
Abstract
The Kirsten rat sarcoma virus transforming protein (KRAS) mutations (predominate in codons 12, 13, and 61) and genomically drive nearly one-third of lung carcinomas. These mutations have complex functions in tumorigenesis, and influence the tumor response to chemotherapy and tyrosine kinase inhibitors resulting in a poorer patient prognosis. Recent attempts using targeted therapies against KRAS alone have met with little success. The existence of specific subsets of lung cancer based on KRAS mutations and coexisting mutations are suggested. Their interactions need further elaboration before newer promising targeted therapies for KRAS mutant lung cancers can be used as earlier lines of therapy. We summarize the existing knowledge of KRAS mutations and their coexisting mutations that is relevant to lung cancer treatment, in this review. We elaborate on the prognostic impact of clinical and pathologic characteristics of lung cancer patients associated with KRAS mutations. We briefly review the currently available techniques for KRAS mutation detection on biopsy and cytology samples. Finally, we discuss the new therapeutic strategies for targeting KRAS-mutant non-small cell lung cancer (NSCLC). These may herald a new era in the treatment of KRASG12Cmutated NSCLC as well as be helpful to develop demographic subsets to predict targeted therapies and prognosis of lung cancer patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer-related deaths among males worldwide [1]. It accounts for 1.38 million cancer deaths per year. It is the fifth common cause of cancer among females [2]. The overall 5-year survival rate of lung cancer remains poor in spite of numerous recent advances in its detection and treatment [3]. Identifying the molecular subsets of lung adenocarcinoma (LADC) and personalized treatment with targeted therapy, is needed to improve patient prognosis and survival [4]. Recent studies have highlighted the need to identify sub-sets of co-existing mutations in the EGFR-mutated LADC, as these may have a major impact on prognosis and newer therapeutic approaches [5].
In lung adenocarcinomas, comprehensive molecular profiling has identified significant mutations in eighteen genes [5], including (Table 1): (i) Oncogenes; EGFR (20–50%) [5, 6, 36], KRAS (33%) [7], BRAF (10%) [8], PI3K (7%), MET (7%) [13], RIT1 (2%), NRB1 [36],ERBB2 [16](ii) tumour suppressor genes; TP53 (46%) [19], STK11(17%), KEAP1(17%), NF1(11%), SETD2(9%), ARID1A(7%),RB1 (4%), CDKN2A (4%), (iii) Gene fusions/splice site mutations causing aberrant RNA transcripts: EML4-ALK [32], CD74-ROS1 [31], KIF5B-RET [33], NTRK [34] and NRG1fusions [35].
Patients with newly diagnosed lung adenocarcinoma commonly undergo sequential molecular testing (for EGFR, ALK, ROS1). They then undergo treatment with EGFR tyrosine kinase inhibitors- erlotinib, gefitinib, etc) and ALK/ROS1 TKIs (crizotinib, ceritinib) [37].KRAS mutations variably occur in LADC in western countries (20–25%) [38, 39] and in Asia (10–15%) [40, 41]. The identification of lung cancer patient subsets based on KRAS mutation analysis before initiation of EGFR targeted therapy needs to be done [42].
KRAS mutations predominantly occur in codon 12, 13 in lung cancer. Codons 10, 61 and 146 are much less frequently mutated. The prevalence of KRAS mutations in early and advanced stage LADCs is similar [7]. A heterogeneous spectrum of KRAS mutations; transversions (80%) or transitions (20%) [43] are identified in lung cancer patients. Patients with transversions, more frequently develop adenocarcinoma while those with transitions more frequently have squamous cell carcinoma [44]. Most KRAS mutations patients are males (60%), current or former smokers (63% and 33%, respectively) with adenocarcinoma (80%) [43]. KRAS mutations are rarely present in small cell lung cancer [45, 46]. The KRAS mutated LADCs grow in a solid pattern with TTF1 positivity (thyroid transcription factor) while the mucinous adenocarcinoma histology lacks TTF-1 [47]. The KRAS mutations are predictive of (i) poor prognosis [48] (ii) resistance to EGFR-TKI therapy in advanced cases [49] (iii) Exclusion of the EGFR and the BRAF mutations [50]. Thus, emphasizing the need to evaluate for KRAS and other coexisting mutations before the initiation of anti-EGFR therapy [51] (Table 1).
Molecular heterogeneity is observed in up to one-third of KRAS-mutant lung cancers and defines their chemotherapy response, tissue spread and prognosis [52]. The co-occurrence of two active mutations either drives oncogenes to functional redundancy [53] or results in cell senescence or death. Smokers with lung adenocarcinomas have concurrent KRAS [54], TP53, STK11, KEAP1 mutations while non-smokers with LADCs commonly have EGFR, TP53 mutations and/or MET alterations [5]. These subsets are associated with varied immune cell restrictions, altered angiogenesis, tumor microenvironment and poor survival [7, 39, 55]: (i) TP53 co-mutations activate the NF-κB pathway [56], increase IFNγ and PD-L1 expression [22] and promote an inflamed tumor immune microenvironment. (ii) LKB1/STK11co-mutations result in infiltration of neutrophils, leading to a pro-inflammatory cytokine milieu [22, 23]. (iii) KEAP1 mutations reduce T and B-lymphocytes infiltration [24] and NRF2 stabilization [24, 25]. (iv) Oncogenic MYC helps in immune evasion by KRAS-driven lung adenocarcinomas. By facilitating (a) IL-23-mediated expulsion of innate immune cells (T, B lymphocytes and NK cells), (b) CCL9-mediated macrophage recruitment and (c) VEGF mediated immunosuppressive microenvironment [57]. (v) PI3KCA co-mutations benefit activation of the BRAF pathway without risk of inducing senescence [58,59,60]. These studies have suggested that targeting the co-mutations and their pathways could be an effective treatment strategy in NSCLC patients [11, 12] (Table 2, Fig. 1).

Mechanism of targeted action of therapeutic agents against KRAS driven carcinomas (i) Direct KRAS inhibitors- targets the RAS proteins activation and prevents the conversion of inactive KRAS to active KRAS (ii) KRAS membrane association - impairs KRAS post-translational modification, lipidation and localization (iii) KRAS downstream signaling pathways- inhibit downstream effector pathways- RAF, MEK, PI3K, mTOR (iv) KRAS synthetic lethality- selective killing of KRAS-mutant cells through inhibition of a second protein (v) Immunotherapy- immune checkpoint inhibitor therapy-inhibit PD-L1 (vi) Inhibition of RAS-regulated metabolic processes- targets mutant KRAS-driven metabolic rewiring
Previous studies using RNASeq data from The Cancer Genome Atlas have identified three subsets of KRAS mutated lung adenocarcinomas based on their dominant co-existing mutations. The three major subsets identified include; KL, KP and KC. These show co-occurring mutations in LKB1/ STK11 (KL), TP53 alterations (KP), CDKN2A/CDKN2B (KC). These biologically distinct subsets have unique intracellular signaling patterns and are susceptible to different therapeutic strategies [7]. KRAS alleles showed enrichment for KRASG12D in the KC subgroup. KL subsets showed enhanced sensitivity to several Hsp90 inhibitor drugs such as ganetespib appeared particularly effective [7] (Table 3).
RAS family and downstream signaling
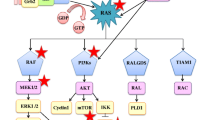
The RAS family of protooncogenes includes three isoforms; Kirsten rat sarcoma virus oncogene (KRAS) (chromosome 12p12.1), Harvey rat sarcoma virus (HRAS) (11p15.5), Neuroblastoma Ras sarcoma virus (NRAS) (1p13.1) [84]. KRAS (85%) is the predominant isoform followed by NRAS (11%) and HRAS (4%). These RAS genes encode a small membrane-localized guanosine triphosphate (GTP)-binding protein with intrinsic GTPase activity. Wild-type RAS proteins exist in an inactive state (GDP-bound) on the plasma membrane. They regulate the protein conformational change between active (GTP bound) and inactive states [85]. This process is regulated by; (i) Guanine Exchange Factors (that promote GDP dissociation and GTP binding), (ii) GAPs-GTPase activating proteins (that stimulate RAS GTPase activity). On mitogenic stimulation, the GEFs recruited to RAS, release GDP and form a transient nucleotide free state (Fig. 1). This nucleotide exchange causes conformational changes in RAS proteins (Switch 1 and Switch 2), which then bind to GTP, engage RAS effector proteins and activate RAS targets (Fig. 1).
KRAS mutations are heterogeneous in their frequency and spectrum in lung cancer and mainly show mutations in codons-12 (89%), 13 (9%), and 61 (1%) [86](Fig. 2).KRAS mutations are categorized into; transitions (a purine-purine, or a pyrimidine-pyrimidine substitution) and transversions (a pyrimidine-purine, or a purine-pyrimidine substitution) [43]. The dominant KRAS mutation patterns are: (i) G → T transversion, in the first base of codon 12(KRASG12C, 40–60%) [87]. In this mutation, glycine is replaced by cysteine and is associated with tobacco smoking [37]. (ii) G → T transversion at the second base of codon 12 replaces glycine by valine (KRASG12V, 20–22%) [87, 88]. (iii) G → A transitions at the second base of codons 12 and 13 (KRASG12D or KRASG13D) are characterized by substitution of glycine with aspartate [89](16–20%) [39]. (iv) G → C transversions at codon 12 with replacement of glycine to alanine, (KRASG12A,7%) or glycine-arginine (KRASG12R,2%) are least frequent.

a Frequency of occurrence of KRAS mutations in exon 2- codons 12, 13, 61 b The spectrum of KRAS mutations in NSCLC, under Codon12, mainly occur as transversions (~ 80%) - G12C, G12V, G12A; G12R. While, transitions occur in ~ 20% cases - G12D, others
Regardless of the site of mutation, RAS point mutations lock the mutant RAS into the GTP-bound oncogenic state, encoding oncoproteins, KRAS4A and KRAS4B [90]. Resulting in the accumulation of constitutively GTP-bound RAS proteins inside the cells. KRAS4A expression is specifically expressed in lung epithelial cells while KRAS4B is ubiquitously expressed. Animals lacking KRAS4A have been found to be highly resistant to the development of lung tumor [91]. Thereby, suggesting the essential role of KRAS4A in KRAS-driven lung tumors and its importance in the design and development of KRAS-targeted therapeutics [92]. These mutated RAS proteins bind to RAS effector proteins based on their unique C-terminal hypervariable region and exhibit specific functions [93,94,95]. The downstream effectors that have been identified include; RAF, MEK, ERK [96], PI3K, AKT, mTOR,Rac1small GTPase and RALGDS/RAL signaling pathways [97, 98](Fig. 1).
Thus RAS oncogenic mutations not only contribute to cancer phenotype, progression and prognosis but are also indicative of their specific downstream signaling pathways (Table 2). For eg. KRAS–G12D preferentially activates AKT signaling whereas KRAS–G12C and G12V preferably activate RAL-A/B signaling [99]. RAF hyperactivation [100] causes MEK1/2 and ERK1/2 phosphorylation and increases their expression in lung cancers [72]. The activated RAS-PI3K-AKT-mTOR pathway promotes cell survival [101] while the activated RAS-RAF-MEK-ERK promotes cell proliferation, survival, and differentiation [102]. These pathways may serve as promising targets to inhibit cancer progression in KRAS mutant lung cancers [96].
Current molecular methods for KRAS mutation detection
The molecular methods of detecting oncogenic KRAS on clinical samples include: nucleic acid sequencing (Sanger/di-deoxy) [61], pyrosequencing [63], real-time PCR with HRManalysis (high-resolution melting) [103] and allele-specific PCR [104], single nucleotide probe extension assays (SNaPshots) [105], or shifted termination assays (STAs) [106] (Table 2). Useful screening methods include conformation-based separation using single-strand conformation polymorphism (SSCP) and denaturing gradient gel electrophoresis (DGGE). The biggest limiting factors in analysis are; small biopsy size, limited amount of DNA and intrinsic KRAS heterozygous status of tumors, (comprising of mutant and wild-type KRAS).
Sanger sequencing
Sanger/dideoxy DNA sequencing method is the gold standard to detect KRAS mutations [61] and their potential variations (substitutions, insertions and deletions). It has a high accuracy of ~ 90% but low analytical sensitivity of ~ 85% as compared to higher sensitivity of other methods such as allele-specific PCR, pyrosequencing, and chip array hybridization (90%, 93%, and 92%, respectively) [107]. The Sanger sequencing method requires at least 30%–40% of neoplastic/ non-neoplastic cells to detect mutations [63]. The detection of gain-of-function mutations in KRAS oncogene is a particular challenge, since thetumor cells can carry one copy of wild-type allele and the non-neoplastic cells in tumor tissue can contribute two wild type alleles.
Whole exome sequencing
Whole exome sequencing (WES) identifies the disease causing variations in protein coding regions of mutated exons in tumor DNA as compared to normal DNA [36, 62, 108]. This method is however limited, if DNA variations are present outside the exon. In this method, whole exome captures and sequencing is performed by using 200 ng of genomic DNA for library preparation. The library is amplified and hybridized to biotinylated oligos specific for exons (baits). The captured libraries are purified using streptavidin magnetic beads and again amplified by PCR. Normalized libraries are pooled and DNA sequenced using paired-end reads and multiplexed. The raw sequence reads are then mapped to the human reference genome. Previous studies have shown NGS sequencing to outperform allele-specific PCR, Sanger sequencing, and pyrosequencing [107]. WES is a cost-effective way of NGS. Using this method, approximately75% of patients received a therapeutic proposal and nearly 23.1% of patients were treated with NGS directed therapy. These included; PI3K/AKT/mTOR inhibitor therapy (27.8%), PARP inhibitors (24.1%), antiangiogenic therapy (21.5%), MEK inhibitor therapy (8.9%) and immunotherapy (6.3%) [108]. However, no differences of progression free survival ratios were observed between patients treated with matched versus standard therapy [108]. Thus, suggesting, the need for multi-omics strategies comprising of circulating cell-free DNA detection, RNA and whole genome sequencing for improving patient outcome.
Pyrosequencing
This sensitive DNA extension sequencing assay can detect < 5% mutant KRAS alleles among wild-type alleles. It measures the release of pyrophosphate moieties during the incorporation of a specific nucleotide into the synthesized DNA. By using the resulting program, the specific nucleic acid sequence for the target region can be detected [109]. Pyrosequencing provides a sensitive method to detect the mutant KRAS alleles from paraffin-embedded tissue [63]. However this method is not economical owing to expensive equipment.
PCR and HRM analysis
PCR methods and high resolution melting curve assays provide a cost-effective, sensitive and reliable mutation analysis using low amounts of DNA [110]. They can discriminate between wild-type and mutant gene in DNA isolated from FFPE tissues. Also they can detect mutations in the commonly mutated genes (KRAS, BRAF, PIK3CA, and AKT1) [64]. Therefore, they are highly applicable to large-scale genotyping. HRM utilizes fluorescent probes complementary to the target amplicon. It is faster in contrast to Sanger sequencing [111, 112]. It distinguishes genetic variants by their differences in melting temperature needed to dissociate probe from target leading to the loss of fluorescence [109]. The disadvantages for HRM include: (i) the need for expensive fluorescently labeled probes. (ii) Additional Sanger sequencing to identify the exact mutational status. (iii) Some rare homozygous mutations might not be detected.
Allele-specific PCR
This common laboratory method characterizes simple genetic variants such as point mutations. It utilizes allele-specific PCR-based K-RAS kits, to detect mutations in KRAS codons 12,13 etc., (G12D, G12V, G12C, G12S, G12A, G12R, G13D). In this method, the targeted alleles are amplified by amplification refractory mutation system (ARMS) and amplification products are detected with Scorpion probes [65]. A fluorescent signal is generated when these probes bind to the PCR amplicon resulting in the separation of the quencher from the fluorophor.
SNaP shot assay
This multiplexed single nucleotide probe extension assay detects point mutations from very small quantity of DNA [113]. These include; (i) EGFR mutations- c.2573 T > G (L858R), c.2369C > T (T790M); (ii) KRAS mutations- c.34G > T (p.Gly12Cys), c.35G > T (p.Gly12Val); (iii) PIK3CA mutations- c.1624G > A (E542K), c.1633G > A (E545K); (iv) BRAF mutation- c.1799 T > A (V600E). The SNaPshot assay is performed using PCR primers, dNTPs labeled with a differential fluorescence and extension primers and products are resolved on a capillary sequencer. The SNaP Shot assay differs from the shifted termination assays (STA) that are based on primer-extension methods to detect a specific mutation. STA incorporates multiple labeled nucleotides to the detection primer as compared to singly labeled nucleotides incorporation in SNaP Shot assay [114].
Screening tests
Single-strand conformation polymorphism (SSCP)
SSCP is a simple and sensitive assay for detection of SNP, based on the conformation of the single-stranded DNA (ssDNA). Any change in base pairs causes conformational change of the ssDNA and shifts DNA migration under non-denaturing electrophoresis conditions. The separated-out DNA bands are then visualized by incorporating radio-isotopes/ fluorescent dyes/ capillary-based or silver staining [115]. SSCP analysis is used as a screening method to detect point mutations, small deletions and insertions. However, it cannot detect the precise nucleotide change. For this DNA sequencing additionally needs to be performed [116].
Denaturing gradient gel electrophoresis (DGGE)
DGGE is another screening method. It separates the PCR products based on sequence differences and the DNA differential denaturing characteristics. In this denaturing gradient gel electrophoresis, PCR products migrate through increasingly higher concentrations of chemical denaturant in the polyacrylamide gel. This results in denaturation of the weaker melting domains of the double-stranded PCR product first and their separation [117].
KRAS mutation directed lung cancer therapy
The KRAS mutated lung cancers are driven by sustained KRAS expression and signaling. These cancers are commonly associated with resistance to therapy and poor prognosis [118]. They are treated with conventional chemotherapy unlike KRAS-wild type lung cancers, where molecular targeted therapy is available [86]. Presently, there is renewed interest in therapeutic strategies inhibiting the functional output of mutated KRAS [89, 119] (Table 3, Fig. 1). The current approaches include, inhibitors of the: (i) KRAS membrane associations, (ii) KRAS downstream signaling pathways, (iii) KRAS synthetic lethality, (iv) direct targeting of KRAS, (v) immunotherapy and (vi) RAS-regulated metabolic processes [119]. Of these approaches, immunotherapy with immune checkpoint inhibitors in KRAS-mutant NSCLC [83] has been considered as one of the most promising treatment approaches (Table 3).
KRAS membrane associations
KRAS protein requires membrane localization to become biologically active. Therefore, impairment of KRAS localization can serve as potential target for KRAS-mutant cancers [120]. For membrane localization, the RAS proteins undergo post-translational modificationand lipidation by enzymatic reactions (prenylation by farnesyltransferase [96] or geranylgeranylation via GGTase-I [118]. Initial studies focused on single-stranded DNA (ssDNA); tipifarnib, lonafarnib, salirasib [67, 68, 121] (Table 3). However, alternative lipidation of RAS proteins by GGTases [122] resulted in failure of FTI therapy of KRAS mutated cancers in clinical trials [123]. Recently, modified FTIswhich specifically react with the CAAX motif of KRAS, and block both its farnesylation and geranylgeranylation are being studied [124]. Another approach to prevent the membrane localization of KRAS protein is by inhibiting the phosphodiesterase 6 delta subunit (PDEδ). Since, PDEδ acts by binding to the farnesylated tail of KRAS and chaperoning its membrane localization [125, 126]. The PDEδ inhibitors can be used to disrupt KRAS:PDEδ binding and disrupting the localization of KRAS in cancer cells [69,70,71].
KRAS downstream signaling pathways
RAF, MEK, PI3K, mTOR are some of the downstream effectors of KRAS signaling. Their inhibitors are used as single agents or as combination therapy for treatment of lung cancer. The single agent therapies available for KRAS mutated LADCs are; BRAF inhibitor (Sorafenib) [73], MEK inhibitors (Selumetinib) [74], mTOR inhibitor (ridafarolimus) [75], focal adhesion kinase inhibitor (defactinib) [76]. However, these have shown disappointing clinical efficacy, so far. Recently, a RAF/MEK inhibitor (RO5126766) has shown to effectively reduce tumours in 60% of patients with a low frequency of higher grade adverse events [127]. The combination therapies inhibit two or more downstream effectors in the RAS pathway. They have been observed to fully block KRAS signaling in several phase I trials of lung cancer [11, 12, 128, 129]. However, their phase III validation studies are awaited or have shown failure. For eg. the Selumetinib and docetaxel combination therapy for KRAS-G12C and KRAS-G12V tumours [98, 130] have failed validation in phase III (SELECT-1) [131]. Similarly phase II trials using combination of a PI3K inhibitor (BKM120) with MEK1/2 inhibitor (MEK162) in patients with NSCLC, has shown little success [11, 72].
KRAS synthetic lethality
The KRAS synthetic lethality approach involves the selective killing of KRAS mutated cancer cells by inhibition of a second protein. In every case, the interactions between mutated KRAS and other proteins on which KRAS mutated cancer cells have become dependent need to be identified first [132]. Then these second hits can be therapeutically targeted resulting in selective death of KRAS-mutant, but not KRAS-wild-type, cells [84]. These include; (i) GATA2 transcription factor (proteasome upregulator) and its inhibitor, bortezomib, which has shown response in KRAS-G12D lung cancers [77]. (ii) Cyclin-dependent kinase 4 (Cdk4) (G1 transition/cell cycle progressor) and its inhibitor proteins, p16INK4A, p15INK4B, p18INK4C and p19INK4D, cause KRAS mutated lung cells to undergo senescence and prevent tumor growth128 (iii) TANK-binding kinase 1 (TBK1), serine-threonine kinase STK33 and polo-like kinase 1(PLK1) are other potential synthetic lethal therapeutic targets that have been identified in cell lines [79,80,81]. Some of these encode protein kinases and may be inhibited by selective TKIs.
KRAS direct targeting
RAS has been perceived to be “undruggable” due to its lack of deep pockets for binding of small molecule inhibitors. However, recent studies have shown some success in the direct inhibition of RAS [120, 133]. This strategy targets the RAS proteins activation and prevents the conversion of inactive KRAS to active KRAS. These include molecules that can, (i) allosterically bind to the Switch-II pocket in GDP-RAS, adjacent to the cyteine residue of KRAS-G12C [134, 135]. (ii) directly bind and impair wild-type KRAS activation by the SOS1-GEF [136]. (ii) selectively recognize and inactivate specific KRAS G12C amino acid substitution [137]. Their results are a majorstep forward in the development of direct KRASG12C inhibitor therapy for lung cancer [133].
The Ras GTPase family inhibitor, sotorasib has recently been approved by US-FDA for treatment of KRAS mutated locally advanced or metastatic solid tumours (NSCLC and colorectal cancer) [138]. Simultaneous approval has been given to the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics. They have recommended that the tumor tissue should be tested if no mutation is detected in plasma specimen. Previously in the clinical trial of sotorasib conducted by Hong et al., 2020 a confirmed objective response in 32.2% patients and disease control (stable disease) in 88.1% NSCLC patients [139] was observed. Adagrasib (MRTX849) is another novel small molecule targeting the KRASG12C mutation that has shown promising activity [140].
However, the mechanisms of acquired resistance to these therapies are currently unknown. Using in vitro deep mutational screening methods diverse genomic and histologic mechanisms imparting resistance to KRASG12C inhibitors have been identified [140]. These acquired KRAS alterations include G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C, and high-level amplification of the KRASG12C allele. Additionally, there exist acquired bypass mechanisms of resistance; (i) MET amplification, (ii) activating mutations in NRAS, BRAF, MAP 2 K1, and RET; (ii) oncogenic fusions involving ALK, RET, BRAF, RAF1, and FGFR3; (iii) and loss-of-function mutations in NF1 and PTEN. These secondary KRAS mutations can cause resistance to sotorasib, adagrasib, or both, in vitro and are suggestive of their sequential use [141]
Immunotherapy
KRAS mutant lung cancers have an immune resistant microenvironment. The immune resistance is caused by smoking that induces T-cell influx and PD-L1 expression by cancer cells and tumor infiltrating lymphocytes (TILs) [119]. PD-L1 positivity is seen in approximately 60–70%TILsand in 20–55%KRAS-mutant tumor cells [142,143,144]. Therefore immune checkpoint inhibitor therapy is being investigated to improve the patient outcome in KRAS mutant cancers [83]. Previously, Falk et al., 2016, have observed hypoxia to significantly increase the PD-L1 expression in KRASG12C and KRASG12D codon subtypes [145]. Identification of the coexisting KRAS mutations subtype may serve as biomarkers of resistance and need to be performed prior to initiation of PD-1/PD-L1 inhibitor therapy [83].
Inhibition of RAS-regulated metabolic processes
Cancer cells harboring KRAS mutation show up-regulation of rate-limiting enzymes, shift of cancer cell metabolism toward anabolic pathways resulting in increased cancer cell growth. Therefore, recent studies are focusing on mutant KRAS-driven metabolic rewiring, including; (i) upregulation of enzymes involved in amino acid, fatty acid or nucleotide biosynthesis. Glucose and glutamine metabolism,(ii) deregulation of scavenging cellular pathways (e.g., autophagy) [146], (iii) PPARγ and WNT/β-catenin, pathways involved in metabolic enzymes changes in cancers [147, 148]. Currently, the metabolic dependencies of oncogenic KRAS driven lung and pancreatic cancers are still in their infancy and hold promise as therapeutic targets [149].
Conclusion
The KRAS driven lung cancers can be categorized into different subsets (such as KL, KP, KC, etc). This categorization is based on tumour histology, type of KRAS mutation (transversions/transitions) and presence of co-existing significant mutations in nearly eighteen genes identified so far. KRAS mutations have heterogeneous spectrum in lung cancer- transversions (80%)/ transitions (20%). KRAS mutations correlate with histology: transversions, more frequently develop adenocarcinoma while transitions more frequently have squamous cell carcinoma. These biologically distinct subsets have unique intracellular signaling patterns and are susceptible to different therapeutic strategies. Thus these subsets can be used to predict new targeted therapeutic strategies and improve the prognosis of lung cancer patients.
A variety of molecular methods are now available for detecting oncogenic KRAS in clinical samples. These entail complete molecular profiling of each patient and identification of KRAS mutations and coexisting mutations. The biggest limiting factors in analysis are; small biopsy size, limited amount of DNA and intrinsic KRAS heterozygous status of tumors.
The G → T transversion, in the first base of codon 12 (KRASG12C) mutation is the commonest in lung adenocarcinomas. Presently, there is renewed interest in therapeutic strategies inhibiting the functional output of mutated KRAS. Direct KRASG12C inhibitor therapy and immunotherapy with immune checkpoint inhibitors are being considered as one of the most promising treatment approaches. These may prove to be a step forward in personalized therapy and in improving prognosis of lung cancer patients.
Availability of data and materials
Not applicable.
Abbreviations
- ARID1A:
-
AT-Rich Interaction Domain 1A
- BRAF:
-
B-Raf Proto-Oncogene
- CCL-9:
-
Chemokine (C-C motif) ligand 9
- CD74:
-
ROS1-Cluster of Differentiation 74-ROS proto-Oncogene 1
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- DGGE:
-
Denaturing gradient gel electrophoresis
- dNTPs:
-
dideoxy nucleotides
- EGFR:
-
Epidermal growth factor receptor
- EML4:
-
ALK-Echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase
- ERBB2:
-
v-erb-b2 avian erythroblastic leukemia viral oncogene homology 2
- ERK:
-
Extracellular signal-regulated kinase
- GEF:
-
Guanine Exchange Factors
- GTP:
-
Guanosine triphosphate
- HRAS:
-
Harvey rat sarcoma viral oncogene
- HRM:
-
High-resolution melting analysis
- HSP-90:
-
Heat Shock Protein-90
- IFNγ:
-
Interferon gamma
- IL-23:
-
Interleukin-23
- KEAP1:
-
Kelch-like ECH-associated protein 1
- KIF5B-RET:
-
Kinesin family member 5B-Ret proto Oncogene
- KRAS:
-
Kirsten rat sarcoma virus transforming protein
- LADC:
-
Lung adenocarcinoma
- MEK:
-
Mitogen extracellular signal-related kinase
- MET:
-
Mesenchymal epithelial transition factor
- mTOR:
-
Mammalian Target of Rapamycin
- MYC:
-
MYC proto Oncogene
- NF1:
-
Neurofibromin 1
- NF-κB:
-
Nuclear Factor kappa-light-chain-enhancer of activated B cells
- NRAS:
-
Neuroblastoma Ras virus oncogene
- NRB1:
-
Neurabin1
- NSCLC:
-
Non-small cell lung cancer
- NTRK:
-
Neurotrophilic tyrosine receptor kinase
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- PPARγ:
-
Peroxisome Proliferator Activated Receptor Gamma
- Rac1small GTPase:
-
Ras-related C3 botulinum toxin substrate 1
- RAF:
-
Rapidly accelerated fibrosarcoma
- RALGDS/RA:
-
Ral guanine nucleotide dissociation stimulator
- RB1:
-
Retinoblastoma 1
- RIT1:
-
Ras-like protein in tissues 1
- SETD2:
-
SET Domain Containing 2
- SSCP:
-
Single-strand conformation polymorphism
- ssDNA:
-
Single-stranded DNA
- STK11:
-
Serine/threonine kinase 11
- TP53:
-
Tumour protein 53
- TTF1:
-
Transcription Termination Factor 1
- WNT/β-catenin:
-
Wingless and Int-1/ beta-catenin
References
Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–53. https://doi.org/10.1002/ijc.31937.
Noronha V, Pinninti R, Patil VM, Joshi A, Prabhash K. Lung cancer in the Indian subcontinent. South Asian. J Cancer. 2016;5(3):95–103. https://doi.org/10.4103/2278-330x.187571.
Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. https://doi.org/10.21037/tlcr.2016.06.07.
Mascaux C, Tomasini P, Greillier L, Barlesi F. Personalised medicine for nonsmall cell lung cancer. Eur Respir Rev. 2017;26:170066. https://doi.org/10.1183/16000617.0066-2017.
Rosell R, Karachaliou N, Arrieta O. Novel molecular targets for the treatment of lung cancer. Curr Opin Oncol. 2020;32(1):37–43. https://doi.org/10.1097/cco.0000000000000590.
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with Clincal response to gefitinib therapy. Science. 2004;304(5676):1497–500. https://doi.org/10.1126/science.1099314.
Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19(9):495–509. https://doi.org/10.1038/s41568-019-0179-8.
Nieto P, Ambrogio C, Esteban-Burgos L, Gómez-López G, Blasco MT, Yao Z, et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature. 2017;548(7666):239–43. https://doi.org/10.1038/nature23297.
Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L, et al. BRAF in non-small cell lung cancer (NSCLC): pick axing another brick in the wall. Cancer Treat Rev. 2018;66:82–94. https://doi.org/10.1016/j.ctrv.2018.04.006.
Baik CS, Myall NJ, Wakelee HA. Targeting BRAF-mutant non-small cell lung Cancer: from molecular profiling to rationally designed therapy. Oncologist. 2017;22(7):786–96. https://doi.org/10.1634/theoncologist.2016-0458.
Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106(43):18351–6. https://doi.org/10.1073/pnas.0907325106.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. https://doi.org/10.1038/nm.1890.
Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers Clincal sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9. https://doi.org/10.1158/2159-8290.Cd-15-0285.
Cavé H, Caye A, Ghedira N, Capri Y, Pouvreau N, Fillot N, et al. Mutations in RIT1 cause Noonan syndrome with possible juvenile myelomonocytic leukemia but are not involved in acute lymphoblastic leukemia. Eur J Hum Genet. 2016;24(8):1124–31. https://doi.org/10.1038/ejhg.2015.273.
Satoh A, Nakanishi H, Obaishi H, WadaM TK, Satoh K, et al. Neurabin-II/spinophilin. An actin filament-binding protein with one pdz domain localized at cadherin-based cell-cell adhesion sites. J Biol Chem. 1998;273(6):3470–5. https://doi.org/10.1074/jbc.273.6.3470.
Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431(7008):525–6. https://doi.org/10.1038/431525b.
Hartung AM, Swensen J, Uriz IE, Lapin M, Kristjansdottir K, Petersen US, et al. The splicing efficiency of activating HRAS mutations can determine Costello syndrome phenotype and frequency in Cancer. PLoS Genet. 2016;12(5):e1006039. https://doi.org/10.1371/journal.pgen.1006039.
Oliveira JB, Bidère N, Niemela JE, Zheng L, Sakai K, Nix CP, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104(21):8953–8. https://doi.org/10.1073/pnas.0702975104.
Aisner DL, Sholl LM, Berry LD, Rossi MR, Chen H, Fujimoto J, et al. LCMC2 investigators. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the lung Cancer mutation consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–47. https://doi.org/10.1158/1078-0432.CCR-17-2289.
Pfeifer GP, Hainaut P. On the origin of G --> T transversions in lung cancer. Mutat Res. 2003;526(1–2):39–43. https://doi.org/10.1016/s0027-5107(03)00013-7.
Fain PR, Barker DF, Goldgar DE, Wright E, Nguyen K, Carey J, et al. Genetic analysis of NF1: identification of close flanking markers on chromosome 17. Genomics. 1987;1(4):340–5. https://doi.org/10.1016/0888-7543(87)90034-6.
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–35. https://doi.org/10.1158/2159-8290.Cd-18-0099.
Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and Proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. https://doi.org/10.1158/0008-5472.Can-15-1439.
Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S, Tull D, et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung Cancer with an altered immune microenvironment. Cell Metab. 2018;27(4):935–43.e4. https://doi.org/10.1016/j.cmet.2018.02.006.
Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat Commun. 2018;9(1):3506. https://doi.org/10.1038/s41467-018-05861-7.
Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391(6667):597–601. https://doi.org/10.1038/35404.
Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94(12):894–903. https://doi.org/10.1093/jnci/94.12.894.
Zhang Y, Xu X, Zhang M, Bai X, Li H, Kan L, et al. ARID1A is downregulated in non-small cell lung cancer and regulates cell proliferation and apoptosis. Tumour Biol. 2014;35(6):5701–7. https://doi.org/10.1007/s13277-014-1755-x.
Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell. 2016;166(4):950–62. https://doi.org/10.1016/j.cell.2016.07.005.
Westcott PM, To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer. 2013;32(2):63–70. https://doi.org/10.5732/cjc.012.10098.
Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70. https://doi.org/10.1200/jco.2011.35.6345.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–703. https://doi.org/10.1056/NEJMoa1006448.
Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18(3):375–7. https://doi.org/10.1038/nm.2644.
Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013;19(11):1469–72. https://doi.org/10.1038/nm.3352.
Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014;4(4):415–22. https://doi.org/10.1158/2159-8290.Cd-13-0633.
Collisson EA, Campbell J, Brooks A, Berger A, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma: the cancer genome atlas research network. Nature. 2014;511(7511):543–50. https://doi.org/10.1038/nature13385.
Bhattacharya S, Socinski MA, Burns TF. KRAS mutant lung cancer: progress thus far on an elusive therapeutic target. ClinTransl Med. 2015;4(1):35. https://doi.org/10.1186/s40169-015-0075-0.
El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and outcomes of patients with metastatic KRAS-mutant lung adenocarcinomas: the lung Cancer mutation consortium experience. J Thorac Oncol. 2019;14(5):876–89. https://doi.org/10.1016/j.jtho.2019.01.020.
Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon JP, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31(17):2173–81. https://doi.org/10.1200/jco.2012.48.1390.
Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24(9):2371–6. https://doi.org/10.1093/annonc/mdt205.
Yoshizawa A, Sumiyoshi S, Sonobe M, Kobayashi M, Fujimoto M, Kawakami F, et al. Validation of the IASLC/ATS/ERS lung adenocarcinoma classification for prognosis and association with EGFR and KRAS gene mutations: analysis of 440 Japanese patients. J Thorac Oncol. 2013;8(1):52–61. https://doi.org/10.1097/JTO.0b013e3182769aa8.
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clincal Oncology Provisional Clincal Opinion: Testing for KRAS Gene Mutations in Patients With Metastatic Colorectal Carcinoma to Predict Response to Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Therapy. J Clin Oncol. 2009;27(12):2091–6. https://doi.org/10.1200/jco.2009.21.9170.
Dumenil C, Vieira T, Rouleau E, Antoine M, Duruisseaux M, Poulot V, et al. Is there a specific phenotype associated with the different subtypes of KRAS mutations in patients with advanced non-small-cell lung cancers? Lung Cancer. 2015;90(3):561–7. https://doi.org/10.1016/j.lungcan.2015.10.012.
Yanagawa N, Tamura G, Oizumi H, Endoh M, Sadahiro M, Motoyama T. Inverse correlation between egfr mutation and fhit,Rassf1a and runx3 methylation in lung adenocarcinoma: relation with smoking status. Anticancer Res. 2011;31:1211–4.
Suzuki Y, Orita M, Shiraishi M, Hayashi K, Sekiya T. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene. 1990;5(7):1037–43.
Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6(8):1353–62.
Rekhtman N, Ang DC, Riely GJ, Ladanyi M, Moreira AL. KRAS mutations are associated with solid growth pattern and tumor-infiltrating leukocytes in lung adenocarcinoma. Mod Pathol. 2013;26(10):1307–19. https://doi.org/10.1038/modpathol.2013.74.
Marabese M, Ganzinelli M, Garassino MC, Shepherd FA, Piva S, Caiola E, et al. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget. 2015;6(32):34014–22. https://doi.org/10.18632/oncotarget.5607.
Cisowski J, Bergo MO. What makes oncogenes mutually exclusive? Small GTPases. 2017;8(3):187–92. https://doi.org/10.1080/21541248.2016.1212689.
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. https://doi.org/10.1038/418934a.
Jakubauskas A, Griskevicius L. KRas and BRaf mutational status analysis from formalin-fixed, paraffin-embedded tissues using multiplex polymerase chain reaction-based assay. Arch Pathol Lab Med. 2010;134(4):620–4. https://doi.org/10.1043/1543-2165-134.4.620.
Lams WT, Yu H, Shyr Y, Patil T, Horn L, McCoach C, et al. First line chemotherapy responsiveness and patterns of metastatic spread identify Clincal syndromes present within advanced KRAS mutant non-small cell lung cancer with different prognostic significance. Clin Lung Cancer. 2018;19(6):531–43. https://doi.org/10.1016/j.cllc.2018.08.011.
Lee T, Lee B, Choi YL, Han J, Ahn MJ, Um SW. Non-small cell lung Cancer with concomitant EGFR, KRAS, and ALK mutation: Clincopathologic features of 12 cases. J Pathol Transl Med. 2016;50(3):197–203. https://doi.org/10.4132/jptm.2016.03.09.
Guibert N, Ilie M, Long E, Hofman V, Bouhlel L, Brest P, et al. KRAS mutations in lung adenocarcinoma: molecular and epidemiological characteristics, methods for detection, and therapeutic strategy perspectives. Curr Mol Med. 2015;15(5):418–32. https://doi.org/10.2174/1566524015666150505161412.
La Fleur L, Falk-Sörqvist E, Smeds P, Berglund A, Sundström M, Mattsson JS, et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer. 2019;130:50–8. https://doi.org/10.1016/j.lungcan.2019.01.003.
Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462(7269):104–7. https://doi.org/10.1038/nature08462.
Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell. 2017;171(6):1301–1315.e14. https://doi.org/10.1016/j.cell.2017.11.013.
Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, Yukiue H, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer. 2006;54(2):209–15. https://doi.org/10.1016/j.lungcan.2006.07.006.
Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther. 2012;11(2):485–91. https://doi.org/10.1158/1535-7163.Mct-11-0692.
Yan J, Roy S, Apolloni A, Lane A, Hancock JF. Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273(37):24052–6. https://doi.org/10.1074/jbc.273.37.24052.
Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, et al. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J Mol Diagn. 2005;7(3):413–21. https://doi.org/10.1016/s1525-1578(10)60571-5.
Anderson SM. Laboratory methods for KRAS mutation analysis. Expert Rev Mol Diagn. 2011;11(6):635–42. https://doi.org/10.1586/erm.11.42.
Amicarelli G, Shehi E, Makrigiorgos GM, Adlerstein D. FLAG assay as a novel method for real-time signal generation during PCR: application to detection and genotyping of KRAS codon 12 mutations. Nucleic Acids Res. 2007;35(19):e131. https://doi.org/10.1093/nar/gkm809.
Franklin WA, Haney J, Sugita M, Bemis L, Jimeno A, Messersmith WA. KRAS mutation: comparison of testing methods and tissue sampling techniques in colon cancer. J Mol Diagn. 2010;12(1):43–50. https://doi.org/10.2353/jmoldx.2010.080131.
Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24(4):381–6. https://doi.org/10.1038/74215.
Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018;285(1):28–41. https://doi.org/10.1111/febs.14125.
Adjei AA, Mauer A, Bruzek L, Marks RS, Hillman S, Geyer S, et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21(9):1760–6. https://doi.org/10.1200/jco.2003.09.075.
Riely GJ, Brahmer JR, Planchard D, Crinò L, Doebele RC, Lopez LAM, et al. A randomized discontinuation phase II trial of ridaforolimus in non-small cell lung cancer (NSCLC) patients with KRAS mutations. J. Clin Oncol. 2012;30:7531. https://doi.org/10.1200/jco.2012.30.15_suppl.7531.
Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638–42. https://doi.org/10.1038/nature12205.
Papke B, Murarka S, Vogel HA, Martín-Gago P, Kovacevic M, Truxius DC, et al. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat Commun. 2016;7:11360. https://doi.org/10.1038/ncomms11360.
Martín-Gago P, Fansa EK, Klein CH, Murarka S, Janning P, Schürmann M, et al. A PDE6δ-KRas inhibitor Chemotype with up to seven H-bonds and Picomolar affinity that prevents efficient inhibitor release by Arl2. Angew Chem Int Ed Engl. 2017;56(9):2423–8. https://doi.org/10.1002/anie.201610957.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19. https://doi.org/10.1038/nrg1879.
Papadimitrakopoulou V, Lee JJ, Wistuba II, Tsao AS, Fossella FV, Kalhor N, et al. The BATTLE-2 study: a biomarker-integrated targeted therapy study in previously treated patients with advanced non-small-cell lung Cancer. J Clin Oncol. 2016;34(30):3638–47. https://doi.org/10.1200/jco.2015.66.0084.
Carter CA, Rajan A, Keen C, Szabo E, Khozin S, Thomas A, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancer. Ann Oncol. 2016;27(4):693–9. https://doi.org/10.1093/annonc/mdw008.
Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J Thorac Oncol. 2011;6(8):1435–7. https://doi.org/10.1097/JTO.0b013e318223c099.
Gerber DE, Camidge DR, Morgensztern D, Cetnar J, Kelly RJ, Ramalingam SS, et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer. 2020;139:60–7. https://doi.org/10.1016/j.lungcan.2019.10.033.
Litvak AM, Drilon AE, Rekhtman N, Pietanza MC, Chaft JE, Woo K, et al. Phase II trial of bortezomib in KRAS G12D mutant lung cancers. J Clin Oncol. 2015;33:e19002. https://doi.org/10.1200/jco.2015.33.15_suppl.e19002.
Puyol M, Martín A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73. https://doi.org/10.1016/j.ccr.2010.05.025.
Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12. https://doi.org/10.1038/nature08460.
Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. https://doi.org/10.1016/j.cell.2009.05.006.
Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34. https://doi.org/10.1016/j.cell.2009.03.017.
Reck M, Carbone DP, Garassino M, Barlesi F. Targeting KRAS in non–small cell lung cancer: recent progress and new approaches. Ann Oncol. 2021;32:1101–10. https://doi.org/10.1016/j.annonc.2021.06.001.
Bai R, Zheng L. Dongsheng Xu, Jiuwei cui. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomarker Res. 2020;8:34. https://doi.org/10.1186/s40364-020-00209-0.
Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21(8):1802–9. https://doi.org/10.1158/1078-0432.Ccr-14-2180.
Nussinov R, Jang H, Tsai CJ, Cheng F. Precision medicine review: rare driver mutations and their biophysical classification. Biophys Rev. 2019;11(1):5–19. https://doi.org/10.1007/s12551-018-0496-2.
Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22(1):235–7. https://doi.org/10.1093/annonc/mdq680.
Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in Cancer. Nucleic Acids Res. 2011;39:D945–50. https://doi.org/10.1093/nar/gkq929.
Karachaliou N, Mayo C, Costa C, Magrí I, Gimenez-Capitan A, Molina-Vila MA, et al. KRAS mutations in lung cancer. Clin Lung Cancer. 2013;14(3):205–14. https://doi.org/10.1016/j.cllc.2012.09.007.
Fujimura K, Wang H, Watson F, Klemke RL. KRAS oncoprotein expression is regulated by a self-governing eIF5A-PEAK1 feed-forward regulatory loop. Cancer Res. 2018;78(6). https://doi.org/10.1158/0008-5472.CAN-17-2873.
To MD, Wong CE, Karnezis AN, Del Rosario R, Di Lauro R, Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40(10):1240–4. https://doi.org/10.1038/ng.211.
Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17–33. https://doi.org/10.1016/j.cell.2017.06.009.
Omerovic J, Laude AJ, Prior IA. Ras proteins: paradigms for compartmentalised and isoform-specific signalling. Cell Mol Life Sci. 2007;64(19–20):2575–89. https://doi.org/10.1007/s00018-007-7133-8.
Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67. https://doi.org/10.1158/0008-5472.Can-11-2612.
Henis YI, Hancock JF, Prior IA. Ras acylation, compartmentalization and signaling nanoclusters (review). Mol Membr Biol. 2009;26(1):80–92. https://doi.org/10.1080/09687680802649582.
Ryan MB, Der CJ, Wang-Gillam A, Cox AD. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1(3):183–98. https://doi.org/10.1016/j.trecan.2015.10.001.
Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21(8):1819–27. https://doi.org/10.1158/1078-0432.Ccr-14-3214.
Gentry LR, Martin TD, Reiner DJ, Der CJ. Ral small GTPase signaling and oncogenesis: more than just 15minutes of fame. Biochim Biophys Acta. 2014;1843(12):2976–88. https://doi.org/10.1016/j.bbamcr.2014.09.004.
Hle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and Clincal outcome. J Natl Cancer Inst. 2012;104(3):228–39. https://doi.org/10.1093/jnci/djr523.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–9.
Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, et al. Constitutive activation of the 41−/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene. 1999;18(3):813–22. https://doi.org/10.1038/sj.onc.1202367.
Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9(9):726–35.
Yunxia Z, Jun C, Guanshan Z, Yachao L, Xueke Z, Jin L. Mutations in epidermal growth factor receptor and K-ras in Chinese patients with colorectal cancer. BMC Med Genet. 2010;11:34. https://doi.org/10.1186/1471-2350-11-34.
Shackelford RE, Whitling NA, McNab P, Japa S, Coppola D. KRAS testing: a tool for the implementation of personalized medicine. Genes Cancer. 2012;3(7–8):459–66. https://doi.org/10.1177/1947601912460547.
Lurkin I, Stoehr R, Hurst CD, van Tilborg AA, Knowles MA, Hartmann A, et al. Two multiplex assays that simultaneously identify 22 possible mutation sites in the KRAS, BRAF, NRAS and PIK3CA genes. PLoS One. 2010;5(1):e8802. https://doi.org/10.1371/journal.pone.0008802.
Ichii-Nakato N, Takata M, Takayanagi S, Takashima S, Lin J, Murata H, et al. High frequency of BRAFV600E mutation in acquired nevi and small congenital nevi, but low frequency of mutation in medium-sized congenital nevi. J Invest Dermatol. 2006;126(9):2111–8. https://doi.org/10.1038/sj.jid.5700366.
Altimari A, de Biase D, De Maglio G, Gruppioni E, Capizzi E, Degiovanni A, et al. 454 next generation-sequencing outperforms allele-specific PCR, sanger sequencing, and pyrosequencing for routine KRAS mutation analysis of formalin-fixed, paraffin-embedded samples. OncoTargetsTher. 2013;6:1057–64. https://doi.org/10.2147/OTT.S42369.
Reda M, Richard C, Bertaut A, Niogret J, Collot T, Fumet JD, et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine. 2020;51:102624. https://doi.org/10.1016/j.ebiom.2019.102624.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. https://doi.org/10.1038/nbt.2514.
Lewandowska MA, Jóźwicki W, Jochymski C, Kowalewski J. Application of PCR methods to evaluate EGFR, KRAS and BRAF mutations in a small number of tumor cells in cytological material from lung cancer patients. Oncol Rep. 2013;30(3):1045–52. https://doi.org/10.3892/or.2013.2579.
Ney JT, Froehner S, Roesler A, Buettner R, Merkelbach-Bruse S. High-resolution melting analysis as a sensitive prescreening diagnostic tool to detect KRAS , BRAF , PIK3CA , and AKT1 mutations in formalin-fixed, paraffin-embedded tissues. Arch Pathol Lab Med. 2012;136(9):983–92. https://doi.org/10.5858/arpa.2011-0176-OA.
Monzon FA, Ogino S, Hammond ME, Halling KC, Bloom KJ, Nikiforova MN. The role of KRAS mutation testing in the management of patients with metastatic colorectal cancer. Arch Pathol Lab Med. 2009;133(10):1600–6. https://doi.org/10.1043/1543-2165-133.10.1600.
Whitehall V, Tran K, Umapathy A, Grieu F, Hewitt C, Evans TJ, et al. A multicenter blinded study to evaluate KRAS mutation testing methodologies in the Clincal setting. J Mol Diagn. 2009;11(6):543–52. https://doi.org/10.2353/jmoldx.2009.090057.
Naser WM, Shawarby MA, Al-Tamimi DM, Seth A, Al-Quorain A, Nemer AM, et al. Novel KRAS gene mutations in sporadic colorectal cancer. PLoS One. 2014;9(11):e113350. https://doi.org/10.1371/journal.pone.0113350.
Srinivasan S, Batra J. Single nucleotide polymorphism typing. J BioinformComput Biol. 2019:432–40. https://doi.org/10.1016/B978-0-12-809633-8.20127-1.
Nikiforova MN, Nikiforov YE. Molecular anatomic pathology: principles, techniques, and application to Immunohistologic diagnosis. In: Diagnostic Immunohistochemistry: WB Saunders; 2011. p. 42–57. https://doi.org/10.1016/b978-1-4160-5766-6.00006-6.
Ridanpää M, Husgafvel-Pursiainen K. Denaturing gradient gel electrophoresis (DGGE) assay for K-ras and N-ras genes: detection of K-ras point mutations in human lung tumour DNA. Hum Mol Genet. 1993;2(6):639–44. https://doi.org/10.1093/hmg/2.6.639.
Forsythe ML, Alwithenani A, Bethune D, Castonguay M, Drucker A, Flowerdew G, et al. Molecular profiling of non-small cell lung cancer. PLoS One. 2020;15(8):e0236580. https://doi.org/10.1371/journal.pone.0236580.
Zhang LL, Kan M, Zhang MM, Yu SS, Xie HJ, Gu Z-H, et al. Multiregion sequencing reveals the intratumor heterogeneity of driver mutations in TP53-driven non-small cell lung cancer. Int J Cancer. 2017;140:103–8. https://doi.org/10.1002/ijc.30437.
Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz-Ares L, Schmid-Bindert G. KRAS-mutant non-small cell lung cancer: from biology to therapy. Lung Cancer. 2018;124:53–64. https://doi.org/10.1016/j.lungcan.2018.07.013.
Wu HZ, Xiao JQ, Xiao SS, Cheng Y. KRAS: a promising therapeutic target for Cancer treatment. Curr Top Med Chem. 2019;19(23):2081–97. https://doi.org/10.2174/1568026619666190905164144.
Ahearn I, Zhou M, Philips MR. Post translational modifications of RAS proteins. Cold Spring Harb Perspect Med. 2018;8(11):a031484. https://doi.org/10.1101/cshperspect.a031484.
Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272(22):14459–64. https://doi.org/10.1074/jbc.272.22.14459.
Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11(11):775–91. https://doi.org/10.1038/nrc3151.
Novotny CJ, Hamilton GL, McCormick F, Shokat KM. Farnesyltransferase-mediated delivery of a covalent inhibitor overcomes alternative Prenylation to Mislocalize K-Ras. ACS Chem Biol. 2017;12(7):1956–62. https://doi.org/10.1021/acschembio.7b00374.
Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, et al. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2011;14(2):148–58. https://doi.org/10.1038/ncb2394.
Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L, et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 2014;157(2):459–71. https://doi.org/10.1016/j.cell.2014.02.051.
Chenard-Poirier M, Kaiser M, Boyd K, Sriskandarajah P, Constantinidou A, Harris SJ, et al. Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. J. Clin Oncol. 2017;35:2506. https://doi.org/10.1200/JCO.2017.35.15_suppl.2506.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The Clincal effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–25. https://doi.org/10.1158/1078-0432.Ccr-11-2381.
Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. 2015;75(1):183–9. https://doi.org/10.1007/s00280-014-2615-5.
Jänne PA, Smith I, McWalter G, Mann H, Dougherty B, Walker J, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer. 2015;113(2):199–203. https://doi.org/10.1038/bjc.2015.215.
Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung Cancer: the SELECT-1 randomized Clincal trial. Jama. 2017;317(18):1844–53. https://doi.org/10.1001/jama.2017.3438.
Carver J, Dexheimer TS, Hsu D, Weng M-T, Smith JL, Guha R, et al. A high-throughput assay for small molecule destabilizers of the KRAS Oncoprotein. PLoS One. 2014;9(8):e103836. https://doi.org/10.1371/journal.pone.0103836.
O'Bryan JP. Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol Res. 2019;139:503–11. https://doi.org/10.1016/j.phrs.2018.10.021.
Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–8. https://doi.org/10.1126/science.aad6204.
Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6(3):316–29. https://doi.org/10.1158/2159-8290.Cd-15-1105.
Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109(14):5299–304. https://doi.org/10.1073/pnas.1116510109.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–51. https://doi.org/10.1038/nature12796.
Blair HA. Sotorasib: first approval. Drugs Springer Nature. 2021;81:1–7. https://doi.org/10.1007/s40265-021-01574-2.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J of Med. 2020;383(13):1207–17. https://doi.org/10.1056/NEJMoa1917239.
Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med. 2021;384(25):2382–93. https://doi.org/10.1056/NEJMoa2105281.
Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, Sotorasib and Adagrasib, and overcoming strategies: insights from the in vitro experiments. J Thorac Oncol. 2021;16(1321–32). https://doi.org/10.1016/j.jtho.2021.04.015.
Calles A, Liao X, Sholl LM, Rodig SJ, Freeman GJ, Butaney M, et al. Expression of PD-1 and its ligands, PD-L1 and PD-L2, in smokers and never smokers with KRAS-mutant lung Cancer. J Thorac Oncol. 2015;10(12):1726–35. https://doi.org/10.1097/jto.0000000000000687.
D'Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br J Cancer. 2015;112(1):95–102. https://doi.org/10.1038/bjc.2014.555.
Huynh TG, Morales-Oyarvide V, Campo MJ, Gainor JF, Bozkurtlar E, Uruga H, et al. Programmed cell death ligand 1 expression in resected lung adenocarcinomas: association with immune microenvironment. J Thorac Oncol. 2016;11(11):1869–78. https://doi.org/10.1016/j.jtho.2016.08.134.
Falk AT, Yazbeck N, Thon L, Guibert N, Hofman V. Zahaf K et al impact of Kras mutant subtypes on PD-L1 expression in lung adenocarcinoma. Ann Oncol. 2016;27(6):15–42. https://doi.org/10.1093/annonc/mdw363.
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. https://doi.org/10.1016/j.tibs.2013.12.004.
Lecarpentier Y, Claes V, Vallée A, Hébert JL. Thermodynamics in cancers: opposing interactions between PPAR gamma and the canonical WNT/beta-catenin pathway. Clin Transl Med. 2017;6:14. https://doi.org/10.1186/s40169-017-0144-7.
Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2015;34:4914–27. https://doi.org/10.1038/onc.2014.416.
Pupo E, Avanzato D, Middonti E, Bussolino F, Lanzetti L. KRAS-driven metabolic rewiring reveals novel actionable targets in Cancer. Front Oncol. 2019;30:848. https://doi.org/10.3389/fonc.2019.00848.
Acknowledgments
Not applicable.
Code availability
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
RK, PK, AS, NK- collected analyzed and interpreted all the data regarding the histological examination and molecular results. SS, PM, RK- analyzed the clinical relevance of the data collected. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts/competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ritu, K., Kumar, P., Singh, A. et al. Untangling the KRAS mutated lung cancer subsets and its therapeutic implications. Mol Biomed 2, 40 (2021). https://doi.org/10.1186/s43556-021-00061-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-021-00061-0