
Abstract
Lung neoplasms are the leading cause of death by cancer worldwide. Non-small cell lung cancer (NSCLC) constitutes more than 80% of all lung malignancies and the majority of patients present advanced disease at onset. However, in the last decade, multiple oncogenic driver alterations have been discovered and each of them represents a potential therapeutic target. Although KRAS mutations are the most frequently oncogene aberrations in lung adenocarcinoma patients, effective therapies targeting KRAS have yet to be developed. Moreover, the role of KRAS oncogene in NSCLC remains unclear and its predictive and prognostic impact remains controversial. The study of the underlying biology of KRAS in NSCLC patients could help to determine potential candidates to evaluate novel targeted agents and combinations that may allow a tailored treatment for these patients. The aim of this review is to update the current knowledge about KRAS-mutated lung adenocarcinoma, including a historical overview, the biology of the molecular pathways involved, the clinical relevance of KRAS mutations as a prognostic and predictive marker and the potential therapeutic approaches for a personalized treatment of KRAS-mutated NSCLC patients.
Similar content being viewed by others
Background
Lung cancer is the most common cancer worldwide both in terms of incidence (1.8 million new cases estimated in 2012) and mortality (1.6 million annual deaths). In fact, lung cancer is the leading cause of death by cancer [1, 2]. Non-small cell lung cancer (NSCLC) comprises about 80% of all lung cancer cases [3]. When patients are diagnosed in early stages of NSCLC the survival rates are relatively higher after surgical resection [4]. However, at the time of diagnosis, the majority of patients have already developed advanced disease and the median survival barely exceeds 18 months from diagnosis [5]. Patients with untreated metastatic NSCLC present an overall survival (OS) rate at one year of only 10%, with a median survival of around 4 to 5 months. Classically, chemotherapy has demonstrated a slight improvement in the survival of patients with advanced NSCLC, reducing symptoms and improving the quality of life. In fact, the effect of the combination of different chemotherapeutic agents with a platinum compound in patients with advanced disease observed no significant differences between the different doublets tested [6]. Those poor results have been significantly improved over the last decade through different therapeutic strategies such as the incorporation of a third antiangiogenic drug to a platinum-based doublet [7], the combination of cisplatin with the antifolate drug pemetrexed [8] and the implementation of pemetrexed maintenance monotherapy after tumor response or stabilization induced by a platinum-based doublet [9].
Also during the last decade, a number of genetic alterations have been described in NSCLC, being Kristen Rat Sarcoma viral oncogene (KRAS), Epidermal Growth Factor Receptor (EGFR) and Anaplastic Lymphoma Kinase (ALK) the most commonly altered oncogenes acting as tumor genomic drivers [10]. The use of targeted therapies in NSCLC individuals with an actionable driver, as EGFR and ALK, has shown high clinical efficacy in comparison with patients in whom no molecular targets for a personalized therapy are identified [11]. In contrast, regarding KRAS oncogene, although the KRAS-MAPK pathway is downstream of EGFR signaling, KRAS-mutation driven lung cancers, which are mainly adenocarcinomas, do not respond to EGFR tyrosine kinase inhibitors (TKIs) [12]. Moreover, KRAS activation is one of the signaling pathways involved in resistance to EGFR TKIs and monoclonal antibodies. In spite of the EGFR inhibition by TKIs, KRAS activation allows the downstream signaling mediated by EGF [13].
Previous studies have reported that the occurrence of EGFR and KRAS mutations is strictly mutually exclusive and each of these genetic alterations is associated with specific clinical characteristics such as pathological features, clinical background and prognostic or predictive implications [14, 15]. Nevertheless, recent studies have described concomitant genetic alterations such as EGFR or Echinoderm microtubule-associated protein-like 4 (EML4) ALK translocation with KRAS (EGFR/KRAS or EML4-ALK/KRAS), most of them associated with an acquired mutation after treatment that promotes drug resistance [16, 17]. Tumor heterogeneity according to which different mutations may coexist in different tumor cells or in the same tumor cell could explain this phenomenon of concomitance. EML4-AKT/KRAS double alteration represents the most common concomitant genomic aberration and is associated with poor prognosis and resistance to anti-ALK agents. Tumors with concomitant EGFR/KRAS mutation usually show the typical histologic patterns and cell characteristics of EGFR-mutated tumors and correlates with a better response to EGFR-TKIs therapy [10, 18, 19].
Effective therapies against KRAS have not been developed yet. Indeed, NSCLC adenocarcinoma patients with tumors harboring KRAS mutations, that account for 25% of the cases, show a shorter median survival (2.41 years) compared to patients candidates to personalized therapies [20, 21]. Despite all clinical advances regarding personalized therapy, there is still a highly remarkable unmet clinical need since a very well-known and highly prevalent tumor driver mutation in NSCLC patients, such as KRAS, still remains refractory to pharmacological inhibition.
Historical overview
The ability of single-stranded murine sarcoma virus, Kirsten and Harvey, to transform normal mammalian genes into potent oncogenes was discovered over four decades ago [22]. These viral oncogenes were only able to generate rat sarcomas for what they were called RAS genes, KRAS and HRAS alluding to its discoverers [23, 24]. It was not until 1982 when new human sequences homologous to the HRAS and KRAS oncogenes were identified in human bladder and lung carcinoma cell lines, respectively [25, 26]. The third member of the human RAS gene family, designated as NRAS, was described in human sarcoma cell lines in 1983 [27, 28].
Mariano Barbacid’s group first established the relationship between RAS genes and lung cancer in 1984. They conducted a landmark study which evidenced the presence of an activating mutation of KRAS oncogene in a human lung cancer specimen that was not observed in normal tissue of the same patient [29]. Soon after, the prevalence of mutational KRAS activation in lung cancers, specifically in NSCLC, was demonstrated [30]. KRAS mutations have been found to be almost an exclusive feature of adenocarcinomas and are more frequent in Western populations. Pooled frequencies of KRAS mutations range from 6.7% to 40.0% for ever/heavy smokers and from 2.9 to 11.4 for never/light smokers [31]. During the following two decades, studies of RAS focused on its biology and biochemical characteristics both in normal and cancer cells, as well as in the signaling cascade in which RAS is involved [32]. Nevertheless, despite the increase in systematic studies of the RAS oncogene, no clinically applicable therapeutic inhibition has proven to be successful for over 30 years. After multiple failed attempts to inhibit RAS either directly or indirectly (downstream effectors and post-transcriptional modifications), ‘The RAS Initiative’ arose (2013), to facilitate connections among RAS researchers to promote new ideas and technologies to bear on RAS. Even so, RAS inhibition and the development of novel therapies remain an unmet clinical need [33,34,35,36,37].
KRAS biology
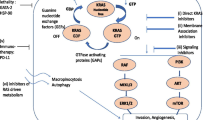
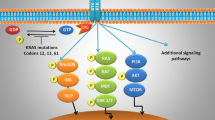
RAS proteins, including KRAS, are intracellular guanine nucleotide binding proteins (G proteins) which belong to the family of small GTPases. G proteins are composed of a G or catalytic domain, which binds guanine nucleotides and activates signaling, and a C-terminal hypervariable region (HVR) that incorporates farnesyl or prenyl groups (post-transcriptional modifications). These modifications diverge in each isoform because of the sequence variability of the HVR and locate RAS proteins to the cell membrane, where they perform their signaling function [38, 39]. The downstream signaling is regulated by two alternative states of RAS proteins: RAS-GTP (active form) and RAS-GDP (inactive form). RAS-GTP complex activates several downstream signaling effectors such as the canonical Raf-MEK-ERK, the PI3K-AKT-mTOR and RalGDS-RalA/B pathways or the TIAM1-RAC1 pathway (Fig. 1), which control multiple cellular functions including proliferation, apoptosis, motility or survival. These signaling cascades are triggered by coupling of several growth factor receptors like EGFR that favor a constitutive activation of KRAS [33, 40,41,42] (Fig. 1). The exchange of GDP-GTP is regulated by additional proteins: Guanine nucleotide exchange factors (GEFs) decrease the affinity of RAS proteins for GDP and favor GTP binding that results in RAS activation, while GTPase-activating proteins (GAPs) accelerate the intrinsic GTPase activity to regulate the RAS cycle [43, 44].

RAS downstream signaling pathways and potential options for therapeutic intervention in lung adenocarcinoma
Most mutations in RAS genes affect exons 2 and 3. These mutations impair the GTPase activity promoting the active GTP-bound state. Generally, the G➔A transition in codons 12 or 13 is dominant in KRAS isoform resulting in G12D or G13D mutations, followed by G➔T transversions that produce G12V [45, 46]. The most frequent mutation in KRAS mutant NSCLC is G12C (41%). It has been proposed that KRAS-mutated tumors behave similarly to the KRAS, EGFR and ALK-native tumors with respect to sites of metastases [47]. However, this may reflect biological heterogeneity, as it has been suggested that the type of point mutation may affect downstream signaling differently, which may translate into different clinical features [48,49,50]. G12C and G12V mutations are usually associated with the RalA/B signaling pathway and both of them present shorter progression free survival (PFS). Patients harboring G12C mutation are more likely to present bone metastases dissemination, while pleuro-pericardial metastases are more frequent in those with G12V mutations. However, KRAS G12D mutations preferably activate PI3K and MEK signaling [51, 52]. Furthermore, concurrent mutations of tumor suppressor genes in KRAS-mutant adenocarcinoma patients (e.g. TP53, LKB1 or KEAP1) should be taken into account because such mutational pattern is related to the control of distinct tumorigenic pathways [53,54,55,56,57]. Thus, tumors initiated with the same oncogenic driver may require different therapeutic approaches. In addition, recent work has established two different groups of KRAS mutant NSCLC, KRAS-dependent or KRAS-independent, according to their requirement for mutant KRAS to maintain viability. Gene expression profiles of NSCLC cell lines show a gene expression signature in KRAS-dependent cells associated with a well-differentiated epithelial phenotype, whereas KRAS independency correlated with an epithelial-mesenchymal transformation (EMT) phenotype. These data suggest that there are specific pathways and activated genes according to the KRAS dependency that have an important role in the different cancer phenotypes and their potential treatments [58, 59].
KRAS mutations as a prognostic factor
Although KRAS mutations have been classically defined as a negative prognostic factor with more undifferentiated tumors having unfavorable survival rates and disease-free survival compared to KRAS wild-type tumors [60], its real clinical significance remains controversial due to heterogeneity amongst studies. Table 1 summarizes the available data of the prognostic value of KRAS status in early and advanced NSCLC.
In view of disparity from individual studies, several meta-analysis have been conducted. A meta-analysis of 28 studies and 3620 patients demonstrated the negative prognosis of KRAS in lung adenocarcinomas, but not in squamous-cell carcinoma histology tumors [61]. However, it must be noted that EGFR-mutations, which are well-known to have a better prognosis and be, in general, mutually exclusive, were not taken into account, leading to a possible overestimation in the control arm.
In 2013, a different meta-analysis conducted by the Lung Adjuvant Cisplatin Evaluation (LACE)-BIO collaborative group that included data from four clinical trials (ANITA, IALT, JBR.10, and CALGB- 9633) was published [62]. No significant differences in the prognostic value neither in the overall group nor when patients were divided by histology were found.
In 2015, pooled data from four trials of EGFR TKIs versus placebo (National Cancer Institute of Canada Clinical Trials Group [NCIC CTG] trial BR.21, TOPICAL, NCIC CTG trial BR.26, and NCIC CTG trial BR.19) including known KRAS status for 1362 of 2624 patients, found no statistically significant differences in OS in the placebo arms between patients harboring KRAS mutations and those with KRAS native status [63].
However, another meta-analysis of 41 studies has described the negative prognostic value of KRAS mutations, showing a worse OS and disease-free survival (DFS) when mutations are present [64]. Furthermore, a recently published meta-analysis exploring the prognostic value of KRAS mutations in circulating tumor DNA indicated a worse PFS and OS in patients harboring KRAS mutated genotypes [65].
Concerning KRAS mutation subtypes, retrospective studies have shown that patients with early stage and advanced NSCLC harboring G12C KRAS mutations had significantly shorter OS compared to other KRAS mutations [66, 67]. In this cohort, there were no differences between both groups for PFS.
In addition to the prognostic impact of the presence of KRAS mutations, concurrent mutations in other genes may have an added prognostic value. On the one hand, EML4-ALK fusion has been proven to be associated with poor prognosis when KRAS mutations are also co-present [19]. On the other hand, KRAS mutated NSCLC patients harboring mutations in the tumor suppressor genes STK11/LKB1 or CDKN2A show a worse prognosis than those with TP53 mutations [53].
KRAS mutations as a predictive factor
Predictive value of KRAS mutations for response to chemotherapy
Most patients with advanced lung cancer receive treatment with chemotherapy regimens based on platinum. KRAS status has been studied in this clinical setting as a biomarker to predict the expected clinical outcome to chemotherapy. However, data to support the predictive value of KRAS mutations in this specific clinical scenario are limited. Table 2 summarizes the predictive value of KRAS mutations for response to therapies.
In 1997, Rodenhuis et al. assessed the influence of KRAS mutations on the response to chemotherapy (carboplatin, ifosfamide and etoposide) in the metastatic setting [68]. Response rate and median OS did not differ according to KRAS status. Neither Schiller et al. found differences in OS when assessing the potential benefit of postsurgical chemotherapy (cisplatin and etoposide) added to thoracic radiation in patients with stage II and IIIA NSCLC according to KRAS status [69]. In 2005, results from the phase III, Tarceva Responses in Conjunction with Paclitaxel and Carboplatin (TRIBUTE) trial in advanced NSCLC comparing first-line carboplatin/paclitaxel plus erlotinib or placebo were published [70]. Response rate, median time to progression and median OS did not differ either between mutant and wild-type tumors.
The results of the JBR10 trial, which studied the effect of postoperative chemotherapy (vinorelbin and cisplatin) in patients with resected stage IB or II NSCLC were reported in 2010 [71]. Significant benefit from chemotherapy was reported in KRAS wild-type patients receiving chemotherapy, whereas there were no differences in the KRAS mutant group. The p value for the interaction analysis was 0.29, showing no statistical significance, meaning that KRAS status has no value as a predictor of survival in patients treated with adjuvant chemotherapy.
The phase III IFCT-0002 trial compared two chemotherapy regimens (carboplatin and paclitaxel vs cisplatin and gemcitabine) and two sequences of chemotherapy (neoadjuvant vs perioperative) in stage I and II NSCLC [72]. Univariate analyses showed that KRAS status was associated with response to chemotherapy. However, this association was not significant in the multivariate analysis.
Nonetheless, in a recently published retrospective analysis of a cohort of patients with advanced NSCLC, patients harboring KRAS activating mutations exhibited a lower proportion of responses to cytotoxic chemotherapy and decreased survival compared to patients harboring native KRAS [73]. Co-mutation of TP53 and KRAS has also been studied, showing worse OS in patients harboring co-mutation versus double wild type tumors [74]. The induction of a different sensitivity pattern depending on the specific KRAS mutation has been studied in the preclinical scenario by generation of NSCLC cell lines overexpressing the three most common amino acid substitutions (G12C, G12V and G12D) leading to the KRAS-mutated proteins [75]. Whereas the expression of G12V shows resistance to paclitaxel and sensitivity to sorafenib, the expression of G12C is related to reduced response to cisplatin and sensitivity to pemetrexed and paclitaxel. G12V mutations resulted in resistance to pemetrexed and sensitivity to cisplatin. There was no correlation between KRAS mutations and response to gemcitabine and EGFR inhibitors. Overall, studies published to date about the predictive value of KRAS mutations show no consistent data and therefore, KRAS status should not be used as a predictive factor to select patients for specific chemotherapy regimens.
Predictive value of KRAS mutations for response to targeted therapy
KRAS has been widely studied as a predictive biomarker for response to targeted agents, in clinical trials involving anti-EGFR therapies in NSCLC. Two meta-analyses evaluating erlotinib and gefitinib have suggested a negative predictive value of KRAS-mutated tumors harboring EGFR activating mutations treated with EGFR TKIs [76, 77]. However, these data are confused by the fact that KRAS mutations result in persistent activation of the EGFR-RAS-RAF-ERK-MEK pathway, even when EGFR is inhibited. When excluding EGFR-mutated tumors from the analyses, data are controversial. While some investigations have not found statistically significant differences in terms of overall response rate (ORR) or PFS according to KRAS mutation status [78], others have found KRAS to be a negative predictor for EGFR-TKI treatment [79].
A potential predictive value of particular KRAS mutation subtypes has also been postulated. Patients harboring the dominant G12C KRAS mutation had shorter PFS and OS than those with non-G12C KRAS mutations in a subgroup of 38 patients harboring a mutated KRAS gene and wild-type EGFR gene who were treated with erlotinib or genitinib [80]. Patients harboring other KRAS mutations than the G12C type showed similar PFS and OS to patients harboring the wild-type-KRAS, wild-type EGFR genotype. Codon 13 mutations also seem to confer a worse outcome than codon 12 mutations [81].
Another recently published pooled analysis including four trials testing EGFR TKIs documented an OS benefit among patients with tumors showing the G12D/S mutation, whereas treatment with EGFR TKIs resulted harmful for those with the G12V mutations [63].
In contrast to KRAS mutant colorectal cancer, where KRAS mutations are predictive of poor response to anti-EGFR monoclonal antibodies, cetuximab and panitumumab, BMS099 and FLEX clinical trials demonstrated no statistically significant association between KRAS status and ORR, PFS, or OS when cetuximab was added to platinum-based chemotherapy in patients with advanced NSCLC [82, 83].
Furthermore, KRAS mutations have also been described as potential biomarkers for response to immune checkpoint inhibitors through alteration of a group of genes involved in cell cycle regulating, DNA replication and damage repair. A remarkable clinical benefit to PD-1 inhibitors has been showed in TP53, KRAS or TP53/KRAS patients [84]. These evidences are related to the positive correlation between KRAS mutation and PD-L1 expression in lung adenocarcinoma, which represents the innate immune resistance. PD-L1 seems to be up-regulated in models of NSCLC with mutation in the KRAS oncogene through p-ERK, hence PD-1 inhibitors as pembrolizumab or ERK inhibitor might recover the tumor immunity of CD3+ T cells, that normally become apoptotic and promote the immune scape [85].
Targeted therapies for KRAS-mutant lung cancer
To date, no efforts at targeting KRAS have proven to be successful (Table 3). Moreover, different strategies for direct inhibition of specific KRAS-mutated proteins using several strategies such as an irreversible allosteric inhibitor of G12C RAS to prevent GTP-KRAS formation [37], compounds that target the guanine nucleotide binding pocket (SML-8-73-1) [86] or allele-specific inhibitors (ARS-853) [87, 88] have been reported. Although these compounds showed great specificity towards inhibition of mutant KRAS tumors in vitro and provide proof-of-concept of direct KRAS inhibition, long-term efficacy as well as toxicity remains as standing hurdles. Therefore, currently non-specific chemotherapy with conventional cytotoxic drugs remains as the standard treatment for KRAS-driven lung cancers.
Indirect strategies for targeting KRAS pathway have been investigated as well. Considering that, KRAS mutations result in activation of the cascade RAF-MEK-ERK and NF-kB, potential targeted therapies for KRAS-mutant lung cancers have focused on inhibiting downstream effectors of this signaling pathway (Fig. 1). Constitutive activation of KRAS leads to the persistent stimulation of downstream signaling pathways that promote tumorigenesis and maintains the oncogenic phenotype, including the PI3K/AKT/mTOR cascade, RHO-FAK pathways and overexpression of MET receptor. Inhibition of these cascades has been tested in preclinical and clinical models.
Farnesyl transfeRASe inhibitors
Given that KRAS ought to be farnesylated to localize in cell membrane, strategies to prevent these post-translational modification have been developed. Despite promising in vitro and in vivo results in preclinical models demonstrated that farnesyl transferase inhibitors (FTI) such as tipifarnib (R115777) or salirasib could prevent the development of lung tumors [89, 90], phase II trials using FTI failed to show clinical activity.
Tipifarnib was tested in 44 patients with advanced NSCLC [91]. Although in vivo activity of tipifarnib in patient tissues was documented, it only translated into a modest clinical activity, since no objective complete or partial responses were seen and only seven patients experienced disease stabilization for at least 6 months.
In a phase II trial testing salirasib, among the 33 patients with advanced lung adenocarcinoma enrolled, 30 showed tumors harboring KRAS mutations (23 previously treated patients and 7 treatment-naïve individuals) [92]. Among the 23 previously treated patients, 30.4% (7/23) showed stable disease at 10 weeks with a median time to progression of 2 months. Median time to progression to first line salirasib was 1 month, with a 40% stable disease rate.
BRaf inhibitors
Clinical attempts to block downstream KRAS signaling pathways through Raf inhibition also yielded disappointing results.
BRaf inhibitors used against BRaf-mutated melanomas, such as vemurafenib or dabrafenib, are unlikely to prove any meaningful clinical effect as targeted agents in KRAS-mutated NSCLC, since KRAS and BRaf activating mutations are mutually exclusive [93]. More importantly, inhibition of activating BRaf mutations in mutant KRAS tumors induces Erk phosphorylation in a Craf-dependent manner to promote tumorigenesis, in what is known as the MAPK paradox [94, 95], thus discouraging the use of BRaf inhibitors in oncogenic KRAS tumors.
Alternatives include inhibition of other Raf members critical for mutant KRAS-driven NSCLC [96]. Sorafenib, an oral multi-tyrosine kinase inhibitor that targets Raf and related transmembrane receptors, was seen to induce CRaf depletion and, secondarily, inhibit cell growth and induce G1 arrest in NSCLC KRAS-mutant cells [97]. However, clinical attempts to inhibit Raf using sorafenib have been disappointing. A phase II clinical trial testing sorafenib in patients with advanced NSCLC who had progressed to at least one platinum-containing regimen showed disease control in 53% of the 57 patients enrolled, but only 9% experienced a documented radiologic response to the treatment [98]. In the MISSION trial, a phase III multicenter, placebo-controlled study that tested sorafenib in patients with relapsed or refractory non-squamous NSCLC after 2 or 3 previous chemotherapy regimens, PFS but not OS was significantly longer in both mutated and wild type-KRAS patients [99]. In the BATTLE trial (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination), sorafenib achieved a better disease control rate in mutant-KRAS patients (61% versus 32%) compared with the combined other treatments (erlotinib, vandetanib or erlotinib) in chemorephractory NSCLC patients. However, these differences were not statistically significant (p = 0.11) [100].
MEK inhibitors
Several agents targeting MEK, which acts downstream of KRAS (Fig. 1), to suppress signaling through the mitogen-activated protein kinase (MAPK) cascade seem to have greater antitumor activity in tumors harboring RAS or BRaf mutations [101], whose proliferation and survival rely on the activation of the RAF-MEK-ERK pathway.
Despite their preclinical activity, first clinical trials using MEK inhibitors as CI-1040 [102], RO5126766 [103, 104] and PD-0325901 [105] in non-selected populations of different tumors types harboring KRAS mutations yielded disappointing results, more likely due to activation of resistance mechanisms through compensatory signaling effectors [106].
Based on their preclinical activity, clinical trials testing more recently developed MEK inhibitors, such as selumetinib and trametinib have been also conducted.
A phase II trial comparing single agent selumetinib (AZD6244 or ARRY-142886) versus pemetrexed in previously treated patients with advanced NSCLC and unreported KRAS status showed no significant clinical benefit in terms of RR or median PFS [107]. Preclinical data demonstrated that AZD6244 has potential to inhibit tumor proliferation, induce differentiation and apoptosis activity in KRAS-mutant xenograft models and that antitumor efficacy was improved by combining with cytotoxic drugs as docetaxel [108]. Based on this, another phase II trial testing the synergistic effect of adding selumetinib to docetaxel in previously treated KRAS-mutant patients was conducted [109]. It showed clinical benefit in PFS and ORR, whereas no improvement in OS and more toxicity was recorded in the selumetinib-docetaxel arm. A subgroup analysis of KRAS-mutations subtypes documented that patients harboring G12V and G12C mutations seemed to experience higher RR and PFS was longer for the combination arm [110]. A non-significant trend toward longer survival was seen in the G12C mutation subgroup. Clinical trials with a G12V and G12C mutations selected population have not been performed yet. The phase III trial did not confirmed the efficacy data, with no improvement in RR, PFS and OS in the combination arm [111].
Combination of selumetinib and erlotinib in NSCLC patients who had progressed to one or prior regimens was also studied in a phase II trial, where patients with neoplasms harboring KRAS mutations were randomized to selumetinib in monotherapy or to the combination arm, whereas patients with KRAS wild-type tumors were randomized to either erlotinib or the combination therapy. In KRAS mutant NSCLC patients, no responses were seen in the monotherapy cohort, and the combination therapy failed to show significant improvement in PFS or ORR and caused more adverse events [112].
Trametinib has also been tested in clinical trials, with similar results. In a phase II trial, the use of trametinib in monotherapy compared to docetaxel in previously treated KRAS driven NSCLC showed similar PFS and RR in both groups [113]. Other combinations of trametinib with gemcitabine, pemetrexed and docetaxel have been tested in phase Ib clinical trials concluding that further investigations are warranted in order to demonstrate their clinical activity [114,115,116,117].
Phase I and II clinical trials using MEK inhibitors in combination with other therapies are still recruiting patients or under evaluation in KRAS-mutated NSCLC (NCT02964689 evaluating binimetinib in addition to standard chemotherapy; NCT01859026 studying MEK162 in combination with erlotinib; NCT02022982 investigating palbociclib and PD-03259019).
NF-kB pathway
Preclinical studies have provided evidence of the dependence of NF-kB pathway of tumor cells harboring KRAS mutations for their viability. Activity modulation of NF-kB by preventing the degradation of NF-kB inhibitor (IkB) using proteasome inhibitors or knocking down TKB1 (an IkB kinase that enhances NK-kB) translates into apoptosis of KRAS-mutated cells [118].
According to these data, a phase II single-institution clinical trial (NCT01833143) testing subcutaneous bortezomib, a downregulator of the NF-kB pathway, in patients with advanced NSCLC harboring KRAS G12D mutation or no past smoking history is ongoing at Memorial Sloan-Kettering Cancer Center. Partial results were presented at 2015 ASCO Annual Meeting, showing modest antineoplastic activity. Indeed, regarding the RR, one partial response and six stabilizations of disease of a total of 16 patients enrolled where reported [119].
MET inhibitors
A phase II trial comparing erlotinib alone or in combination with the MET inhibitor tivantinib (ARQ 197) in previously treated EGFR TKI-naïve unselected advanced NSCLC failed to demonstrate clinical benefit in PFS in this cohort [120]. Nevertheless, exploratory analyses demonstrated significant differences in PFS in the subgroup of patients harboring KRAS mutations. A phase III clinical trial named MARQUEE, compared erlotinib and tivantinib with erlotinib and placebo in patients with locally advanced or metastatic, nonsquamous NSCLC and stratified the cohort by KRAS and EGFR status [121]. Despite an improvement in PFS was shown, the Data Monitoring Committee closed the study prematurely, because the data had crossed the futility boundary. Subgroup results concerning the cohort of KRAS-mutant patients have not been reported yet.
Onartuzumab, a monoclonal antibody that targets MET receptor, has also been tested in combination with erlotinib in molecularly unselected recurrent NSCLC patients [122]. In this phase II clinical trial, although there were no statistically significant differences in RR, PFS or OS between both arms (onartuzumab plus erlotinib vs. o placebo plus erlotinib), significant differences in PFS and OS between groups were observed in favor of the MET-positive group. In an exploratory subgroup analysis concerning potential predictive biomarkers, no responses were observed in the group of patients harboring KRAS mutations [123].
Targeting PI3K-AKT-mTOR pathway
It has been demonstrated that KRAS mutations can coexist with PI3K activation in tumors at the same time [124]. However, based on preclinical data, monotherapy with PI3K inhibitors seems to be insufficient in tumors harboring KRAS mutations as the RAF-MEK-ERK pathway hijacks tumor growth through compensatory mechanisms.
The blockade of the pathway using mTOR inhibitors, arresting tumor cells in G1 phase, has also been tested [125]. Ridaforolimus, an mTOR inhibitor, has been tested in a phase II trial in advanced NSCLC harboring KRAS mutations [126]. Although PFS was significantly improved, RR was only 1% in the ridaforolimus arm in compared with the placebo arm and no significant differences in OS were identified.
In order to block KRAS signaling completely, preclinical studies had suggested dual inhibition of PI3K/AKT/mTOR and BRAF/MEK/ERK pathways as an effective approach [127]. This modality has also been studied in the clinic [128]. Phase I trials in unselected advanced solid tumors using PI3K combined with either MEK or mTOR inhibitors are now under evaluation [129,130,131,132]. Although no preliminary data in KRAS-mutated population have been reported yet, important toxic effects could be anticipated given the importance of these two signaling pathways in normal cells homeostasis.
Targeting FAK
The RHOA-FAK pathway, involved in cell migration, has also proved to play an important role in some KRAS-mutated tumors, in which the mutation of KRAS added to inactivation of the tumor suppressor genes INK4a/ARF/p16, leads to hyperactivation of the GTPase RHOA by MEK1/2 and ERK1/2 [133]. Despite the absence of specific drugs targeting RHOA, FAK inhibitors have been developed. Defactinib, a FAK inhibitor V2–6063, is being tested in heavily pretreated patients with KRAS-mutant NSCLC in an ongoing clinical trial. Partial results were presented at the 16th World Conference on Lung Cancer in 2015, showing a 12-weeks PFS of 36%, but efficacy did not appear to correlate with INK4a/ARF/p16 status [134].
Targeting HSP90
Inhibition of heat shock proteins has been tested as another potential therapeutic strategy in the KRAS-mutated NSCLC scenario. The molecular chaperone Hsp90 is required for proteins’ stability and maturation and protection from proteasomal degradation. Many of these proteins are signaling transduction proteins, such as EGFR, RAF, AKT or products of mutated overexpressed oncogenes that maintain the oncogenic phenotype. Therefore, inhibition of heat shock proteins results in blockade of multiple oncogenic signaling pathways in tumor cells [135]. Treatment with ganetespib, an Hsp90 inhibitor, of KRAS-mutated cells resulted in decrease levels of EGFR, MET and CRAF, leading to inactivation of the RAS/RAF/MEK/ERK and PI3K/AKT pathways resulting consequently in apoptosis [136].
Clinical trials using ganetespib in monotherapy or in combination with other drugs, such as chemotherapy, MEK inhibitors, PI3K/mTOR inhibitors or mTOR inhibitors have been tested as well, with disappointing results in the KRAS-mutated setting [136,137,138].
Other strategies
Additional therapeutic strategies for mutant KRAS NSCLC such as reovirus type 3 [139], docetaxel nanoparticles (NCT02283320) or abemaciclib (a cell cycle inhibitor selective for the cyclin-dependent kinases CDK4 and CDK6) [140] are currently under development. Preliminary data analysis of a phase III trial testing abemaciclib in monoteraphy in KRAS-mutated advanced NSCLC did not meet its primary endpoint of OS (not published yet) .
Preclinical data raises interest in some of these therapies, as CDK4 had been identified as a synthetic lethal partner with KRAS oncogene in a study that shows genetic and pharmacological evidence demonstrating the role of CDK4 in proliferation of KRAS-mutant lung cells [141].
In addition, with the advent of checkpoint inhibitors, and given the high burden of neo-antigens associated to KRAS-mutated NSCLC, the use of immunotherapy in KRAS-mutated NSCLC appears as a novel therapeutic option with promising results. In fact, KRAS mutations, in conjunction with TP53 mutations, have been recently proposed as biomarkers to predict clinical benefit from PD-1/PD-L1 blockade [84]. Moreover, the ineffectiveness of immunotherapy in KRAS/LKB1 patients has been described and associated with a marked increase in inflammatory cytokines that recruit neutrophils and block T cells [142]. Complementary to the previous therapeutic strategies, many preclinical investigations have been carried out or are under way with the aim of discovering potential therapeutic targets for the treatment of KRAS-activated NSCLC adenocarcinoma patients. Among them, loss-of-functions screens have spearheaded the identification of KRAS dependencies or synthetic lethal interactions in the last decade. These have unveiled molecular targets potentially amenable to therapeutic intervention such as PLK1 [143], TBK1 [118]], BCL-XL [144], FAS [145] and XPO1 [146]. Other approaches have focused on gene-expression analyses of early events in oncogenesis, building upon the premise that inhibition of such events could attenuate tumor growth and relapse [147]. These studies led to the identification of the kinase receptor DDR1 [148] and the transcription factor FOSL1 [149] as KRAS vulnerabilities in mutant KRAS tumors. Notably, these studies provided the rationale for combinatorial approaches involving either inhibition of DDR1 and Notch signaling [148] or inhibition of the FOSL1 target AURKA and MEK [148], both of which blocked tumor initiation and progression as well as induced tumor regression. Additionally, chemical screens have unveiled further options to treat mutant KRAS cancers using combinatorial strategies, which included the combination of IGFR1 and MEK inhibitors [150], TNKR and MEK inhibitors [151], or PLK1 and ROCK inhibitors [152]. Lastly, diverse research lines are currently open in this field, leading to the identification of promising unconventional therapeutic targets such as miR-1298 that inhibits tumor growth in KRAS-driven tumors [153] or the Inhibitor of Differention-1 (Id1) [154] that may have chemopreventive and therapeutic efficacy in KRAS-mutated lung tumors.
Conclusions
In conclusion, agents targeting driver oncogenic mutations in the advanced NSCLC setting have already changed the treatment paradigm. Given the high incidence of KRAS mutations in patients with NSCLC, this is a promising therapeutic target. However, KRAS is a heterogeneous entity and other coexisting alterations may be crucial for its role and biologic impact. Even though attempts to target KRAS pathway have shed little light so far, new molecules or new therapeutic strategies may revolutionize outcomes in patients with KRAS-driven NSCLC in the near future. Further investigations to better understand the pathways involved, to identify possible synthetic lethal partners and for a better patient selection are needed.
References
Ferlay J, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–6.
Blandin Knight S, et al. Progress and prospects of early detection in lung cancer. Open Biol. 2017;7(9):170070.
Wang T, et al. Five-year lung cancer survival: which advanced stage nonsmall cell lung cancer patients attain long-term survival? Cancer. 2010;116(6):1518–25.
Schiller JH, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346(2):92–8.
Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–50.
Scagliotti GV, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–51.
Paz-Ares L, et al. Maintenance therapy with pemetrexed plus best supportive care versus placebo plus best supportive care after induction therapy with pemetrexed plus cisplatin for advanced non-squamous non-small-cell lung cancer (PARAMOUNT): a double-blind, phase 3, randomised controlled trial. Lancet Oncol. 2012;13(3):247–55.
Lee T, et al. Non-small cell lung cancer with concomitant EGFR, KRAS, and ALK mutation: Clinicopathologic features of 12 cases. J Pathol Transl Med. 2016;50(3):197–203.
Kwak EL, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703.
Murray S, et al. Somatic mutations of the tyrosine kinase domain of epidermal growth factor receptor and tyrosine kinase inhibitor response to TKIs in non-small cell lung cancer: an analytical database. J Thorac Oncol. 2008;3(8):832–9.
Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–400.
Gainor JF, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–81.
Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010;29(1):49–60.
Wang S, et al. Potential clinical significance of a plasma-based KRAS mutation analysis in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16(4):1324–30.
Li S, et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br J Cancer. 2014;110(11):2812–20.
Zhang H, et al. Clinical outcome of epidermal growth factor receptor-tyrosine kinase inhibitors therapy for patients with overlapping kirsten rat sarcoma 2 viral oncogene homolog and epidermal growth factor receptor gene mutations. Thorac Cancer. 2016;7(1):24–31.
Ulivi P, et al. Nonsquamous, non-small-cell lung cancer patients who carry a double mutation of EGFR, EML4-ALK or KRAS: frequency, clinical-pathological characteristics, and response to therapy. Clin Lung Cancer. 2016;17(5):384–90.
Gao W, et al. KRAS and TP53 mutations in bronchoscopy samples from former lung cancer patients. Mol Carcinog. 2017;56(2):381–8.
Kris MG, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006.
Scolnick EM, et al. Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. J Virol. 1973;12(3):458–63.
Harvey JJ. An unidentified virus which causes the rapid production of tumours in mice. Nature. 1964;204:1104–5.
Kirsten WH, Mayer LA. Morphologic responses to a murine erythroblastosis virus. J Natl Cancer Inst. 1967;39(2):311–35.
Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses. Proc Natl Acad Sci U S A. 1982;79(11):3637–40.
Parada LF, et al. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297(5866):474–8.
Hall A, et al. Identification of transforming gene in two human sarcoma cell lines as a new member of the ras gene family located on chromosome 1. Nature. 1983;303(5916):396–400.
Shimizu K, et al. Three human transforming genes are related to the viral ras oncogenes. Proc Natl Acad Sci U S A. 1983;80(8):2112–6.
Santos E, et al. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223(4637):661–4.
Rodenhuis S, et al. Mutational activation of the K-ras oncogene. A possible pathogenetic factor in adenocarcinoma of the lung. N Engl J Med. 1987;317(15):929–35.
Dearden S, et al. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24(9):2371–6.
Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459–65.
Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21(8):1819–27.
Kilgoz HO, et al. KRAS and the reality of personalized medicine in non-small cell lung cancer. Mol Med. 2016;22
Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6(7):541–55.
Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–42.
Ostrem JM, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51.
Tetlow AL, Tamanoi F. The Ras superfamily G-proteins. Enzyme. 2013;33 Pt A:1–14.
Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294(5545):1299–304.
Takashima A, Faller DV. Targeting the RAS oncogene. Expert Opin Ther Targets. 2013;17(5):507–31.
Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15(14):R563–74.
Hingorani SR, Tuveson DA. Ras redux: rethinking how and where Ras acts. Curr Opin Genet Dev. 2003;13(1):6–13.
Hennig A, et al. Ras activation revisited: role of GEF and GAP systems. Biol Chem. 2015;396(8):831–48.
Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–77.
Karachaliou N, et al. KRAS mutations in lung cancer. Clin Lung Cancer. 2013;14(3):205–14.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67.
Doebele RC, et al. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer. 2012;118(18):4502–11.
Kim EY, et al. KRAS oncogene substitutions in Korean NSCLC patients: clinical implications and relationship with pAKT and Ra1GTPases expression. Lung Cancer. 2014;85(2):299–305.
Renaud S, et al. Specific KRAS amino acid substitutions and EGFR mutations predict site-specific recurrence and metastasis following non-small-cell lung cancer surgery. Br J Cancer. 2016;115(3):346–53.
Biernacka A, et al. The potential utility of re-mining results of somatic mutation testing: KRAS status in lung adenocarcinoma. Cancer Genet. 2016;209(5):195–8.
Ihle NT, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104(3):228–39.
Jia Y, et al. Characterization of distinct types of KRAS mutation and its impact on first-line platinum-based chemotherapy in Chinese patients with advanced non-small cell lung cancer. Oncol Lett. 2017;14(6):6525–32.
Skoulidis F, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860–77.
Schabath MB, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35(24):3209–16.
Krall EB, et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife. 2017;6:e18970.
Kottakis F, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539(7629):390–5.
Romero R, Sayin VI. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. 2017;23(11):1362–8.
Pass HI, et al. Reconstructing targetable pathways in lung cancer by integrating diverse omics data. Nat Med. 2013;4:2617.
Singh A, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500.
Slebos RJ, et al. K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. N Engl J Med. 1990;323(9):561–5.
Mascaux C, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92(1):131–9.
Shepherd FA, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31(17):2173–81.
Zer A, et al. Pooled analysis of the prognostic and predictive value of KRAS mutation status and mutation subtype in patients with non-small cell lung cancer treated with epidermal growth factor receptor tyrosine Kinase inhibitors. J Thorac Oncol. 2016;11(3):312–23.
Pan W, et al. KRAS mutation is a weak, but valid predictor for poor prognosis and treatment outcomes in NSCLC: a meta-analysis of 41 studies. Oncotarget. 2016;7(7):8373–88.
Fan G, et al. Prognostic value of EGFR and KRAS in circulating tumor DNA in patients with advanced non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(20):33922–32.
Svaton M, et al. The prognostic role of KRAS mutation in patients with advanced NSCLC treated with second- or third-line chemotherapy. Anticancer Res. 2016;36(3):1077–82.
Nadal E, et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J Thorac Oncol. 2014;9(10):1513–22.
Rodenhuis S, et al. Mutational activation of the K-ras oncogene and the effect of chemotherapy in advanced adenocarcinoma of the lung: a prospective study. J Clin Oncol. 1997;15(1):285–91.
Schiller JH, et al. Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small-cell lung cancer on E4592: a laboratory ancillary study on an eastern cooperative oncology group prospective randomized trial of postoperative adjuvant therapy. J Clin Oncol. 2001;19(2):448–57.
Eberhard DA, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900–9.
Tsao MS, et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol. 2007;25(33):5240–7.
Zalcman, G. et al. Use of RAS effector rAssF1A promoter gene methylation and chromosome 9p loss of heterozygosity (LOH) to predict progression-free survival (PFs) in perioperative chemotherapy (CT) phase iii trial iFCT-0002 in resectable non-small cell lung cancer [abstract].. ASCO Meeting Abstracts, 2008. 26.
Hames ML, et al. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer. 2016;92:29–34.
Shepherd FA, et al. Pooled analysis of the prognostic and predictive effects of TP53 Comutation status combined with KRAS or EGFR mutation in early-stage Resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2017;35(18):2018–27.
Garassino MC, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22(1):235–7.
Mao C, et al. KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies. Lung Cancer. 2010;69(3):272–8.
Linardou H, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962–72.
Garassino MC, et al. Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial. Lancet Oncol. 2013;14(10):981–8.
Ludovini V, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6(4):707–15.
Fiala O, et al. The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Genet. 2013;206(1–2):26–31.
Metro G, et al. Impact of specific mutant KRAS on clinical outcome of EGFR-TKI-treated advanced non-small cell lung cancer patients with an EGFR wild type genotype. Lung Cancer. 2012;78(1):81–6.
Khambata-Ford S, et al. Analysis of potential predictive markers of cetuximab benefit in BMS099, a phase III study of cetuximab and first-line taxane/carboplatin in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(6):918–27.
O'Byrne KJ, et al. Molecular biomarkers in non-small-cell lung cancer: a retrospective analysis of data from the phase 3 FLEX study. Lancet Oncol. 2011;12(8):795–805.
Dong ZY, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung Adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–24.
Chen N, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66(9):1175–87.
Hunter JC, et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A. 2014;111(24):8895–900.
Lito P, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–8.
Patricelli MP, et al. Selective inhibition of Oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6(3):316–29.
Gunning WT, et al. Chemoprevention of benzo(a)pyrene-induced lung tumors in mice by the farnesyltransferase inhibitor R115777. Clin Cancer Res. 2003;9(5):1927–30.
Zundelevich A, et al. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Mol Cancer Ther. 2007;6(6):1765–73.
Adjei AA, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21(9):1760–6.
Riely GJ, et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6(8):1435–7.
Heideman DA, et al. KRAS and BRAF mutation analysis in routine molecular diagnostics: comparison of three testing methods on formalin-fixed, paraffin-embedded tumor-derived DNA. J Mol Diagn. 2012;14(3):247–55.
Heidorn SJ, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–21.
Poulikakos PI, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30.
Blasco RB, et al. C-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19(5):652–63.
Takezawa K, et al. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009;69(16):6515–21.
Dingemans AM, et al. A phase II study of sorafenib in patients with platinum-pretreated, advanced (stage IIIb or IV) non-small cell lung cancer with a KRAS mutation. Clin Cancer Res. 2013;19(3):743–51.
Paz-Ares L, et al. Monotherapy Administration of Sorafenib in patients with non-small cell lung cancer (MISSION) trial: a phase III, multicenter, placebo-controlled trial of Sorafenib in patients with relapsed or refractory predominantly Nonsquamous non-small-cell lung cancer after 2 or 3 previous treatment regimens. J Thorac Oncol. 2015;10(12):1745–53.
Kim ES, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53.
Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439(7074):358–62.
Rinehart J, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–62.
Martinez-Garcia M, et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res. 2012;18(17):4806–19.
Honda K, et al. Phase I and pharmacokinetic/pharmacodynamic study of RO5126766, a first-in-class dual Raf/MEK inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;72(3):577–84.
Haura EB, et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16(8):2450–7.
Manchado E, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534(7609):647–51.
Hainsworth JD, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5(10):1630–6.
Davies BR, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209–19.
Janne PA, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14(1):38–47.
Janne PA, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer. 2015;113(2):199–203.
Janne PA, et al. Selumetinib plus Docetaxel compared with Docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: the SELECT-1 randomized clinical trial. JAMA. 2017;317(18):1844–53.
Carter CA, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann Oncol. 2016;27(4):693–9.
Blumenschein GR Jr, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol. 2015;26(5):894–901.
Infante JR, et al. A phase 1b study of trametinib, an oral Mitogen-activated protein kinase kinase (MEK) inhibitor, in combination with gemcitabine in advanced solid tumours. Eur J Cancer. 2013;49(9):2077–85.
Gandara DR, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with docetaxel in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. J Clin Oncol. 2013;31(15):1.
Kelly K, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with pemetrexed for KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. J Clin Oncol. 2013;31(15):1.
Gandara DR, et al. A phase 1/1b study evaluating Trametinib plus Docetaxel or Pemetrexed in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12(3):556–66.
Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12.
Litvak AM, et al. Phase II trial of bortezomib in KRAS G12D mutant lung cancers. J Clin Oncol. 2015;33(15):1.
Sequist LV, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–15.
Scagliotti GV, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer. 2012;13(5):391–5.
Spigel DR, et al. Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4105–14.
Koeppen H, et al. Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib+/−onartuzumab in advanced non-small cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res. 2014;20(17):4488–98.
Ihle NT, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69(1):143–50.
Wallin JJ, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10(12):2426–36.
Riely GJ, et al. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. J Clin Oncol. 2012;30(15):1.
Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6.
Simmons BH, et al. Combination of a MEK inhibitor at sub-MTD with a PI3K/mTOR inhibitor significantly suppresses growth of lung adenocarcinoma tumors in Kras(G12D-LSL) mice. Cancer Chemother Pharmacol. 2012;70(2):213–20.
LoRusso P, et al. A first-in-human phase Ib study to evaluate the MEK inhibitor GDC-0973, combined with the pan-PI3K inhibitor GDC-0941, in patients with advanced solid tumors. J Clin Oncol. 2012;30(15_suppl):2566.
Heist RS, et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: results of a phase Ib dose-escalation trial. J Clin Oncol. 2013;(15):31, 2.
Ramanathan RK, et al. A phase 1b trial of PI3K inhibitor copanlisib (BAY 80-6946) combined with the allosteric-MEK inhibitor refametinib (BAY 86-9766) in patients with advanced cancer. J Clin Oncol. 2014;32(15):1.
Juric D, et al. A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. J Clin Oncol. 2014;32(15):1.
Konstantinidou G, et al. RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov. 2013;3(4):444–57.
D.E. Gerber, R C., D. Morgensztern, J. Cetnar, R. Kelly, S.S. Ramalingam, D.R. Spigel, W. Jeong, P. Scaglioni, M. Li, M. Keegan, J. Horobin, T.F. Burns, Phase II Study of Defactinib, VS-6063, a Focal Adhesion Kinase (FAK) Inhibitor, in Patients with KRAS Mutant Non-Small Cell Lung Cancer (NSCLC). Paper presented at: 16th World Conference on Lung Cancer, Denver 2015.
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–72.
Acquaviva J, et al. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol Cancer Ther. 2012;11(12):2633–43.
Socinski MA, et al. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013;19(11):3068–77.
Ramalingam S, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann Oncol. 2015;26(8):1741–8.
Villalona-Calero MA, et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer. 2016;122(6):875–83.
Goldman JW, et al. Treatment rationale and study design for the JUNIPER study: a randomized phase III study of Abemaciclib with best supportive care versus Erlotinib with best supportive Care in Patients with Stage IV non-small-cell lung cancer with a detectable KRAS mutation whose disease has progressed after platinum-based chemotherapy. Clin Lung Cancer. 2016;17(1):80–4.
Puyol M, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73.
Koyama S, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008.
Luo J, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48.
Corcoran RB, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–8.
Mou H, et al. Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis. Proc Natl Acad Sci U S A. 2017;114(14):3648–53.
Kim J, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature. 2016;538(7623):114–7.
Burrell RA, Swanton C. The evolution of the unstable cancer genome. Curr Opin Genet Dev. 2014;24:61–7.
Ambrogio C, et al. Combined inhibition of DDR1 and notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. 2016;22(3):270–7.
Vallejo A, et al. An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer. Nat Commun. 2017;8:14294.
Molina-Arcas M, et al. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3(5):548–63.
Schoumacher M, et al. Inhibiting Tankyrases sensitizes KRAS-mutant cancer cells to MEK inhibitors via FGFR2 feedback signaling. Cancer Res. 2014;74(12):3294–305.
Wang J, et al. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat Commun. 2016;7:11363.
Zhou Y, et al. miR-1298 inhibits mutant KRAS-driven tumor growth by repressing FAK and LAMB3. Cancer Res. 2016;76(19):5777–87.
Roman-Moreno M, V S., Lopez I, Baraibar Argota I, Fraile E, Gil Bazo I. High chemopreventive and therapeutic efficacy of Id1 inhibition in KRAS-mutant (KM) adenocarcinoma (AD) non-small cell lung cancer (NSCLC). Paper presented at: European Society of Medical Oncology Annual Meeting, Madrid 2017. Annals of Oncology 2017. 28(Supplement 5).
Acknowledgements
MR was supported by ADA and FPU15/00173 fellowships. SV was supported by the Spanish Ministry of Economy and Competitiveness (MINECO, SAF2013-46423-R), the European Commission (S.V., 618312 KRASmiR FP7-PEOPLE-2013-CIG), the Worldwide Cancer Research (16-0224) and the Fundación La Caixa-FIMA agreement. SV and IGB were supported by a grant (RD12/0036/0040) from Red Temática de Investigación Cooperativa en Cáncer, Instituto de Salud Carlos III, Spanish Ministry of Economy and Competitiveness & European Regional Development Fund “Una manera de hacer Europa” and cofunded by FEDER funds/European Regional Development Fund (ERDF). IGB was also supported by two grants from Instituto de Salud Carlos III (PI11/00976 and PI15/02223).
Author information
Authors and Affiliations
Contributions
MR and IB performed the scientific literature search, designed the review structure, elaborated the figures and tables and wrote the manuscript. EN and CR helped with the design of the manuscript, contributed to the discussion and drafted the manuscript. SV and IGB conceived the manuscript idea, directed the scientific search, helped with the manuscript writing and corrected the final version of the text. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Román, M., Baraibar, I., López, I. et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer 17, 33 (2018). https://doi.org/10.1186/s12943-018-0789-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0789-x