro. This finding highlights the need for combinatorial approaches to fully exploit the ability of autologous NK cells [108].
In conclusion, both allogeneic and autologous NK cell therapy have demonstrated clinical efficacy either alone or in combination with conventional therapies. Table 3 summarizes the findings of clinical trials where NK cells have been infused into cancer patients. There is ongoing research on autologous NK cells and some 50 clinical trials are presently underway. Furthermore, studies on the efficacy of allogeneic NK cell transfer are presently underway and exhibiting promising clinical potential (e.g., NCT00720785, NCT03068819, NCT02782546, NCT01898793, NCT03081780, NCT03319459, NCT03213964, NCT03019640, NCT01729091, NCT01787474, NCT02809092, NCT02271711, and NCT03579927). Adoptive cell treatment with allogeneic NK cells has similar disadvantages to autologous NK cells, such as timely ex vivo expansion as well as the activation of clinical-grade NK cells and in vivo persistence after infusion. In the future, scientists should invest more effort in this area to refine NK cell treatments.
CAR-NK cell therapy
Background
The recent approval by the U.S. Food and Drug Administration (FDA) of CAR-T cell therapy targeting CD19 is a critical breakthrough in the design of genetically modified cell treatments for cancer. This has also stimulated much attention in the preparation of CAR-NK cells for tumor immune therapy [143]. In clinical research, the transfusion of unmodified allogeneic NK cells has demonstrated slight efficacy, specifically in AML. However, their short lifespan (2–3 weeks after transfusion) has somewhat limited their success. Allogeneic NK cells, despite their ability to exert allo-reactivity, do not cause acute GVHD. Hence, relative to CAR-T cells, allogeneic NK cells are potential therapeutic agents that require no other genetic modifications. Thus, the novel biological properties of NK cells render them a more attractive origin of genetically modified immune cell-based immune therapy.
Study overview
Progress in research has contributed to a gradual improvement in the design of CARs. Several generations of CARs now exist: 1st-generation CARs comprise a basic structure with one signaling region [144]; 2nd-generation CARs harbor an extra co-stimulatory domain, such as CD28 or 4-1BB [145, 146]; 3rd-generation CARs have multiple co-stimulatory domains [147, 148]; and 4th-generation CARs have multiple co-stimulatory domains and cytokines signals (Fig. 4C). Many co-stimulatory domains have been explored, consisting of immune globulin superfamily members (CD28 and ICOS), TNF receptor superfamily members (4-1BB, CD27, OX40, and CD40), and others (e.g., CD40L and TLR) [149]. At the very beginning, the CAR constructs used for CAR-NK cells were optimized for T-cell signaling and function. Although certain signaling/co-stimulatory domains used in CAR design (e.g., CD3ζ and 4-1BB) are shared between T and NK cells, the role of other co-stimulatory molecules (e.g., CD28) in NK cells is elusive [150]. To date, numerous reports have been published on the greater specificity for NK-cell signaling, DAP10, DAP12, and 2B4 [151, 152]. DAP10 and DAP12 are activation motifs harboring immunoreceptor tyrosine-based activation and deliver stimulation signals to NK cells. DAP10 signals the activating receptor NKG2D, whereas DAP12 mediates signaling via NKG2C, NKp44, and the activating killer immunoglobulin receptor (KIR). Another activating receptor is 2B4, which belongs of signaling lymphocyte activating molecule (SLAM) family; when bound to its natural ligand CD48, 2B4 recruits articulator molecules such as SLAM-associated protein (SAP) (ITSM) to mediate signal transduction via its immunoreceptor tyrosine-based switch motif [153]. More recently, 4th-generation CARs were investigated [154]. These vectors incorporate transgenic “payloads” designed to promote the growth, persistence, and anti-tumor activity of CAR-engineered NK cells. To date, results have been published that justify the superior activity of these latest CAR design revisions, and the expansion the function of CAR-modified NK cells within the existing approach to cancer immunotherapy is promising [155, 156].
NK cell sources
In addition, scientists have explored different cell origins for the production of CAR-expressing NK cells. These cell sources include peripheral blood [157,158,159], umbilical cord blood [134, 156, 160,161,162,163], stem cells (including hematopoietic and stimulated pluripotent stem cells) [66, 164, 165], and NK cell lines [162, 166, 167]. Notably, each source has its unique advantages and disadvantages (Table 4).
Clinical efficacy of CAR-NK in cancers
Currently, CAR-NK cell therapy has yielded preclinical anti-malignant activity both in hematological cancers and solid tumors, including leukemia, lymphoma, myeloma, ovarian cancer, and glioblastoma [168]. In some clinical trials, this therapy has exhibited efficacy.
AML
In 2018, Jianhua Yu’s group reported a phase I first-in-human clinical trial involving CD33-CAR NK cells for individuals with relapsed as well as refractory AML [169]. In their study of CD33-CAR-NK cells, they enrolled three patients and tested their safety, but no adverse events were reported. Notably, the author revealed that CAR NK-92 cells could be generated at a much lower cost than CAR-T cells, and believed that even after optimization they would be broadly accessible for cancer management [169].
Lymphoma
In 2020, Rezvani et al. demonstrated that allogeneic CAR-NK cells originated from cord blood were beneficial for high-risk B cell lymphoma and CD19+ CLL [156]. The use of CAR-NK cells exhibited no association with the onset of cytokine release syndrome, neurotoxicity, or GVHD. Furthermore, the quantities of inflammatory cytokines, comprising IL-6, did not exceed baseline levels and the maximum tolerated dose was not attained. Eight (73%) of the 11 patients treated responded, of which seven patients (four with lymphoma as well as three with CLL) were in complete remission, whereas one individual had Richter transformation in partial remission but persistent CLL. Their reactions were prompt and were reported for 30 days post-infusion for all dose levels. Infused CAR-NK cells were expanded and then persisted at low quantities for at least one year [156]. The company Fate Therapeutics recently announced clinical data on FT596, an allogeneic iPSC-derived CAR-NK cell. FT596 monotherapy demonstrated durable tumor clearance and extended in vivo survival, demonstrating the promise of iPSC-derived NK cell therapy as a novel cancer immunotherapy for development [170]. In addition, FT596 exhibited enhanced killing of CD20+ lymphoma cells in vivo when combined with rituximab compared with rituximab alone. This is a preliminary indication that the combination of antibody drugs and NK cell therapy can have a synergistic effect and is another new direction for CAR-NK cell development [170].
MM
MM is a malignancy caused by genetic mutations that occur during the differentiation of B lymphocytes into plasma cells. Common tumor antigens in MM cell lines include cell surface glycoprotein CD2 subset 1 (CS1) [171], CD138 [172], and BCMA [173]. Relatively little research has been conducted on CAR-NK cell products for the treatment of MM. Most of the CAR-NK cell products currently used to treat MM use the NK92 cell line, and the tumor antigens used are mostly CS1 [174] and CD138 [175]. CS1 is a highly expressed protein on the surface of MM cells and is mostly involved in MM cell adhesion and growth [174]. CD138 is involved in the adhesion, growth, and maturation of MM cells and is a major diagnostic marker for MM. In vitro studies have demonstrated that CS1-CAR-NK92 cells and CD138-IFNα-CAR-NK92 cells, designed using CS1 and CD138 as targets, respectively, successfully inhibited MM cell growth and prolonged the survival of myeloma mice [174].
Solid tumors
CAR-T cells are subject to PD-1/PD-L1-mediated immunosuppression in the fight against solid tumors. NK cells with very low surface PD-1 expression and relatively little immunosuppression by the tumor microenvironment may be good candidates for fighting solid tumors. In solid tumor research, CAR-NK cells are mostly targeted at metastatic solid malignancies expressing tumor-associated antigens such as HER2, PSMA, mesothelin, ROBO1, or MUC1, including prostate cancer, ovarian cancer, breast cancer, pancreatic cancer and non-small-cell lung cancer. In addition, PD-L1 is upregulated in the TME and immunosuppressive cells in several cancer types. A new NK-92 cell line was designed to target PD-L1, ER-retained IL-2, and a high-affinity CD16 CAR called PD-L1-targeted haNK (t-haNK). Exciting preclinical data suggest that these cells have specific anti-tumor effects against 15 tumor cell lines in vitro and strong anti-tumor effects against triple negative breast, bladder, and lung cancers in vivo. The QUITL3.064 phase I clinical trial (NCT04050709) is currently underway with PD-L1 t-haNK in combination with other agents for assessing safety and efficacy in patients with locally advanced or metastatic pancreatic cancer (NCT0439099).
Given the safety and efficacy of CAR-NK cell therapy, numerous clinical trials on hematologic cancers and solid tumors are currently underway. Table 5 presents CAR-NK cell-based therapy clinical trials for hematologic malignancies and solid tumors.
Advantages and challenges of CAR-NK cells
NK cells have some powerful therapeutic advantages. (1) They are more widely available, can be derived from allogeneic cells, and do not need to rely on the patient’s own specific immune cells. (2) NK cells do not require MHC molecules for antigen presentation or antigen activation and can target a wide range of pathogenic antigens with greater cytotoxicity. (3) NK cells do not secrete the major cytokines that trigger CRS and can greatly mitigate the risk of adverse effects. (4) Allogeneic NK cells also do not cause graft-versus-host reactions. However, CAR-NK cell therapy still entails some difficulties and dilemmas. (1) In vitro expansion of NK cells is the first hurdle for CAR-NK cell immunotherapy. The number of NK cells from a single donor is not sufficient for therapy, which makes the expansion and activation of NK cells critical. This production process usually takes two to three weeks to culture NK cells. Therefore, obtaining enough NK cells remains a challenge. (2) Selecting the appropriate method for transducing CARs into NK cells is the key to CAR-NK cell immunotherapy [57]. Thus far, both viral and nonviral vectors have been used to transform CARs. Although retroviral vectors have high transfection efficiency, they may cause insertional mutations, carcinogenesis, and other adverse effects. By contrast, lentiviral vectors, despite exhibiting a low incidence of insertional mutations, have transfection efficiencies as low as 20% for peripheral blood NK cells [57]. The transfection of CAR-NK cells with mRNA is also considered a safe and practical transfection method. A study demonstrated that in xenograft tumor models, mRNA-transfected NK cells exhibit significant cytotoxicity after 24 h of electroporation, with receptor expression levels exceeding 80% [57]. Furthermore, it has recently been demonstrated that the transfection of mRNA can effectively avoid “targeted non-tumor” toxicity, which is a critical factor limiting the clinical application of CAR-modified immunotherapy. However, the anti-tumor effect of CAR-NK cells transfected with mRNA through electroporation is transient as the expression level of CARs does not exceed 3 days. (3) Another challenge for CAR-NK therapy is the impact of the TME. (4) The primary barrier to reliable preclinical evaluation of solid cancer CAR-NK therapy is the lack of clinically relevant models of animals that encapsulate the complexity of interactions in the TME [168]. Most studies have relied on human tumor cell lines derived from immunocompromised NOD scid γ null (NSG) mice, which lack an effective immune system [168]. While existing NSG models can be applied to rapidly assess the function and persistence of CAR effector, they cannot establish a clinically relevant TME or accurately estimate CAR-NK cell function and persistence. Furthermore, such models can explore the cross-talk between different immune cells and tumors.
NK cell-based immune checkpoint
KIRs
The KIR family (also known as CD158) is a diverse and polymorphic group of NK cell receptor subtypes containing both inhibitory and activating KIRs, each of which recognizes a specific HLA class I congener (HLA-A, -B, or -C) as a ligand [176]. IPH2101 and lirilumab (IPH2102/BMS-986015) are IgG4 mAbs against the KIR2DL1/2/3 NK cell inhibitory receptor, while IPH4102 is a humanized anti-KIR3DL2 IgG1 mAb. The IPH2101 blockade of KIR improves survival in vivo, and preclinical evidence also suggests its effectiveness in AML cells [177]. A phase I study of IPH2101 in elderly patients with AML in first complete remission demonstrated that the overall and relapse-free survival compared favorably with reports in comparable patient populations [177]. Phase II trial results of lirilumab indicated poor efficacy of monotherapy for MM [178]. However, lirilumab in combination with full-dose azacitidine was well-tolerated in patients with heavily pretreated/relapsed AML [179]. A recent study reported the efficacy and tolerability of lirilumab as a single agent or in combination with azacitidine in patients with myelodysplastic syndrome (MDS) [180]. IPH4102, also known as lacutamab, was well-tolerated in a phase I clinical evaluation in relapsed/refractory cutaneous T-cell lymphoma, with the most common adverse effects including edema, fatigue, and lymphopenia [181]. The clinical activity was also encouraging, with ORR achieved in 16 of 44 patients (36%) [181]. Patients with relapsed/refractory cutaneous T-cell lymphoma with Sézary syndrome exhibited a better clinical response (43%) [181].
Several clinical trials are still ongoing in cisplatin-ineligible muscle-invasive bladder cancer (NCT03532451), relapsed or refractory tumors (NCT02813135), locoregionally recurrent squamous cell carcinoma of the head and neck (NCT03341936), and advanced T cell lymphoma (NCT03902184).
NGK2A and CD94
NKG2A (also known as CD159) and CD94 are heterodimeric inhibitory receptors of the C-type lectin family, which recognize the nonclassical MHC-I molecule HLA-E as a ligand [182]. The results of in vitro and in vivo studies have suggested that humanized anti-NKG2A/CD94 (IPH2201, monalizumab) antibodies are safe and effective for use in hematological malignancies and solid tumors [183]. In in-vitro trials, monalizumab improved NK cell dysfunction in chronic lymphocytic leukemia [184]. Monalizumab was well-tolerated as a single agent for the treatment of gynecologic malignancies (up to 10 mg/kg administered intravenously or by SC) [185]. A preliminary evaluation of the safety and efficacy of monalizumab in combination with cetuximab in previously treated, recurrent, and/or metastatic squamous cell carcinoma (SCC) of the head and neck revealed an ORR of 27.5%, a median PFS of 5 months, and a median overall survival (OS) of 10 months with the combination [186]. This is an encouraging result when compared with the historical record of cetuximab efficacy alone in previous studies (ORR 12.6%, PFS 2.3 m, OS 5.6 m). The combination therapy had similar adverse effects to cetuximab alone. Overall, blocking NKG2A/CD94 represents an exciting therapeutic approach; in particular, its combination with other immuno-oncology therapeutics is the way forward and warrants further exploration. In addition, clinical trials are currently evaluating the efficacy of monalizumab in combination with a variety of other targeted agents for the treatment of multiple tumors (NCT02643550, NCT02671435, NCT03822351, NCT03833440, and NCT03088059).
TIGIT and CD96
TIGIT is an immunosuppressive receptor expressed on NK and T cells [187]. CD96 belongs to the same immunoglobulin superfamily as TIGIT and has similar inhibitory effects, but it binds with lower affinity to CD155, its ligand. CD155 (mainly) and CD112 act as ligands for TIGIT and CD96 binding to suppress T-cell- and NK-cell-mediated immunity [188]. It is hardly expressed in normal human tissues, but many tumor cell lines and primary malignancies highly express CD155 [189,190,191]. Among the functions of CD155, immunomodulation through its interaction with the inhibitory receptors TIGIT and CD96 and the activating receptor CD226 is of particular interest. Various cancers exhibit upregulation of CD155 and corresponding upregulation of NK and T cell expression of TIGIT and CD96 to evade anti-tumor immunity through inducing T cell or NK cell suppression [189,190,191].
Animal experiments revealed that TIGIT intrinsic expression inhibits NK and CD8+ T cell function, thereby aiding colorectal cancer cell growth in vivo [192]. TIGIT is associated with NK cell depletion in tumor-bearing mice and patients with colon cancer, and this depletion is restored by its blockade, thereby stimulating strong anti-tumor immunity [187]. The presence of NK cells is crucial for the therapeutic efficacy of both checkpoints of TIGIT and/or PD-L1 blockade or dual blockade, as NK cell deficiency is associated with a lower frequency of IFN-γ or TNF-secreting TIL (CD8+) and a higher frequency of PD-1-expressing TIL (CD8+) [187]. NK cells account for 25–50% of hepatic lymphocytes, which indicates their importance for liver immunity. Furthermore, the survival and prognosis of patients with hepatocellular carcinoma (HCC) are positively correlated with the number of NK cells in blood and tumor tissue [193, 194]. Cancer progression in HCC patients is associated with dysfunctional NK cell infiltration, mainly in the CD11b−CD27− subpopulation [193, 194]. Sun et al. identified depleted tumor-infiltrating CD96+ NK cells and found that their expression was correlated with poor clinical outcomes in HCC patients [195]. NK cell depletion was reversed when CD96–CD155 interactions or TGF-β1 were blocked [195].
In recent years, the combination of checkpoint inhibitors has received increasing attention for achieving synergistic effects. Enhancing CD8+ T-cell activation has been reported to improve the survival rate of hormonal mice with dual targeting of PD-1 and TIGIT. Dixon et al. reported that dual blockade of TIGIT and PD-1 produced a synergistic anti-tumor effect leading to complete tumor regression in the MC38 colon cancer model [196]. In melanoma patients, dual blockade of TIGIT and PD-1 synergistically increased tumor infiltration and tumor antigen-specific CD8+ T cell proliferation, degranulation, and cytokine secretion, indicating the potential for dual blockade [197]. Hong et al. suggested that PD-1 and TIGIT could also be potential targets for the treatment of RCC [198]. In patients with GBM, this dual blocker also improved anti-tumor immunity and survival [199]. While these studies reflect the efficacy of double checkpoint blockade in various cancers by exploring the role of T cells, some studies have also suggested that the efficacy of the double checkpoint is also dependent on NK cells. Anti-TIGIT plus anti-PD-L1 blocker prevented NK cell depletion in hormonal mice and colon cancer patients [187]. On the other hand, anti-CD96 combined with adriamycin chemotherapy, anti-CTLA-4, or anti-PD-1 exhibited more effective inhibition of tumor metastasis in three different tumor models [200]. Bladder cancer patients with failing NK cells exhibited upregulation of TIM-3 and TIGIT both in the periphery and in the tumor [201]. Indeed, the roles of TIGIT and CD96 in NK cell depletion in various cancers are still under investigation, and further revelations are required to determine their potential as monotherapy or in combination with other checkpoints. Clinical trials are ongoing regarding TIGIT blockade as a monotherapy or in combination with other therapies for treating various hematological malignancies and solid tumors (e.g., NCT04818619, NCT04150965, NCT04656535, NCT04500678, NCT04732494, NCT0435383, NCT04693234, NCT04570839, NCT04354246, NCT04047862, NCT04952597, NCT04457778, NCT04543617, NCT03563716, and NCT04746924). Overall, TIGIT monoclonal antibody is in the early stage of clinical trials, and the results are subject to further observation.
Future perspectives
Many strategies have been developed for exploiting the anti-tumor properties of NK cells. Researchers are testing IL-2 and IL-15, two cytokines that promote NK cell activity, but exacerbating the immune response poses a safety concern. NK cells from healthy donors are stimulated in vitro by IL-2 and IL-15 and then transfused back into the blood of cancer patients. This strategy can take advantage of the mismatch between donor KIR and patient HLA to reduce the suppression of NK cell function and promote their anti-tumor activity. Since the introduction of CAR engineering technologies, the paradigm guiding the field of cell therapy has shifted. As the first immune effector cells engineered by CAR with promising results in the clinic, CAR-T cells have set the pace for the future design of CAR-based immunotherapy. NK cells characterize a specialized population of immune effector cells with a rapid response and powerful anti-tumor capacity. Despite their success, CAR-T cells still have significant drawbacks that have fueled research into other immune effector cells as an alternate approach for CAR engineering. In the past decade, clinical research on hematological cancers has pioneered the concept of peripatetic NK cell immunotherapy. Evidence suggests that NK cells have high safety and efficacy. Some clinical efficacy has also been demonstrated for allogeneic as well as autologous NK cell therapy, either alone or in combination with conventional therapies. Crucially, tumor antigen-expressing CAR-NK cell therapy increases anti-tumor activities. Thus, NK cell transfer presents an effective method of fighting cancer. In addition, antibodies that directly target NK cell inhibitory receptors, such as those targeting KIRs, NKG2A, and TIGIT, can enhance NK cell responses and thus kill tumor cells, and some are currently being validated in clinical trials. Therefore, based on the pan-specific recognition property of NK cells, NK cell-based multiple immune combination therapy is a strategy for further improving anti-tumor efficacy and deserves further exploration.
Availability of data and materials
Not applicable.
Abbreviations
- NK cells:
-
Natural killer cells
- SCC:
-
Squamous cell carcinoma
- OS:
-
Overall survival time
- HCC:
-
Hepatocellular carcinoma
- MDS:
-
Myelodysplastic syndrome
- SLAM:
-
Signaling lymphocyte activating molecule
- DCs:
-
Dendritic cells
- TME:
-
Tumor microenvironment
- TLR:
-
Toll-like receptor
- mAbs:
-
Monoclonal antibodies
- CLPs:
-
Common lymphocyte progenitors
- LNs:
-
Lymph nodes
- PB:
-
Peripheral blood
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Canfell K, Kim JJ, Brisson M, et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):591–603.
Hutchinson MKND, Mierzwa M, D’Silva NJ. Radiation resistance in head and neck squamous cell carcinoma: dire need for an appropriate sensitizer. Oncogene. 2020;39(18):3638–49.
Garcia-Mayea Y, Mir C, Masson F, Paciucci R, Leonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2020;60:166–80.
Min HY, Lee HY. Mechanisms of resistance to chemotherapy in non-small cell lung cancer. Arch Pharm Res. 2021;44(2):146–64.
Fujitani T, Takahara T, Hattori H, Imajo Y, Ogasawara H. Radiochemotherapy for non-Hodgkin’s lymphoma in palatine tonsil. Cancer. 1984;54(7):1288–92.
Laughney AM, Hu J, Campbell NR, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med. 2020;26(2):259–69.
Póvoa V, Rebelo de Almeida C, Maia-Gil M, et al. Innate immune evasion revealed in a colorectal zebrafish xenograft model. Nat Commun. 2021;12(1):1–15.
Möckl L. The emerging role of the mammalian glycocalyx in functional membrane organization and immune system regulation. Front Cell Dev Biol. 2020;8:253.
Chulpanova DS, Kitaeva KV, Green AR, Rizvanov AA, Solovyeva VV. Molecular aspects and future perspectives of cytokine-based anti-cancer immunotherapy. Front Cell Dev Biol. 2020;8:402.
Panda A, Arjona A, Sapey E, et al. Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol. 2009;30(7):325–33.
Germic N, Frangez Z, Yousefi S, Simon HU. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019;26(4):715–27.
Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10(3):230–52.
Borghaei H, Smith MR, Campbell KS. Immunotherapy of cancer. Eur J Pharmacol. 2009;625(1–3):41–54.
Kim N, Lee HHHJ, Lee HHHJ, et al. Natural killer cells as a promising therapeutic target for cancer immunotherapy. Arch Pharm Res. 2019;42(7):591–606.
Rothlin CV, Ghosh S. Lifting the innate immune barriers to antitumor immunity. J Immunother Cancer. 2020;8(1):e000695.
Liang W, Ferrara N. Iron metabolism in the tumor microenvironment: contributions of innate immune cells. Front Immunol. 2021;11:626812.
Yi M, Xu L, Jiao Y, et al. The role of cancer-derived microRNAs in cancer immune escape. J Hematol Oncol. 2020;13(1):25.
Crispen PL, Kusmartsev S. Mechanisms of immune evasion in bladder cancer. Cancer Immunol Immunother. 2020;69(1):3–14.
Vago L, Gojo I. Immune escape and immunotherapy of acute myeloid leukemia. J Clin Invest. 2020;130(4):1552–64.
Bruschini S, Ciliberto G, Mancini R. The emerging role of cancer cell plasticity and cell-cycle quiescence in immune escape. Cell Death Dis. 2020;11(6):1–3.
Ge Z, Wu S, Zhang Z, Ding S. Mechanism of tumor cells escaping from immune surveillance of NK cells. Immunopharmacol Immunotoxicol. 2020;42(3):187–98.
Wu Y, Biswas D, Swanton C. Impact of cancer evolution on immune surveillance and checkpoint inhibitor response. Semin Cancer Biol. 2021. https://doi.org/10.1016/j.semcancer.2021.02.013.
Dersh D, Hollý J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol. 2021;21(2):116–28.
Sapski S, Beha N, Kontermann RE, Müller D. Influence of antigen density and immunosuppressive factors on tumor-targeted costimulation with antibody-fusion proteins and bispecific antibody-mediated T cell response. Cancer Immunol Immunother. 2020;69:2291–303.
Close HJ, Stead LF, Nsengimana J, et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin Exp Immunol. 2020;200(1):33–44.
Eladl E, Tremblay-Lemay R, Rastgoo N, et al. Role of CD47 in hematological malignancies. J Hematol Oncol. 2020;13(1):96.
Ratta M, Fagnoni F, Curti A, et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood. 2002;100(1):230–7.
Demangel C, Bertolino P, Britton WJ. Autocrine IL-10 impairs dendritic cell (DC)-derived immune responses to mycobacterial infection by suppressing DC trafficking to draining lymph nodes and local IL-12 production. Eur J Immunol. 2002;32(4):994–1002.
Mimura K, Kono K, Takahashi A, Kawaguchi Y, Fujii H. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother. 2007;56(6):761–70.
Carlier J, Martin H, Mariamé B, et al. Paracrine inhibition of GM-CSF signaling by human cytomegalovirus in monocytes differentiating to dendritic cells. Blood. 2011;118(26):6783–92.
Peng DH, Rodriguez BL, Diao L, et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8+ T cell exhaustion. Nat Commun. 2020;11(1):1–18.
Li W, Wang Y, Zhao H, et al. Identification and transcriptome analysis of erythroblastic island macrophages. Blood. 2019;134(5):480–91.
Li W, Wang Y, Chen L, An X. Erythroblast island macrophages: recent discovery and future perspectives. Blood Sci. 2019;1(1):61–4.
Li W, Guo R, Song Y, Jiang Z. Erythroblastic island macrophages shape normal erythropoiesis and drive associated disorders in erythroid hematopoietic diseases. Front Cell Dev Biol. 2021;8:1858.
Yang H, Yan M, Li W, Xu L. SIRPα and PD1 expression on tumor-associated macrophage predict prognosis of intrahepatic cholangiocarcinoma. J Transl Med. 2022;20(1):140.
Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity. 2020;52(5):742–52.
Sayitoglu EC, Georgoudaki AM, Chrobok M, et al. Boosting natural killer cell-mediated targeting of sarcoma through DNAM-1 and NKG2D. Front Immunol. 2020;11:40.
Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32(9):456–65.
Martin JD, Cabral H, Stylianopoulos T, Jain RK. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat Rev Clin Oncol. 2020;17(4):251–66.
Ishihara J, Ishihara A, Sasaki K, et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci TranslMed. 2019;11(487):eaau3259.
Berraondo P, Sanmamed MF, Ochoa MC, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6–15.
Zahavi D, Weiner L. Monoclonal antibodies in cancer therapy. Antibodies. 2020;9(3):34.
Sahin U, Türeci Ö. Personalized vaccines for cancer immunotherapy. Science. 2018;359(6382):1355–60.
Jahanafrooz Z, Baradaran B, Mosafer J, et al. Comparison of DNA and mRNA vaccines against cancer. Drug Discov Today. 2020;25(3):552–60.
Liu S, Jiang Q, Zhao X, et al. A DNA nanodevice-based vaccine for cancer immunotherapy. Nat Mater. 2021;20(3):421–30.
Zhao J, Song Y, Liu D. Clinical trials of dual-target CAR T cells, donor-derived CAR T cells, and universal CAR T cells for acute lymphoid leukemia. J Hematol Oncol. 2019;12(1):1–11.
Shah NN, Ahn KW, Litovich C, et al. Is autologous transplant in relapsed DLBCL patients achieving only a PET+ PR appropriate in the CAR T-cell era? Blood. 2021;137(10):1416–23.
Huang R, Li X, He Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86.
Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61.
Amor C, Feucht J, Leibold J, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–32.
Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–67.
Castenmiller C, Keumatio-Doungtsop BC, van Ree R, de Jong EC, van Kooyk Y. Tolerogenic immunotherapy: targeting DC surface receptors to induce antigen-specific tolerance. Front Immunol. 2021;12:422.
Wculek SK, Cueto FJ, Mujal AM, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24.
Wang Y, Xiang Y, Xin VW, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13(1):107.
Zhang C, Liu Y. Targeting NK cell checkpoint receptors or molecules for cancer immunotherapy. Front Immunol. 2020;11:1295.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100.
Wolf BJ, Choi JE, Exley MA. Novel approaches to exploiting invariant NKT cells in cancer immunotherapy. Front Immunol. 2018;9:384.
Bae EA, Seo H, Kim IK, Jeon I, Kang CY. Roles of NKT cells in cancer immunotherapy. Arch Pharm Res. 2019;42(7):543–8.
Lee SN, Jin SM, Shin HS, Lim YT. Chemical strategies to enhance the therapeutic efficacy of toll-like receptor agonist based cancer immunotherapy. Acc Chem Res. 2020;53(10):2081–93.
Chuang YC, Tseng JC, Huang LR, et al. Adjuvant effect of toll-like receptor 9 activation on cancer immunotherapy using checkpoint blockade. Front Immunol. 2020;11:1075.
Huang X, Zhang X, Lu M. Recent trends in the development of Toll-like receptor 7/8-targeting therapeutics. Expert Opin Drug Discov. 2021;16(8):869–80.
Huntington ND, Cursons J, Rautela J. The cancer–natural killer cell immunity cycle. Nat Rev Cancer. 2020;20(8):437–54.
Herberman RB, Nunn ME, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. I. Distribution of reactivity and specificity. Int J Cancer. 1975;16(2):216–29.
Kiessling R, Klein E, Wigzell H. „Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5(2):112–7.
Zhu H, Blum RH, Bernareggi D, et al. Metabolic reprograming via deletion of CISH in human iPSC-derived NK cells promotes in vivo persistence and enhances anti-tumor activity. Cell Stem Cell. 2020;27(2):224-237.e6.
Naujoks W, Quandt D, Hauffe A, et al. Characterization of surface receptor expression and cytotoxicity of human NK Cells and NK cell subsets in overweight and obese humans. Front Immunol. 2020;11:573200.
Goodier MR, Wolf A, Riley EM. Differentiation and adaptation of natural killer cells for anti-malarial immunity. Immunol Rev. 2020;293(1):25–37.
Yu J, Mao HC, Wei M, et al. CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK-cell subsets. Blood. 2010;115(2):274–81.
Yang C, Malarkannan S. Transcriptional regulation of NK cell development by mTOR complexes. Front Cell Dev Biol. 2020;8:1280.
Wagner JA, Wong P, Schappe T, et al. Stage-specific requirement for eomes in mature NK cell homeostasis and cytotoxicity. Cell Rep. 2020;31(9): 107720.
Yang C, Siebert JR, Burns R, et al. Single-cell transcriptome reveals the novel role of t-bet in suppressing the immature NK gene signature. Elife. 2020;9:1–23.
Carotta S, Pang SHM, Nutt SL, Belz GT. Identification of the earliest NK-cell precursor in the mouse BM. Blood. 2011;117(20):5449–52.
Bi J, Wang X. Molecular regulation of NK cell maturation. Front Immunol. 2020;11:1945.
Ma Q, Dong X, Liu S, et al. Hepatitis B e antigen induces NKG2A+ natural killer cell dysfunction via regulatory T cell-derived interleukin 10 in chronic hepatitis B virus infection. Front Cell Dev Biol. 2020;8:421.
El-Deeb NM, El-Adawi HI, El-wahab AEA, et al. Modulation of NKG2D, KIR2DL and cytokine production by Pleurotus ostreatus glucan enhances natural killer cell cytotoxicity toward cancer cells. Front Cell Dev Biol. 2019;7:165.
Wu SY, Fu T, Jiang YZ, Shao ZM. Natural killer cells in cancer biology and therapy. Mol Cancer. 2020;19(1):120.
Björkström NK, Ljunggren HG, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. 2016;16(5):310–20.
Mandelboim O, Lieberman N, Lev M, et al. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409(6823):1055–60.
Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80-AICL interaction. Nat Immunol. 2006;7(12):1334–42.
Klimosch SN, Bartel Y, Wiemann S, Steinle A. Genetically coupled receptor–ligand pair NKp80-AICL enables autonomous control of human NK cell responses. Blood. 2013;122(14):2380–9.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–40.
Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47(5):820–33.
Geiger TL, Sun JC. Development and maturation of natural killer cells. Curr Opin Immunol. 2016;39:82–9.
Abel AM, Yang C, Thakar MS, Malarkannan S. Natural killer cells: development, maturation, and clinical utilization. Front Immunol. 2018;9:1869.
Wu C, Espinoza DA, Koelle SJ, et al. Clonal expansion and compartmentalized maintenance of rhesus macaque NK cell subsets. Sci Immunol. 2018;3(29):eaat9781.
Grégoire C, Chasson L, Luci C, et al. The trafficking of natural killer cells. Immunol Rev. 2007;220(1):169–82.
Sun H, Sun C, Tian Z, Xiao W. NK cells in immunotolerant organs. Cell Mol Immunol. 2013;10(3):202–12.
Cooley S, Xiao F, Pitt M, et al. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood. 2007;110(2):578–86.
Anfossi N, André P, Guia S, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25(2):331–42.
Menasche BL, Davis EM, Wang S, et al. PBRM1 and the glycosylphosphatidylinositol biosynthetic pathway promote tumor killing mediated by MHC-unrestricted cytotoxic lymphocytes. Sci Adv. 2020;6(48):eabc3243.
Ni J, Wang X, Stojanovic A, et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α unleashes NK cell activity. Immunity. 2020;52(6):1075-1087.e8.
Beĺanger S, Tu MM, Rahim MMA, et al. Impaired natural killer cell self-education and “missing-self” responses in Ly49-deficient mice. Blood. 2012;120(3):592–602.
Kärre K. Natural killer cell recognition of missing self. Nat Immunol. 2008;9(5):477–80.
Shifrin N, Raulet DH, Ardolino M. NK cell self tolerance, responsiveness and missing self recognition. Semin Immunol. 2014;26(2):138–44.
Rahim MMA, Chen P, Mottashed AN, et al. The mouse NKR-P1B:Clr-b recognition system is a negative regulator of innate immune responses. Blood. 2015;125(14):2217–27.
Assarsson E, Kambayashi T, Schatzle JD, et al. NK cells stimulate proliferation of T and NK cells through 2B4/CD48 interactions. J Immunol. 2004;173(1):174–80.
Pozo D, Valés-Gómez M, Mavaddat N, et al. CD161 (human NKR-P1A) signaling in NK cells involves the activation of acid sphingomyelinase. J Immunol. 2006;176(4):2397–406.
Fauriat C, Ivarsson MA, Ljunggren HG, Malmberg KJ, Michaëlsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood. 2010;115(6):1166–74.
Boyington JC, Riaz AN, Patamawenu A, et al. Structure of CD94 reveals novel C-type lectin fold: Implications for the NK cell-associated CD94/NKG2 receptors. Immunity. 1999;10(1):75–82.
Baychelier F, Sennepin A, Ermonval M, et al. Identification of a cellular ligand for the natural cytotoxicity receptor NKp44. Blood. 2013;122(17):2935–42.
Ferlazzo G, Tsang ML, Moretta L, et al. Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J Exp Med. 2002;195(3):343–51.
López-Botet M, Llano M, Navarro F, Bellon T. NK cell recognition of non-classical HLA class I molecules. Semin Immunol. 2000;12(2):109–19.
Lanier LL, Corliss B, Wu J, Phillips JH. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity. 1998;8(6):693–701.
Pupuleku A, Costa-García M, Farré D, et al. Elusive role of the CD94/NKG2C NK cell receptor in the response to cytomegalovirus: novel experimental observations in a reporter cell system. Front Immunol. 2017;8:1317.
Eagle RA, Trowsdale J. Promiscuity and the single receptor: NKG2D. Nat Rev Immunol. 2007;7(9):737–44.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–10.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–36.
Oka N, Markova T, Tsuzuki K, et al. IL-12 regulates the expansion, phenotype, and function of murine NK cells activated by IL-15 and IL-18. Cancer Immunol Immunother. 2020;69(9):1699–712.
Romee R, Schneider SE, Leong JW, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120(24):4751–60.
Hood SP, Foulds GA, Imrie H, et al. Phenotype and function of activated natural killer cells from patients with prostate cancer: patient-dependent responses to priming and IL-2 activation. Front Immunol. 2019;9:3169.
Burns LJ, Weisdorf DJ, DeFor TE, et al. IL-2-based immunotherapy after authologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transpl. 2003;32(2):177–86.
Steele N, Anthony A, Saunders M, et al. A phase 1 trial of recombinant human IL-21 in combination with cetuximab in patients with metastatic colorectal cancer. Br J Cancer. 2012;106(5):793–8.
Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82.
Pérez-Martínez A, Fernández L, Valentín J, et al. A phase I/II trial of interleukin-15-stimulated natural killer cell infusion after haplo-identical stem cell transplantation for pediatric refractory solid tumors. Cytotherapy. 2015;17(11):1594–603.
Ljunggren HG, Kärre K. In search of the “missing self”: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–44.
Zwirner NW, Domaica CI, Fuertes MB. Regulatory functions of NK cells during infections and cancer. J Leukoc Biol. 2021;109(1):185–94.
Hatjiharissi E, Xu L, Santos DD, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood. 2007;110(7):2561–4.
Smyth MJ, Crowe NY, Pellicci DG, et al. Sequential production of interferon-γ by NK1.1+ T cells and natural killer cells is essential for the antimetastatic effect of α-galactosylceramide. Blood. 2002;99(4):1259–66.
Bryceson YT, March ME, Ljunggren H-G, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev. 2006;214(1):73–91.
Martín-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH1 priming. Nat Immunol. 2004;5(12):1260–5.
Smyth MJ, Hayakawa Y, Takeda K, Yagita H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat Rev Cancer. 2002;2(11):850–61.
Nguyen-Pham TN, Yang DH, Nguyen TAT, et al. Optimal culture conditions for the generation of natural killer cell-induced dendritic cells for cancer immunotherapy. Cell Mol Immunol. 2012;9(1):45–53.
Kelly JM, Darcy PK, Markby JL, et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol. 2002;3(1):83–90.
Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19(3):200–18.
Imai H, Saio M, Nonaka K, et al. Depletion of CD4 + CD25 + regulatory T cells enhances interleukin-2-induced antitumor immunity in a mouse model of colon adenocarcinoma. Cancer Sci. 2007;98(3):416–23.
Bachanova V, Cooley S, Defor TE, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123(25):3855–63.
Marçais A, Cherfils-Vicini J, Viant C, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15(8):749–57.
Ranson T, Vosshenrich CAJ, Corcuff E, et al. IL-15 is an essential mediator of peripheral NK-cell homeostasis. Blood. 2003;101(12):4887–93.
Wrangle JM, Velcheti V, Patel MR, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19(5):694–704.
Romee R, Cooley S, Berrien-Elliott MM, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131(23):2515–27.
Leong JW, Chase JM, Romee R, et al. Preactivation with IL-12, IL-15, and IL-18 induces cd25 and a functional high-affinity il-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transpl. 2014;20(4):463–73.
Seo H, Jeon I, Kim BS, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. 2017;8(1):1–14.
Liu E, Ang SOT, Kerbauy L, et al. GMP-compliant universal antigen presenting cells (uAPC) promote the metabolic fitness and antitumor activity of armored cord blood CAR-NK cells. Front Immunol. 2021;12:330.
Hodgins JJ, Khan ST, Park MM, Auer RC, Ardolino M. Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Invest. 2019;129(9):3499–510.
Olson JA, Leveson-Gower DB, Gill S, et al. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115(21):4293–301.
Shah NN, Baird K, Delbrook CP, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–92.
Bishara A, De Santis D, Witt CC, et al. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor-alloreactive T cells causing GVHD. Tissue Antigens. 2004;63(3):204–11.
Porrata LF, Inwards DJ, Ansell SM, et al. Early lymphocyte recovery predicts superior survival after autologous stem cell transplantation in non-hodgkin lymphoma: a prospective study. Biol Blood Marrow Transpl. 2008;14(7):807–16.
Rueff J, Medinger M, Heim D, Passweg J, Stern M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol Blood Marrow Transpl. 2014;20(6):896–9.
Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17(19):6287–97.
Sakamoto N, Ishikawa T, Kokura S, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. 2015;13(1):277.
Xie G, Dong H, Liang Y, et al. CAR-NK cells: a promising cellular immunotherapy for cancer. EBioMedicine. 2020;59: 102975.
Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transpl. 2010;16(9):1245–56.
Maher J, Brentjens RJ, Gunset G, Rivière I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat Biotechnol. 2002;20(1):70–5.
Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4–1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–84.
Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106(9):3360–5.
Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer. 2011;129(12):2935–44.
Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8(5): e1049.
Lang S, Vujanovic NL, Wollenberg B, Whiteside TL. Absence of B7.1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur J Immunol. 1998;28(3):780–6.
Lanier LL, Cortiss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391(6668):703–7.
Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell-mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003;4(6):557–64.
Nakajima H, Colonna M. 2B4: An NK cell activating receptor with unique specificity and signal transduction mechanism. Hum Immunol. 2000;61(1):39–43.
Chmielewski M, Abken H. TRUCKs: The fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–54.
Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Karadimitris A. Cord blood CAR-NK cells: favorable initial efficacy and toxicity but durability of clinical responses not yet clear. Cancer Cell. 2020;37(4):426–7.
Wang X, Jasinski DL, Medina JL, et al. Inducible MyD88/CD40 synergizes with IL-15 to enhance antitumor efficacy of CAR-NK cells. Blood Adv. 2020;4(9):1950–64.
Liu D, Sun X, Du Y, Kong M. Propofol promotes activity and tumor-killing ability of natural killer cells in peripheral blood of patients with colon cancer. Med Sci Monit. 2018;24:6119–28.
Herrera L, Juan M, Eguizabal C. Purification, culture, and CD19-CAR lentiviral transduction of adult and umbilical cord blood NK cells. Curr Protoc Immunol. 2020;131(1): e108.
Daher M, Basar R, Gokdemir E, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Vacca P, Pietra G, Tumino N, et al. Exploiting human NK cells in tumor therapy. Front Immunol. 2020;10:3013.
Gang M, Marin ND, Wong P, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136(20):2308–18.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181-192.e5.
Zhu H, Blum RH, Bjordahl R, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135(6):399–410.
Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123-357ra123.
Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy—advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91.
Daher M, Rezvani K. Outlook for new car-based therapies with a focus on car nk cells: what lies beyond car-engineered t cells in the race against cancer. Cancer Discov. 2021;11(1):45–58.
Tang X, Yang L, Li Z, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Goodridge JP, Mahmood S, Zhu H, et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood. 2019;134(Supplement_1):301–301.
Zhang Y, Chen L, Wang Y, et al. Combination therapy with daratumumab and CAR-NK targeting CS1 for multiple myeloma. Blood. 2016;128(22):1342–1342.
Jiang H, Zhang W, Shang P, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2):297–310.
Martín EM, Encinas J, García-Ortiz A, et al. Exploring NKG2D and BCMA-CAR NK-92 for adoptive cellular therapy to multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019;19(10):e24–5.
Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Luanpitpong S, Poohadsuan J, Klaihmon P, Issaragrisil S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J Immunol Res. 2021;2021:1–16.
Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos Trans R Soc B BiolSci. 2012;367(1590):800–11.
Vey N, Bourhis JH, Boissel N, et al. Aphase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood. 2012;120(22):4317–23.
Nijhof IS, Van Bueren JJL, Van Kessel B, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica. 2015;100(2):263–8.
Daver N, Garcia-Manero G, Basu S, et al. Phase IB/II study of lirilumab in combination with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML). Blood. 2016;128(22):1641–1641.
Yalniz FF, Daver N, Rezvani K, et al. A pilot trial of lirilumab with or without azacitidine for patients with myelodysplastic syndrome. Clin Lymphoma Myeloma Leuk. 2018;18(10):658–63.
Bagot M, Porcu P, Marie-Cardine A, et al. IPH4102, a first-in-class anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019;20(8):1160–70.
Braud VM, Allan DSJ, O’Callaghan CA, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A. B and C Nature. 1998;391(6669):795–9.
Seymour L, Tinker A, Hirte H, Wagtmann N, Dodion P. Phase I and dose ranging, phase II studies with IPH2201, a humanized monoclonal antibody targeting HLA-E receptor CD94/NKG2A. Ann Oncol. 2015;26:ii3.
McWilliams EM, Mele JM, Cheney C, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. 2016;5(10): e1226720.
Tinker AV, Hirte HW, Provencher D, et al. Dose-ranging and cohort-expansion study of monalizumab (IPH2201) in patients with advanced gynecologic malignancies: a trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin Cancer Res. 2019;25(20):6052–60.
Cohen RB, Lefebvre G, Posner MR, et al. Monalizumab in combination with cetuximab in patients (pts) with recurrent or metastatic (R/M) head and neck cancer (SCCHN) previously treated or not with PD-(L)1 inhibitors (IO): 1-year survival data. Ann Oncol. 2019;30: v460.
Zhang Q, Bi J, Zheng X, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Chan CJ, Martinet L, Gilfillan S, et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15(5):431–8.
O’Donnell JS, Madore J, Li XY, Smyth MJ. Tumor intrinsic and extrinsic immune functions of CD155. Semin Cancer Biol. 2020;65:189–96.
Masson D, Jarry A, Baury B, et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49(2):236–40.
Lupo KB, Matosevic S, Matosevic S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J Hematol Oncol. 2020;13(1):1–10.
Liang R, Zhu X, Lan T, et al. TIGIT promotes CD8+T cells exhaustion and predicts poor prognosis of colorectal cancer. Cancer Immunol Immunother. 2021;70(10):2781–93.
Chew V, Tow C, Teo M, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol. 2010;52(3):370–9.
Chew V, Chen J, Lee D, et al. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut. 2012;61(3):427–38.
Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. 2015;36(10):1191–9.
Dixon KO, Schorer M, Nevin J, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol. 2018;200(8):3000–7.
Chauvin JM, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–58.
Hong X, Wang X, Wang T, Zhang X. Correlation of T cell immunoglobulin and ITIM domain (TIGIT) and programmed death 1 (PD-1) with clinicopathological characteristics of renal cell carcinoma may indicate potential targets for treatment. Med Sci Monit. 2018;24:6861–72.
Hung AL, Maxwell R, Theodros D, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769.
Blake SJ, Stannard K, Liu J, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016;6(4):446–59.
Attalla K, Farkas AM, Anastos H, et al. TIM-3 and TIGIT are possible immune checkpoint targets in patients with bladder cancer. Urol Oncol Semin Orig Investig. 2020. https://doi.org/10.1016/j.urolonc.2020.06.007.
Acknowledgements
We thank all of the authors listed in this manuscript.
Funding
This work was supported by the Natural Science Foundation of China (81970183).
Author information
Authors and Affiliations
Contributions
JC, FG, and MY wrote the manuscript, collected the related literature, and finished the figures and tables. BS and YL revised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest relating to the publication of this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chu, J., Gao, F., Yan, M. et al. Natural killer cells: a promising immunotherapy for cancer. J Transl Med 20, 240 (2022). https://doi.org/10.1186/s12967-022-03437-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03437-0