
Abstract
Background
Inflammatory Bowel Disease (IBD) is a multifactorial chronic disease. Understanding only one aspect of IBD pathogenesis does not reflect the complex nature of IBD nor will it improve its clinical management. Therefore, it is vital to dissect the interactions between the different players in IBD pathogenesis in order to understand the biology of the disease and enhance its clinical outcomes.
Aims
To provide an overview of the available omics data used to assess the potential mechanisms through which various players are contributing to IBD pathogenesis and propose a precision medicine model to fill the current knowledge gap in IBD.
Results
Several studies have reported microbial dysbiosis, immune and metabolic dysregulation in IBD patients, however, this data is not sufficient to create signatures that can differentiate between the disease subtypes or between disease relapse and remission.
Conclusions
We summarized the current knowledge in the application of omics in IBD patients, and we showed that the current knowledge gap in IBD hinders the improvements of clinical decision for treatment as well as the prediction of disease relapse. We propose one way to fill this gap by implementing integrative analysis of various omics datasets generated from one patient at a single time point.
Similar content being viewed by others


Background
Inflammatory Bowel Disease (IBD) is an inflammatory disorder of the gastrointestinal (GI) tract, resulting from the complex interactions between genetic make-up, microbiome composition, environmental factors, and mucosal immune response [1]. IBD is characterized by the repeated alternating cycles of clinical relapse and remission [2] and in the absence of an adequate treatment, a chronic inflammation leading to irreversible intestinal damages [3]. Based on the disease manifestation, IBD is classified into three major subtypes [4]: Ulcerative Colitis (UC), which primarily affects the colon, Crohn’s disease (CD) which affects various GI sites [5], and a third subtype where histology assessments done on patients do not categorize to either UC or CD. This subtype is defined as “Inflammatory Bowel Disease, type unclassified” or “Undetermined” (IBD-U) [6, 7]. IBD is a lifelong disease that substantially reduces the quality of life for the patients and their families [8].
Although the first case of UC was reported in Europe in 1875 [9] and CD was first reported in USA in 1932 [10], IBD was still a rare disease until the second half of the 20th century. Post-World War II, a rapid increase in the incidence of UC and CD had been reported, with more than 5 million people affected worldwide [11,12,13]. This drastic increase in IBD patterns suggest that other factors aside from industrialization must be involved in driving the changes observed in the prevalence of IBD [13, 14]. Recent studies have found a number of environmental factors including modern diet, increasing body mass index, glycemic response, medications and gut microbiota can trigger the host immune response, and have been linked to increasing IBD prevalence [15]. In addition, early childhood exposure to antibiotics, birth mode and limited childhood exposure to environmental microorganisms can also influence susceptibility to IBD development [16]. Although a great progress has been made in our understanding of IBD pathogenesis, translating this knowledge into a personalized clinical decision is still far from being achieved [17, 18]. Progress to date indicates that IBD is a multifactorial disease, therefore, a systems biology approach aiming to integrate biological omics and non-omics datasets can be a solution to resolve the complexity of the disease etiology and its heterogenous clinical outcomes. Such comprehensive approach is not only critical to provide the vast information needed for developing the best therapy or interventional strategies to IBD patients, but also for discovering clinical biomarkers that can characterize IBD pathogenesis in a subtype-specific manner. Moreover, a systems biology approach will also help with the prediction and interception of the disease and will promote personalized treatment for IBD patients.
In this review, we will summarize the results generated from various omics platforms including genomics, microbiomics, immune-proteomics, immune-transcriptomics, lipidomics and metabolomics. We will also discuss the efforts made to delineate IBD pathogenesis using these datasets and propose a framework to improve the current understanding of IBD.
IBD etiology and diagnosis: what do we know?
While the etiology of IBD remains exclusive, evidence indicates that the genetic make-up, mode of birth, mode of feeding at a young age, hygiene, exposure to infections, diet and stress among others are the key factors for developing IBD [15, 16]. IBD is usually suspected when the patient present with specific symptoms including diarrhea, abdominal pain, anemia and weight loss [19]. The multifaceted triggering factors of IBD and the major disease symptoms are summarized in Fig. 1. The mechanisms underlying the disease pathogenesis are not fully understood, and there is difficulty in understanding which and how-many triggering factors are involved. However, an overactive mucosal immune response and a dysbiotic gut microbiome are commonly observed in all the IBD subtypes [20, 21].

The multifaceted triggering factors for IBD and major disease symptoms. IBD develops at the intersection of host genetic predisposition, environmental influences, immune dysregulation and dysbiosis of the gut microbiota (left side). The major symptoms reported in the IBD patients are summarized on the right side
Until recently, the diagnosis of IBD seemed straightforward, as it mainly required the presence of a chronic inflammation in the GI tract with the exclusion of other causes of inflammation due to an infectious disease, vasculitides or others [22]. The current diagnostic method consists of a combination of a detailed history assessment, physical and laboratory examination, esophagogastroduodenoscopy, ileo-colonoscopy combined with histology, and imaging of the small bowel using video capsule endoscopy or enteroscopy [23,24,25]. Mucosal biopsies often show a characteristic appearance of UC or CD [26,27,28]. Small bowel imaging is recommended in all suspected cases of IBD at diagnosis; however, it can be delayed in a typical UC presentation, based on endoscopy and histology [19]. A typical UC presentation is identified by a continuous mucosal inflammation of the colon, starting from the rectum, without involving the small bowel, and with the presence of a characteristic crypt architecture disruption [4, 5, 19, 29, 30]. The inflammation is usually more severe distally and if a reverse gradient is observed, a reconsideration for the diagnosis should be prompted [4, 5, 19, 30]. It is also worth noting, that five atypical variants of UC are identified, which make the disease diagnosis and treatment more complex and often unsatisfactory [19]. The diagnosis of CD is usually based on the presence of aphthous or linear ulcers in the ileum or colon, although they can also be detected in any area of the GI tract [29]. The presence of deep serpentine ulcers along the bowel lining, and epithelioid granulomas detected in a biopsy from any area of the GI tract are sufficient to define the diagnosis of CD [4]. Endoscopy and colonoscopy are currently used for the differential diagnosis of CD and UC however, they always carry risks of bowel perforation [31].
On the other hand, the non-invasive routine laboratory investigations currently include blood testing for C-reactive protein, albumin, transaminases and erythrocyte sedimentation rate in addition to fecal testing for calprotectin and lactoferrin [30, 31]. Nonetheless, these investigations can only identify a systemic inflammation and are complementary to the invasive tests conventionally used for detecting GI tract specific flares. Serology testing can be used to subtype IBD patients: the anti-Saccharomyces cerevisiae antibody is found more often in CD than in UC patients and is usually associated with more severe forms of the disease. Whereas, the perinuclear antineutrophil cytoplasmic antibody is more common in UC (60–70%) as compared to CD patients [32]. However, serum positivity may be associated with other diseases which makes it harder for IBD diagnosis.
When features used to differentiate UC from CD in patients with IBD remain uncertain even after a complete workup, patients will be referred to as IBD-U until in some cases the disease develops its characteristic subtype features over time [6, 7, 33]. It is nowadays challenging to choose the best diagnostic tests and correctly classify IBD patients, especially with the increased frequency of disease heterogeneity and atypical phenotypes [19]. Moreover, clinicians are often faced with a difficult clinical decision for the IBD-U patients and frequently resort to mixing treatment protocols in an anticipation for the development of either UC or CD over time [6, 7, 33]. Such approaches often result in unsatisfactory patient outcomes, unnecessary treatment or, in some cases, inappropriate clinical care. Therefore, it is critical to understand the disease signature specific to each subtype in order to provide the most appropriate and personalized care for patients suffering from IBD.
Application of omics: a step towards a better understanding of IBD pathogenesis
Genomics in IBD pathogenesis
In the past two decades, technological advances in genomics and availability of large consortia genomic data have significantly contributed to our understanding of the link between specific gene loci and their relative contributions to IBD susceptibility [34, 35]. Genome-wide association studies in IBD patients have identified more than 300 genetic variants affecting various host functions—including intestinal homeostasis, epithelial barrier function, microbial composition, autophagy, production and secretion of anti-microbial peptides, and regulation of adaptive immunity [36].
Both CD and UC share around 30% of the IBD-related genetic loci [34,35,36], indicating that, despite being considered as two distinct IBD-subtypes with different clinical presentation, there are several common disease-related pathways such as those implicated in host immune functions, including cytokine, chemokine signaling and T helper (Th) cell responses. For example, caspase recruitment domain 9 (CARD9), IL-12 receptor (IL-12R), IL-23R, IL-2, IL-10, IL-21, interferon (IFN)-γ are shown to be associated with both CD and UC [36] (Table 1). On the other hand, mutation in genes such as the nucleotide-binding oligomerization domain-containing protein 2(NOD2) and autophagy related gene (Atg16l1) among others have been shown to be specific to patients with CD [37], while others like IL1R1/IL1R2 genes are specific to patients with UC [35, 38].
Taken together, these findings suggest that genetic predisposition plays an important role in IBD pathogenesis. However, there is still a critical knowledge gap in understanding the IBD etiology, as patients without known genetic predisposition can still develop the disease, suggesting that genomics alone is not enough to reveal the complex IBD puzzle.
Microbiomics in IBD: from postulated theories to known differential microbial signatures
Our understanding of the human microbiome in health and disease has significantly expanded owing to the establishment of the 16S rRNA gene sequencing of the microbial genomes. The technology has propelled research on microbiome composition and function, as well as it allowed us to understand the effect of various factors in modifying the microbiome composition [39,40,41,42,43].
The human GI tract is densely populated by trillions of microbes including bacteria, viruses, fungi, and protozoa [39, 44]. The microbiota is continuously shaped by the exposure to a wide array of antigens found in the GI community. A healthy gut microbiota is composed of four predominant bacterial phyla, with Firmicutes and Bacteroides accounting for more than 87% of the GI microbial communities [45]. The number and composition of the microbial communities also vary in different parts of the GI tract [46, 47]. The microbiota plays an important role in maintaining the integrity of the gut epithelial barrier, food digestion, synthesis of vitamins and biomolecules, and development of mucosal immune cells among many other functions [41, 48]. In turn, the GI tract environment supports the growth, reproduction, and longevity of the gut microbial communities to maintain a state of symbiosis [49].
In the healthy state, the gut homeostasis is maintained [50]. The intestinal immune responses are regulated in order to provide a protective immunity against potential invading pathogens, while limiting any immune reaction in response to innocuous microbes and dietary antigens [50, 51]. Changes in the gut microbial compositions or microbial dysbiosis, is defined as a decrease in the intestinal microbial diversity resulting in an imbalance between commensal “protective” versus potential pathogens “harmful”, thus promoting an excessive intestinal inflammation [52]. When persistent, this response can induce a chronic, unregulated intestinal inflammation that is observed in various human diseases such as IBD, irritable bowel syndrome, asthma, obesity, cardiovascular diseases, kidney diseases, to name a few, from the wide array of diseases known to present with gut dysbiosis [39, 41, 53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72].
Role of the microbiome in IBD: postulated theories
An increasing amount of evidence supports that microbial imbalance in the GI tract influences the development and progression of IBD [61, 73,74,75,76,77,78,79,80]. Considering the key roles of the intestinal microbiota in the pathogenesis of IBD, the following theories have been postulated:
-
A.
Imbalance between protective versus harmful microbes: The role of dysbiosis in IBD pathogenesis has been described in many studies assessing IBD patients. A decrease in Firmicutes and an expansion in Proteobacteria was observed in patients with CD, compared with the healthy controls in multiple studies [81, 82]. Similarly, a decrease in butyrate-producing species, such as Faecalibacterium prausnitzii and Roseburia hominis, has been reported in patients with UC or CD [83, 84]. In addition, studies comparing members of the same family (including twins), who are discordant for IBD, have postulated that dysbiosis is the primary causative agent of IBD [82, 85]. A summary of the microbial signatures known to be associated with CD or UC is summarized in Fig. 2.
Fig. 2 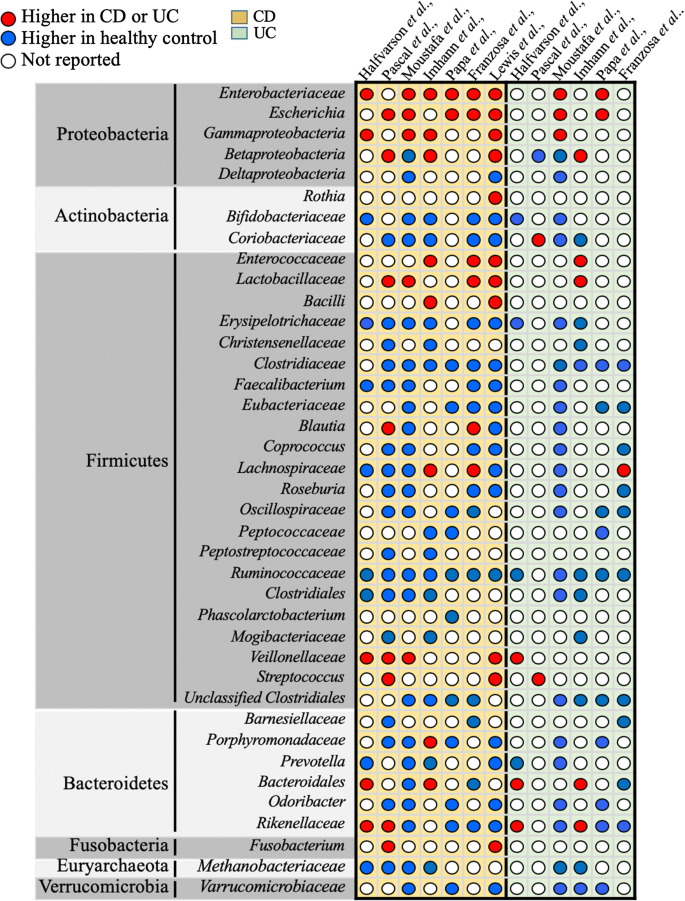
Gut microbiota dysbiosis in CD or UC patients. Qualitative comparison of relative microbial dysbiosis in CD and UC patients, retrieved from different original studies (Halfvarson et al., Pascal et al., Moustafa et al., Imhann et al., Papa et al., Franzosa et al., and Lewis et al.,). The relative increase or decrease in microbial levels is represented by red or blue dots respectively. White dots represent data not reported
-
B.
Presence of a potential pathogen: IBD may be driven by a persistent pathogen (such as members of Proteobacteria) that contributes and exacerbates the disease pathogenesis. Members of the phylum Proteobacteria, specifically Escherichia coli, are frequently found at higher ratios in IBD patients as compared with the healthy individuals [86,87,88]. Strains of the adherent-invasive E. coli (AIEC) were isolated from the ileal mucosa in patients with CD, along with an increase in TNFα secretion [89,90,91,92,93,94,95,96,97,98,99]. On the other hand, AIEC is usually considered a commensal microbe present in healthy individuals, which suggests that facultative pathogens can cause disease in susceptible hosts [81, 86, 91, 100,101,102,103]. Mycobacterium avium subspecies, Paratuberculosis, and Pseudomonas aeruginosa have also been investigated as potential causes of CD, owing to their ability to induce chronic granulomatous enteritis and colitis in animals [104,105,106,107]. Fusobacterium nucleatum has been postulated to play a major role in the association between IBD and the development of colorectal cancer [108, 109]. Rather than acting as individual infectious agents, it is more likely that the interactions between the reported pathobionts (Table 2) and the rest of the GI microbiota underlie the disease pathogenesis [110].
Table 2 Presence of pathogenic bacteria (pathobionts) in CD or UC patients -
C.
Dysregulated immune response: Loss of the mucosal membrane integrity observed in IBD patients results in a dysregulated immune response caused by an excessive bacterial translocation combined with a continuous immune cells exposure to the microbial antigens. The integrity of the intestinal epithelial tract is established by tight cell junctions (TJ) that exist between GI epithelial cells and create a barrier against microbes [21]. The mucosal layer on the surface of the GI lining acts as the first line of defense against invading pathogens, while the epithelial cells act as the second level of surveillance [21]. The development and organization of the mucosal interface in the GI tract, are intimately linked with the gut microbiota, without which the immune system is immature and defective [111,112,113,114]. While dysbiosis promotes the growth of invasive pathogenic microbes, it also induces inflammation in the GI lining, leading to microbial translocation through the intestinal mucosal barrier to the mesenteric lymph nodes [111]. Changes in the continuity or the number of TJ strands in the intestinal epithelial cells are considered a hallmark in IBD patients [115]. An impairment in the signaling pathways responding to microbial components such as autophagy, IL23/IL17, and Paneth cells function have also been associated with IBD pathogenesis [116].

Microbiota signature for CD and UC diagnosis: are we there yet?
As discussed in the preceding section, the diagnosis of CD or UC patients primarily depend on endoscopy or colonoscopy; however, there are cases in which the characteristic morphology of either subtypes is absent, such as in the case of IBD-U patients [31].
The number of studies reporting gut microbial dysbiosis in IBD patients, particularly in CD patients, continues to grow exponentially and raise the possibility of using microbial signature as a diagnostic tool to distinguish between IBD subtypes [84, 117, 118]. However, inconsistent microbial profiles across studies and huge inter- and intra-individual variations, emphasize the need of longitudinal studies for a better understanding of the microbial pattern associated with IBD subtypes [19, 42]. To this end, many extensive longitudinal IBD cohorts were initiated with the aim to identify microbial signature for CD and UC. For instance, Pascal et al., used a frequent sampling every 3 months, and revealed eight microbial groups including Fusobacterium, Escherichia, Faecalibacterium, Collinsella, Anaerostipes, Methanobrevibacter, an unknown Peptostreptococcaceae, and an unknown Christensenellaceae that were differentially present in CD and UC patients [119, 120]. Similarly, in recent years many studies have combined cutting-edge methodologies to characterize differentially abundant gut microbial composition in CD and UC patients, but inconsistency in microbial signature across studies [16, 119,120,121,122,123,124] hinders the identification of universal microbial biomarker for disease prediction (Fig. 2). The discrepancy in microbial composition could be due to the complexity of interaction between the fluctuating gut microbiota and host features during disease course, in which the gut microbiota either influences other host functions or is being influenced by other factors- such as host genetic, diet, drugs, disease, life-style can shape the composition of gut microbiota [125]. Although, these factors and interactions are common but certainly not identical among patients.
To further establish a comprehensive insight into the host-microbial interaction and other intrinsic as well as extrinsic factors in IBD, Lloyd-Prince et al. recently provided the most detailed view of the microbiome, metabolome and host response in IBD patients [126]. Due to high inter-individual variations, which contributed to the majority of data variance, the researchers could not identify consistent microbial signature. However, researchers presented dynamic view of the complex interaction during active disease, which was well beyond the host-microbial interaction, involving multiple other downstream components including the metabolome, proteome and transcriptome [126, 127]. Despite the promising findings, the key challenges in these studies still hinder future progress for developing biomarkers. Including data reproducibility due to the lack of standard protocols for sample collection, storage, DNA extraction and sequencing methods [19]. More work is still needed to address the challenges in order to define CD and UC-specific microbial signature and to improve clinical decision making, especially in cases where a diagnosis is hard to be reached.
Apart from bacteria, the role of other microorganisms in IBD pathogenesis have been widely overlooked. These microorganisms include fungi, archaea, and viruses. Development of culture-independent methods such as sequencing of the 18S ribosomal subunit or the internal transcribed spacer region has allowed a comprehensive assessment of the mycobiome in human disease [128]. Gut mycobiome dysbiosis is observed in IBD patients and is characterized by an increase in Basidiomycota to Ascomycota ratio, a decrease in the proportion of Saccharomyces cerevisiae to Malassezia sympodialis and an increase in the abundance of Candida albicans (Table 2) [118]. Although, Candida remains asymptomatic in many IBD patients, immune-suppression and/or antibiotic treatment, which is commonly used in CD patients, are independently associated with the expansion of C. albicans [129].
Similarly, changes in bacteriophage composition in the gut of IBD patients have been described, particularly an increase in Caudovirales numbers was detected in ileal biopsy collected from CD patients [130, 131]. However, a direct contribution of the virome to IBD pathogenesis remains to be investigated.
Immuno-proteomics in IBD pathogenesis
Recent studies have highlighted a complex interplay between host genetics and environmental factors in the perturbation of the host epithelial barrier function. Thereby allowing the translocation of microbial antigens into the bowel wall, which results in aberrant immune response in the mucosal layer [132, 133]. Excessive cytokines production in the mucosal layer, not only induces intestinal inflammation and associated clinical symptoms of IBD, but also induces the systemic effects of IBD [134, 135]. For example, a reduced expression of antimicrobial peptides in the mucus layer and epithelial junction proteins like E-cadherin and claudins has been observed in IBD patients [136,137,138]. This in turn supports the excessive microbial growth in the mucus layer and helps the microbial translocation through the impaired epithelial barrier. The ingested microbial components can result in an overactivation of the different immune functions, leading to intestinal inflammation. CpG oligodeoxynucleotides in inflamed bowel have been shown to stimulate TLR9 signaling and induce the production of IFN by mucosal dendritic cells [116, 139]. IFNs can promote epithelial impairment or production of pro-inflammatory cytokines (Fig. 3). Following activation, macrophages residing in the lamina propria produce a large amount of IL-6, IL-18 and IL-23 into the impaired epithelium [140]. These cytokines can activate nearby antigen presenting cells (APC) and T cells (Fig. 3); where both T helper (Th)1 and Th2 cell subsets have been shown to play a crucial role in the inflamed bowel wall by secreting large amounts of pro-inflammatory cytokines [141]. Different cytokine profiles have also been observed between IBD subtypes: CD patients have a more pronounced Th1 response and produce larger amount of IL-2 and INF-gamma compared with UC patients [142]. Several interleukins are now targeted for IBD therapy [143] but with inconsistent efficacy [144], suggesting the importance of immuno-proteomic profiling in IBD patients. It is becoming clear that there is a complex network of cytokine production/activation in the inflamed bowel [145] which, in turn, is affected by its interplay with the microbial communities, host genetics, and environmental factors.

Current understanding of the Microbial–Immune interaction models in IBD. Intestinal homeostasis involves a cross-talk between the epithelial barrier functions, the immune system and the gut microbiota. The balance between pro- and anti-inflammatory cytokines in the intestinal mucosa regulates the epithelial barrier functions. In IBD, various initiating factors such as genetic susceptibility, environmental factors and microbial dysbiosis have been shown to impair the epithelial barrier functions. This results in leaky epithelial barrier resulting in microbial invasion/translocation. The translocated microbes stimulate the immune cells such as dendritic cells (DC) and macrophages leading to the activati on of an inflammatory cascade. The key cytokines produced by activated macrophages and DC (IL-12, IL-27, IL-4, 6, IL-23, TGFb) stimulate various T helper cell subsets (Th1, Th2, Th17, Th9) resulting in the release of cytokines that contribute to defining the immune phenotypes of CD or UC. Activated macrophages secrete IL-12 that in turn activates the innate lymphoid cell (ILC3) and ILC1 and the release of IL-17A, IL-17F, IL-22 and IFN-γ (yellow box). Translocated microbes result in the activation of Natural Killer T (NKT) cells. NKT-cells proliferate and differentiate into Th2 type cells via the secretion of IL-13. In homeostasis, Panet cells, located at the small intestinal crypt secrete various antimicrobial peptides (AMPs), defensins, transforming tumor necrosis factor α (TNF-α), growth factor β1 (TGF-β1) and retinoic acid. In IBD, the dysfunction of Paneth cells results in reduced AMP production and reduced signaling to regulatory T cells (Treg) resulting in a decrease of anti-inflammatory mediators. Infiltration of mucosal plasma cells is also observed in IBD patients. Black arrows indicate the direction of change in IBD. Red arrows indicate the signaling sequencing of events. IL interleukin, IgA immunoglobulin A, AMPs antimicrobial peptides, DC dendritic cells, ILC innate lymphoid cell, Abs antibodies, TGF transforming growth factor, TNF tumor necrosis factor, IFN interferon, Th T helper, CD Crohn’s disease, UC ulcerative colitis
Immuno-transcriptomics in IBD pathogenesis: how to find disease signatures?
Robust immune-transcriptomic signatures that can differentiate between healthy subjects and IBD patients or between the different IBD subtypes can serve as reliable, clinical prognostic or diagnostic biomarkers. However, such signatures are still not well-defined. The large number of genes involved in IBD pathogenesis makes the identification of a targeted, manageable list of genes for defining a signature extremely difficult. The number of potential target genes has been growing consistently; a look at the network of signaling pathways that regulates the immune responses in IBD patients clearly highlights its complexity. Immune-gene network consists of hundreds of immune cells, including their subpopulations, that are involved in the disease pathogenesis along with their highly complex gene signatures and associated functions. Therefore, the role of immune-transcriptomics can only be understood in the context of stimulations received from other factors of IBD pathogenesis [125]. The integration of genetic information, immuno-transcriptomic signature, and proteomic profiles of IBD patients, particularly on how they change during the disease remission and relapse or in response to treatment, has a great potential to uncover novel disease-specific pathways and potential biomarkers.
Other omics: lipidomics and metabolomics in IBD pathogenesis
Previous studies have showed differences in the metabolic and lipidomic profiles of IBD patients when compared with the healthy controls [146] or when comparing CD and UC patients [147]. Agouridis et al. showed that total cholesterol and high-density lipoprotein (HDL) cholesterol levels were lower in IBD patients as compared with the healthy controls, while low density lipoprotein (LDL) cholesterol levels were higher in the IBD patients [148] Fan et al. compared the serum lipid profile of healthy controls with those of CD and UC patients [149] and identified 33 specific lipidome signatures negatively associated with CD patients and 5 lipid species significantly correlated with UC patients. Moreover, Santoru et al. reported increased levels of diacylglycerol and n-acylphosphatidylethanolamines in IBD patients, when compared with the healthy individuals, while urobilin, phosphatidylcholine, urobilinogen, phosphatidic acid phosphatidylserine, phosphatidylcholine and ceramide were decreased [77]. Recent analysis of the lipidome profiles of IBD patients also reflected the role of several pathways that are crucial for epithelial homeostasis, including barrier function and innate immune response [150]. Moreover, Scoville et al. showed that the serum metabolomic profile in IBD patients reflected differences in a number of lipid, amino acid, and tricarboxylic acid cycle related metabolites when compared to the healthy controls [151]. In a more recent study, Murgia et al. showed that using metabolomics and lipidomics, they were able to differentiate between IBD patients and healthy controls, and more importantly between CD and UC patients [152]. These studies illustrate the value of lipidomics and metabolomics in identifying potential biomarkers, however they remain limited and require validation in bigger cohorts.
Conclusion
A significant progress has been achieved across multiple omics layers ranging from the genome, transcriptome, proteome, metabolome, microbiome, etc.…. Nonetheless, despite this advancement, multiple studies still assess those omics independently without taking into consideration of their complex interactions in health or disease. Hence, stemming from a single focal point, one omics approach performed at a time can only explain one aspect of any complex disease.
Multifactorial chronic diseases, such as IBD, are considered challenging as they have limited treatment options. However, they exhibit a great variety of molecular interactions involving a complex interplay between genetics, microbiome and the immune system among others [153, 154]. Classical reductionist approaches have identified key genes or pathways when trying to characterize the cause and progression of IBD and data collected from IBD patients using single omics such as microbiomics [61, 75, 79, 97, 109, 118, 155, 156] and genomics [18, 157, 158] are also available. However, they offer an incomplete overview of the complex etiology of the disease. IBD is considered a great example for systems biology approaches and multi-omics data integration, where using multi-layered analyses to combine various omics dataset is ideal to unravel the biological complexity of the disease, along with comprehensive modelling of the interactions between host factors and the microbial functional feature, in order to define personalized treatment options. While urgently needed, an integration of all omics that can be utilized on IBD patients from a systems biology perspective is still far from being achieved.
An in-depth molecular reclassification of the IBD subtypes is required and a “deep dive” assessment of the IBD pathogenesis and progression is needed in order to achieve a comprehensive analysis of the disease. Large-scale cohorts of IBD patients are needed for systems biology applications, along with a standardization of all the protocols ranging from disease index calculation methods, time points for sample collection, collection kits, sample storage, frequency of sampling, etc.…
In this review, we propose that integrating datasets generated at various biological layers will create a landscape, that will explain the mechanisms of interactions between those layers through the identification of key biological components from each dataset along with their functional characterization and followed by integration of each constituent using integrative analysis to identify personalized signature (Fig. 4). Moreover, this will also allow us to remove abnormal signatures across multiple molecular dimensions (Fig. 4). This unbiased and comprehensive strategy will open entirely new avenues for developing novel personalized therapeutic interventions, and help identify biomarkers that can be non-invasive, highly specific, reliable and easy to assess by clinicians in routine practices.

A proposed model for addressing the knowledge gap in IBD. A systems biology approach is one way to understand the complex IBD pathogenesis. Sample collection at various stages of the disease, a detailed disease index calculation, sample frequency and collection methods have to be optimized. High throughput technologies allowed processing of the samples using various omics approaches including (genomics, transcriptomics, proteomics, microbiomics, metabolomics, lipidomics and others). On the other hand, non-omics data like environmental factors, dietary information or others can also be generated. Integration of all types of omics and non-omics data will allow us to understand the phenotype-genotype interactions and help generate signatures associated with the disease. These signatures will pave the ways towards personalized treatment
Availability of data and materials
Not applicable.
Abbreviations
- IBD:
-
Inflammatory Bowel Disease
- GI:
-
Gastrointestinal
- UC:
-
Ulcerative colitis
- CD:
-
Crohn’s disease
- IBD-U:
-
Inflammatory Bowel Disease, type unclassified
- Th:
-
T helper cells
- AIEC:
-
Adherent-invasive E. coli
- Tj:
-
Tight cell junctions
References
Mehta M, Ahmed S, Dryden G. Immunopathophysiology of inflammatory bowel disease: how genetics link barrier dysfunction and innate immunity to inflammation. Innate Immun. 2017;23:497–505.
Lee SH, Kwon JE, Cho ML. Immunological pathogenesis of inflammatory bowel disease. Intest Res. 2018;16:26–42.
Rieder F, Fiocchi C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–35.
Yu YR, Rodriguez JR. Clinical presentation of Crohn’s, ulcerative colitis, and indeterminate colitis: symptoms, extraintestinal manifestations, and disease phenotypes. Semin Pediatr Surg. 2017;26:349–55.
Feakins RM. Ulcerative colitis or Crohn’s disease? Pitfalls and problems. Histopathology. 2014;64:317–35.
Tontini GE, Vecchi M, Pastorelli L, et al. Differential diagnosis in inflammatory bowel disease colitis: state of the art and future perspectives. World J Gastroenterol. 2015;21:21–46.
Monteiro S, Dias de Castro F, Boal Carvalho P, et al. Essential role of small bowel capsule endoscopy in reclassification of colonic inflammatory bowel disease type unclassified. World J Gastrointest Endosc. 2017;9:34–40.
Oliveira SB, Monteiro IM. Diagnosis and management of inflammatory bowel disease in children. BMJ. 2017;357:j2083.
Samuel Wilks WM. Lectures on pathological anatomy: Lindsay and Blakiston. 1875.
Smith MS, Wakefield AJ. Crohn’s disease: ancient and modern. Postgrad Med J. 1994;70:149–53.
Kim HJ, Hann HJ, Hong SN, et al. Incidence and natural course of inflammatory bowel disease in Korea, 2006–2012: a nationwide population-based study. Inflamm Bowel Dis. 2015;21:623–30.
Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42 (quiz e30).
Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720–7.
Sykora J, Pomahacova R, Kreslova M, et al. Current global trends in the incidence of pediatric-onset inflammatory bowel disease. World J Gastroenterol. 2018;24:2741–63.
Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65:330–9.
Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67:108–19.
Weersma RK, Xavier RJ, Consortium IBDMO, et al. Multiomics analyses to deliver the most effective treatment to every patient with inflammatory bowel disease. Gastroenterology. 2018;155:e1–4.
Fiocchi C. Integrating omics: the future of IBD? Dig Dis. 2014;32(Suppl 1):96–102.
Levine A, Koletzko S, Turner D, et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr. 2014;58:795–806.
Xu XR, Liu CQ, Feng BS, et al. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol. 2014;20:3255–64.
Shi N, Li N, Duan X, et al. Interaction between the gut microbiome and mucosal immune system. Mil Med Res. 2017;4:14.
Dilauro S, Crum-Cianflone NF. Ileitis: when it is not Crohn’s disease. Curr Gastroenterol Rep. 2010;12:249–58.
Tharian B, George N, Navaneethan U. Endoscopy in the diagnosis and management of complications of inflammatory bowel disease. Inflamm Bowel Dis. 2016;22:1184–97.
Taylor S, Lobo AJ. Diagnosis and treatment of inflammatory bowel disease. Practitioner. 2016;260:19–23.
Padoan A, D’Inca R, Scapellato ML, et al. Improving IBD diagnosis and monitoring by understanding preanalytical, analytical and biological fecal calprotectin variability. Clin Chem Lab Med. 2018;56:1926–35.
Koksal AR, Boga S, Alkim H, et al. How does a biopsy of endoscopically normal terminal ileum contribute to the diagnosis? Which patients should undergo biopsy? Libyan J Med. 2014;9:23441.
Magro F, Langner C, Driessen A, et al. European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013;7:827–51.
Surawicz CM, Haggitt RC, Husseman M, et al. Mucosal biopsy diagnosis of colitis: acute self-limited colitis and idiopathic inflammatory bowel disease. Gastroenterology. 1994;107:755–63.
Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17:1314–21.
Kornbluth A, Sachar DB, Practice Parameters Committee of the American College of G. Ulcerative colitis practice guidelines in adults (update): American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2004;99:1371–85.
Lichtenstein GR, Loftus EV, Isaacs KL, et al. ACG clinical guideline: management of Crohn’s disease in adults. Am J Gastroenterol. 2018;113:481–517.
Bartunkova J, Kolarova I, Sediva A, et al. Antineutrophil cytoplasmic antibodies, anti-Saccharomyces cerevisiae antibodies, and specific IgE to food allergens in children with inflammatory bowel diseases. Clin Immunol. 2002;102:162–8.
Matsui T, Yao T, Sakurai T, et al. Clinical features and pattern of indeterminate colitis: Crohn’s disease with ulcerative colitis-like clinical presentation. J Gastroenterol. 2003;38:647–55.
Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25.
Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17.
Shaw KA, Cutler DJ, Okou D, et al. Genetic variants and pathways implicated in a pediatric inflammatory bowel disease cohort. Genes Immun. 2019;20:131–42.
Homer CR, Richmond AL, Rebert NA, et al. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139:1630–41, 1641 e1–2.
Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78.
Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51.
Althani AA, Marei HE, Hamdi WS, et al. Human microbiome and its association with health and diseases. J Cell Physiol. 2016;231:1688–94.
Human Microbiome Project C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Singh P, Kumar M, Al Khodor S. Vitamin D deficiency in the gulf cooperation council: exploring the triad of genetic predisposition, the gut microbiome and the immune system. Front Immunol. 2019;10:1042.
Mathur T, Barman TK, Kumar M, et al. In vitro and in vivo activities of DS-2969b, a novel GyrB inhibitor, against Clostridium difficile. Antimicrob Agents Chemother. 2018;62(4). https://doi.org/10.1128/AAC.02157-17.
Lederberg J, McCray AT. ‘Ome Sweet’ omics—a genealogical treasury of words. Scientist. 2001;15:8.
Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol. 2017;8:1162.
Barko PC, McMichael MA, Swanson KS, et al. The gastrointestinal microbiome: a review. J Vet Intern Med. 2018;32:9–25.
Harmsen HJ, de Goffau MC. The human gut microbiota. Adv Exp Med Biol. 2016;902:95–108.
Hoffmann C, Dollive S, Grunberg S, et al. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE. 2013;8:e66019.
Lozupone CA, Stombaugh JI, Gordon JI, et al. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30.
van de Guchte M, Blottiere HM, Dore J. Humans as holobionts: implications for prevention and therapy. Microbiome. 2018;6:81.
Sirisinha S. The potential impact of gut microbiota on your health: current status and future challenges. Asian Pac J Allergy Immunol. 2016;34:249–64.
Weiss GA, Hennet T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci. 2017;74:2959–77.
Peng J, Xiao X, Hu M, et al. Interaction between gut microbiome and cardiovascular disease. Life Sci. 2018;214:153–7.
Mangiola F, Nicoletti A, Gasbarrini A, et al. Gut microbiota and aging. Eur Rev Med Pharmacol Sci. 2018;22:7404–13.
Liu J, Wu M, He J, et al. Antibiotic-induced dysbiosis of gut microbiota impairs corneal nerve regeneration by affecting CCR2-negative macrophage distribution. Am J Pathol. 2018;188:2786–99.
Li N, Wang Q, Wang Y, et al. Oral probiotics ameliorate the behavioral deficits induced by chronic mild stress in mice via the gut microbiota-inflammation axis. Front Behav Neurosci. 2018;12:266.
Jorgensen SF, Fevang B, Aukrust P. Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J Clin Immunol. 2019;39(1):30–6. https://doi.org/10.1007/s10875-018-0574-z.
Joossens M, Faust K, Gryp T, et al. Gut microbiota dynamics and uraemic toxins: one size does not fit all. Gut. 2019;68:2257–60.
Bell JS, Spencer JI, Yates RL, et al. From nose to gut—the role of the microbiome in neurological disease. Neuropathol Appl Neurobiol. 2018.
Baim AD, Movahedan A, Farooq AV, et al. The microbiome and ophthalmic disease. Exp Biol Med. 2018. https://doi.org/10.1177/1535370218813616.
Dickson I. Gut microbiota: diagnosing IBD with the gut microbiome. Nat Rev Gastroenterol Hepatol. 2017;14:195.
Flint HJ, Scott KP, Louis P, et al. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–89.
Al Khodor S, Shatat IF. Gut microbiome and kidney disease: a bidirectional relationship. Pediatr Nephrol. 2017;32:921–31.
Al Khodor S, Reichert B, Shatat IF. The microbiome and blood pressure: can microbes regulate our blood pressure? Front Pediatr. 2017;5:138.
Tseng CH, Wu CY. The gut microbiome in obesity. J Formos Med Assoc. 2019;118(Suppl 1):S3–9. https://doi.org/10.1016/j.jfma.2018.07.009.
Qin Y, Roberts JD, Grimm SA, et al. An obesity-associated gut microbiome reprograms the intestinal epigenome and leads to altered colonic gene expression. Genome Biol. 2018;19:7.
Murugesan S, Nirmalkar K, Hoyo-Vadillo C, et al. Gut microbiome production of short-chain fatty acids and obesity in children. Eur J Clin Microbiol Infect Dis. 2018;37:621–5.
Luo Y, Blackledge WC. Microbiome-based mechanisms hypothesized to initiate obesity-associated rheumatoid arthritis. Obes Rev. 2018;19:786–97.
Leong KSW, Derraik JGB, Hofman PL, et al. Antibiotics, gut microbiome and obesity. Clin Endocrinol. 2018;88:185–200.
Langan EA, Gratz V, Billmann F, et al. Does the gastrointestinal microbiome contribute to the ‘obesity paradox’ in melanoma survival? Br J Dermatol. 2018;179:225–6.
Cheng M, Zhang X, Zhu J, et al. A metagenomics approach to the intestinal microbiome structure and function in high fat diet-induced obesity mice fed with oolong tea polyphenols. Food Funct. 2018;9:1079–87.
Castaner O, Goday A, Park YM, et al. The gut microbiome profile in obesity: a systematic review. Int J Endocrinol. 2018;2018:4095789.
Fedorak RN, Ismond KP. Practical considerations and the intestinal microbiome in disease: antibiotics for IBD therapy. Dig Dis. 2016;34:112–21.
Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: current results and future approaches. Curr Treat Options Gastroenterol. 2015;13:105–20.
Lavelle A, Sokol H. Gut microbiota: beyond metagenomics, metatranscriptomics illuminates microbiome functionality in IBD. Nat Rev Gastroenterol Hepatol. 2018;15:193–4.
Rehman A, Rausch P, Wang J, et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut. 2016;65:238–48.
Santoru ML, Piras C, Murgia A, et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci Rep. 2017;7:9523.
Sitkin S, Vakhitov T, Pokrotnieks J. How to increase the butyrate-producing capacity of the gut microbiome: do IBD patients really need butyrate replacement and butyrogenic therapy? J Crohns Colitis. 2018;12:881–2.
Wu GD. Diet, the gut microbiome and the metabolome in IBD. Nestle Nutr Inst Workshop Ser. 2014;79:73–82.
Kumar M, Mathur T, Joshi V, et al. Effect of DS-2969b, a novel GyrB inhibitor, on rat and monkey intestinal microbiota. Anaerobe. 2018;51:120–3.
Martinez-Medina M, Garcia-Gil LJ. Escherichia coli in chronic inflammatory bowel diseases: an update on adherent invasive Escherichia coli pathogenicity. World J Gastrointest Pathophysiol. 2014;5:213–27.
Lopez-Siles M, Martinez-Medina M, Busquets D, et al. Mucosa-associated Faecalibacterium prausnitzii and Escherichia coli co-abundance can distinguish Irritable Bowel Syndrome and Inflammatory Bowel Disease phenotypes. Int J Med Microbiol. 2014;304:464–75.
Takahashi K, Nishida A, Fujimoto T, et al. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in Crohn’s disease. Digestion. 2016;93:59–65.
Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–83.
Willing BP, Dicksved J, Halfvarson J, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139(1844–1854):e1.
Conte MP, Longhi C, Marazzato M, et al. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn’s disease patients: phenotypic and genetic pathogenic features. BMC Res Notes. 2014;7:748.
Man SM, Zhang L, Day AS, et al. Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm Bowel Dis. 2010;16:1008–16.
Rowan F, Docherty NG, Murphy M, et al. Desulfovibrio bacterial species are increased in ulcerative colitis. Dis Colon Rectum. 2010;53:1530–6.
Negroni A, Costanzo M, Vitali R, et al. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:913–24.
Campos N, Magro F, Castro AR, et al. Macrophages from IBD patients exhibit defective tumour necrosis factor-alpha secretion but otherwise normal or augmented pro-inflammatory responses to infection. Immunobiology. 2011;216:961–70.
Sasaki M, Sitaraman SV, Babbin BA, et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab Investig. 2007;87:1042–54.
Zhang S, Fu J, Dogan B, et al. 5-Aminosalicylic acid downregulates the growth and virulence of Escherichia coli associated with IBD and colorectal cancer, and upregulates host anti-inflammatory activity. J Antibiot. 2018;71:950–61.
Renouf MJ, Cho YH, McPhee JB. Emergent behavior of IBD-associated Escherichia coli during disease. Inflamm Bowel Dis. 2019;25(1):33–44.
Ummarino D. Spondyloarthropathies: E. coli links IBD to spondyloarthritis. Nat Rev Rheumatol. 2017;13:198.
Kotlowski R. Use of Escherichia coli Nissle 1917 producing recombinant colicins for treatment of IBD patients. Med Hypotheses. 2016;93:8–10.
Bucker R, Schulz E, Gunzel D, et al. alpha-Haemolysin of Escherichia coli in IBD: a potentiator of inflammatory activity in the colon. Gut. 2014;63:1893–901.
Ray K. IBD: fertilizing the gut: host-derived nitrate contributes to Escherichia coli bloom in colitis. Nat Rev Gastroenterol Hepatol. 2013;10:197.
Darfeuille-Michaud A, Colombel JF. Pathogenic Escherichia coli in inflammatory bowel diseases: proceedings of the 1st international meeting on E. coli and IBD, June 2007, Lille, France. J Crohns Colitis. 2008;2:255–62.
La Ferla K, Seegert D, Schreiber S. Activation of NF-kappaB in intestinal epithelial cells by E. coli strains isolated from the colonic mucosa of IBD patients. Int J Colorectal Dis. 2004;19:334–42.
O’Brien CL, Bringer MA, Holt KE, et al. Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut. 2017;66:1382–9.
Zhang Y, Rowehl L, Krumsiek JM, et al. Identification of candidate adherent-invasive E. coli signature transcripts by genomic/transcriptomic analysis. PLoS ONE. 2015;10:e0130902.
Borruel N, Carol M, Casellas F, et al. Increased mucosal tumour necrosis factor alpha production in Crohn’s disease can be downregulated ex vivo by probiotic bacteria. Gut. 2002;51:659–64.
Glasser AL, Boudeau J, Barnich N, et al. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001;69:5529–37.
Wagner J, Short K, Catto-Smith AG, et al. Identification and characterisation of Pseudomonas 16S ribosomal DNA from ileal biopsies of children with Crohn’s disease. PLoS ONE. 2008;3:e3578.
Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease-associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567–75.
Horing E, Gopfert D, Schroter G, et al. Frequency and spectrum of microorganisms isolated from biopsy specimens in chronic colitis. Endoscopy. 1991;23:325–7.
Mottawea W, Chiang CK, Muhlbauer M, et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat Commun. 2016;7:13419.
Bashir A, Miskeen AY, Hazari YM, et al. Fusobacterium nucleatum, inflammation, and immunity: the fire within human gut. Tumour Biol. 2016;37:2805–10.
Strauss J, Kaplan GG, Beck PL, et al. Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis. 2011;17:1971–8.
Hall AB, Yassour M, Sauk J, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9:103.
Alipour M, Zaidi D, Valcheva R, et al. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J Crohns Colitis. 2016;10:462–71.
Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut. 2013;62:1653–64.
Gordon HA. Morphological and physiological characterization of germfree life. Ann N Y Acad Sci. 1959;78:208–20.
Ahmad R, Sorrell MF, Batra SK, et al. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. 2017;10:307–17.
Martini E, Krug SM, Siegmund B, et al. Mend your fences: the epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol. 2017;4:33–46.
Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24.
Kang S, Denman SE, Morrison M, et al. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–42.
Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039–48.
Pascal V, Pozuelo M, Borruel N, et al. A microbial signature for Crohn’s disease. Gut. 2017;66:813–22.
Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004.
Franzosa EA, Sirota-Madi A, Avila-Pacheco J, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4:293–305.
Moustafa A, Li W, Anderson EL, et al. Genetic risk, dysbiosis, and treatment stratification using host genome and gut microbiome in inflammatory bowel disease. Clin Transl Gastroenterol. 2018;9:e132.
Papa E, Docktor M, Smillie C, et al. Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS ONE. 2012;7:e39242.
Lewis JD, Chen EZ, Baldassano RN, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18:489–500.
de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017;14:739–49.
Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–62.
Metwaly A, Haller D. Multi-omics in IBD biomarker discovery: the missing links. Nat Rev Gastroenterol Hepatol. 2019;16:587–8. https://doi.org/10.1038/s41575-019-0188-9.
Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–16.
Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21:334–41.
Norman JM, Handley SA, Baldridge MT, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–60.
Wagner J, Maksimovic J, Farries G, et al. Bacteriophages in gut samples from pediatric Crohn’s disease patients: metagenomic analysis using 454 pyrosequencing. Inflamm Bowel Dis. 2013;19:1598–608.
Blumberg RS. Environment and genes: what is the interaction? Dig Dis. 2016;34:20–6.
McCole DF. IBD candidate genes and intestinal barrier regulation. Inflamm Bowel Dis. 2014;20:1829–49.
Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549.
Ruffolo C, Scarpa M, Faggian D, et al. Subclinical intestinal inflammation in patients with Crohn’s disease following bowel resection: a smoldering fire. J Gastrointest Surg. 2010;14:24–31.
Gassler N, Rohr C, Schneider A, et al. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am J Physiol Gastrointest Liver Physiol. 2001;281:G216–28.
Zeissig S, Burgel N, Gunzel D, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72.
Di Narzo AF, Brodmerkel C, Telesco SE, et al. High-throughput identification of the plasma proteomic signature of inflammatory bowel disease. J Crohns Colitis. 2019;13:462–71.
Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Investig. 2005;115:695–702.
Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14:329–42.
Neurath MF, Finotto S, Glimcher LH. The role of Th1/Th2 polarization in mucosal immunity. Nat Med. 2002;8:567–73.
Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389:266–75.
Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14:269–78.
Lichtenstein GR. Comprehensive review: antitumor necrosis factor agents in inflammatory bowel disease and factors implicated in treatment response. Therap Adv Gastroenterol. 2013;6:269–93.
Friedrich M, Pohin M, Powrie F. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity. 2019;50:992–1006.
Lu K, Knutson CG, Wishnok JS, et al. Serum metabolomics in a Helicobacter hepaticus mouse model of inflammatory bowel disease reveal important changes in the microbiome, serum peptides, and intermediary metabolism. J Proteome Res. 2012;11:4916–26.
Storr M, Vogel HJ, Schicho R. Metabolomics: is it useful for inflammatory bowel diseases? Curr Opin Gastroenterol. 2013;29:378–83.
Agouridis AP, Elisaf M, Milionis HJ. An overview of lipid abnormalities in patients with inflammatory bowel disease. Ann Gastroenterol. 2011;24:181–7.
Fan F, Mundra PA, Fang L, et al. Lipidomic profiling in inflammatory bowel disease: comparison between ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2015;21:1511–8.
Titz B, Gadaleta RM, Lo Sasso G, et al. Proteomics and lipidomics in inflammatory bowel disease research: from mechanistic insights to biomarker identification. Int J Mol Sci. 2018;19:2775.
Scoville EA, Allaman MM, Brown CT, et al. Alterations in lipid, amino acid, and energy metabolism distinguish Crohn’s disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics. 2018;14:17.
Murgia A, Hinz C, Liggi S, et al. Italian cohort of patients affected by inflammatory bowel disease is characterised by variation in glycerophospholipid, free fatty acids and amino acid levels. Metabolomics. 2018;14:140.
Contijoch EJ, Britton GJ, Yang C, et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. Elife. 2019;8:e40553.
Kumar M, Singh P, Murugesan S, et al. Microbiome as an immunological modifier. Methods Mol Biol. 2020;2055:595–638.
Ni J, Wu GD, Albenberg L, et al. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14:573–84.
Waldschmitt N, Metwaly A, Fischer S, et al. Microbial signatures as a predictive tool in IBD-pearls and pitfalls. Inflamm Bowel Dis. 2018;24:1123–32.
Bercovici S, Meek C, Wexler Y, et al. Estimating genome-wide IBD sharing from SNP data via an efficient hidden Markov model of LD with application to gene mapping. Bioinformatics. 2010;26:i175–82.
Fang K, Grisham MB, Kevil CG. Application of comparative transcriptional genomics to identify molecular targets for pediatric IBD. Front Immunol. 2015;6:165.
Acknowledgements
We thank Dr. Susie Huang and Dr. Mamoun Elawad for reviewing the manuscript.
Funding
This work was made possible by grant# NPRP10-0125-170242 received from the Qatar National Research Fund (a member of Qatar Foundation).
Author information
Authors and Affiliations
Contributions
All authors have contributed equally to writing this manuscript. Guarantor of the article: SAK. Specific author contributions: SAK had the idea for the review, MK and MG wrote the first draft. SAK critically revised the last version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors reviewed and approved the final version for submission.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kumar, M., Garand, M. & Al Khodor, S. Integrating omics for a better understanding of Inflammatory Bowel Disease: a step towards personalized medicine. J Transl Med 17, 419 (2019). https://doi.org/10.1186/s12967-019-02174-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-02174-1