
Abstract
Lung cancer is the leading cause of mortality among all cancer types worldwide. The latest available global statistics of the World Health Organization report 1.59 million casualities in 2012. Worldwide, 1 in 5 cancer deaths are caused by lung cancer. In 2016, in the United States alone, there are an estimated 224,390 new cases of lung cancer, of which 158,080 are expected to result in death, as reported by the National Cancer Institute. Non-small cell lung cancer (NSCLC), a histological subtype, comprises about 85% of all cases, which is nearly 9 out of 10 lung cancer patients. Efforts are under way to develop and improve targeted therapy strategies. Certain mutations are being clinically targeted, such as those in EGFR and ALK genes. However, one of the most frequently mutated genes in NSCLC is the Kirsten rat sarcoma viral oncogene homolog (KRAS), which is currently not targetable. Approximately 25% of all types of NSCLC tumors contain KRAS mutations, which remain as an undruggable challenge. These mutations are indicative of poor prognosis and show negative response to standard chemotherapy. Furthermore, tumors harboring KRAS mutations are unlikely to respond to currently available targeted treatments such as tyrosine kinase inhibitors. Therefore, there is a definitive, urgent need to generate new targeted therapy approaches for KRAS mutations. Current strategies have major limitations and revolve around targeting molecules upstream and downstream of KRAS. Direct targeting is not available in the clinic. Combination therapies using multiple agents are being sought. Concentrated efforts are needed to accelerate basic research and consecutive clinical trials to achieve effective targeting of KRAS.
Similar content being viewed by others

Introduction
Statistics from 2013 onward indicate more than 200,000 new lung cancer cases in the United States alone, of which more than 150,000 are estimated to result in death. Worldwide statistics show lung cancer as the third most common cancer type. (1–4) In 2012, approximately 1,825,000 new cases were diagnosed globally. Incidence rates are shown to be highest in North America. (2,4) In Europe, lung cancer is the fourth most common cancer type, and 410,000 new cases were diagnosed in 2012. (2,4)
Lung cancer is divided into 2 major histological subtypes: small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). This review focuses on NSCLC. Early diagnosis for NSCLC has been difficult. Cancer Research UK reports the following diagnosis percentages: in stage I, 14.5%; in stage II, 7.3%; in stage III, 31.8%; and in stage IV, 35.8%. (2) This is mostly due to symptoms not immediately providing clues to the disease, such as persistent cough, which is already present in most smokers. This can lead to misdiagnosis of infection. For patients diagnosed at stage IV, where NSCLC is highly metastatic, the median overall survival (OS) is less than a year. (2,3)
Recently, there has been considerable progress in the survival of patients who are able to receive targeted therapy based on molecular profiling of their tumors. For such cases, the median OS was measured to be more than 3 years. Examples include erlotinib targeting EGFR mutations and crizotinib targeting ALK mutations. (1) Lung cancers very frequently harbor somatic KRAS mutations. (5) Mutations in this particular gene comprise about 25% of NSCLC tumors. (6,7) However, these mutations are not targetable and are indicative of poor prognosis. (8) In addition, tumors harboring KRAS mutations are unlikely to respond to any currently available targeted treatment. (1,3,7,9) In this review, KRAS genotypes in NSCLC are discussed with respect to the lack of and the need for novel molecular agents as targeted therapies.
Types of NSCLC
NSCLC consists of subtypes grouped according to their histological distinctions: lung adenocarcinomas (LACs), which arise in cells lining the alveoli, account for about 40%; squamous cell carcinomas (SCCs) account for about 25% to 30%; and large-cell carcinomas (LCCs) account for about 10% to 15% of NSCLC cases. (3,10) SCC is reported to start in the early stages of squamous cells, which are mostly found near the bronchus in the middle of the lungs. Carcinoma starting in the early stages of squamous cells is generally linked to smoking history. (3) Adenocarcinoma is the most common subtype and is characterized by cells that normally secrete mucus. This type is mostly seen in current or former smokers. Interestingly, it is also the most common type in nonsmokers. (3) LCC can start in any part of the lungs. In comparison with other types, it tends to grow and spread faster, thus it becomes harder to follow and apply treatment. Aside from histological distinctions, molecular subtypes of NSCLC are continually expanding and are distinct per patient. Importantly, each individual case could result from various different molecular profiles, which is a challenge for treatment. (11) In this regard, tumor sequencing becomes useful.
Tumor Sequencing and Mutations
Revolution in sequencing technologies has had major impact on identification and understanding of molecular variations within tumor genomes. Current developments in next-generation sequencing (NGS) enable scientists to analyze whole genomes in less than 2 wks. (12,13) The steadily decreasing cost of NGS is making tumor sample sequencing more accessible. Comparison of tumor genome and normal genome sequences of patients is in clinical use and is expected to become a standard clinical tool in identifying genome-wide cancer variations. Based on sequencing results, a better understanding of the molecular underpinnings of the tumor becomes possible, which is significant in guiding patients for mutation-specific treatment.
There are several challenges in NGS application. First, sequencing DNA from tumor tissue is challenging in itself. Tumor tissue samples are often heterogeneous, which leads to further difficulty in interpretation of driver mutation(s). (12–14) NSCLC tumors display high heterogeneity. However, as noted by Alsdorf et al. in 2013, in NSCLC tumor heterogeneity regarding the KRAS mutation is rare. KRAS mutation appears to be an early event in tumorigenesis and is a true driver mutation. (15) Furthermore, in most cases, biopsy is performed only once, leading to a single tumor sample from a specific body part. However, cancer keeps evolving and the tumor molecular profile keeps changing, presenting an additional challenge in identifying mutation-specific therapy options. (14)
Additionally, there are technical challenges due to elementary sequencing platform differences and error rates. This accuracy problem of NGS presents a challenge and can be overcome by resequencing the same genome several times. (12,13) Moreover, NGS coverage is also of importance. Simply put, higher levels of coverage indicate higher degrees of confidence. Differences in testing methods have been linked with identification of different molecular findings within the same NSCLC patient. (13) Comprehensive studies need to be designed to cover multiple platforms in cross-comparative analyses to improve detection rates overall.
Finally, rapid evolution of high-throughput technology has greatly moved forward the field of cancer biology. However, the main challenge in tumor sequencing is bioinformatics analysis. Without bioinformatics interpretation, molecular findings will not be of clinical use. Translation of NGS into clinics will only be plausible by validation and assembly of data through computational tools. (12,13,16) Future work is likely to shift in a direction to integrate pathway data, showing gene interactions specific to patients. Pathway knowledge is expected to prove useful for targeted treatment strategies. (17)
Mutations and Personalized Targeted Treatment
Nowadays, genotype-driven therapy is a standardized treatment method for patients diagnosed with a prominent subtype of NSCLC. (18) A significant amount of attention has been paid to developing precision treatment strategies targeting EGFR and ALK mutations for adenocarcinoma. NSCLC driver mutations occur in many other oncogenes, such as AKT1, BRAF, HER2, KRAS, MEK1, MET, NRAS, PIK3CA, RET and ROS1. Figure 1 shows the distribution of driver mutation frequencies in NSCLC tumors. As can be seen, KRAS mutations comprise a very large proportion of the driver genotypes.

Frequency of driver mutations in NSCLC. Red: KRAS genotypes; blue: other genotypes. (Data obtained from https://mycancergenome.org/ on 2 June 2016.)
Thus far, mutations in EGFR and translocations and rearrangements in ALK are the most effectively targeted oncogenes using specific tyrosine kinase inhibitors (TKIs) such as afatinib, crizotinib, erlotinib and gefinitib. (1) However, currently there is no targeted drug treatment for KRAS mutations.
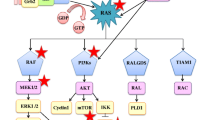
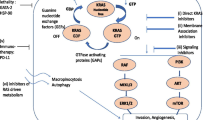
KRAS oncogene. KRAS functions by propagating signal transduction pathways upon activation and is involved in cell differentiation, cell growth, apoptosis, cell survival and cell proliferation. KRAS is one of the initially defined oncogenes and belongs to the Ras family with other genes including HRAS and NRAS. Different Ras proteins are involved in different cancer types. Ras molecules are downstream of growth factor receptors and upstream of pathways such as RAF-MEK-ERK, critical for cellular proliferation, and PI3K-AKT-mTOR, critical for cell survival (1,5) (Figure 2).

KRAS-related pathways. Targeted agents for pathogenic mutations involved in NSCLC are shown. (Comprehensive information available at: https://mycancergenome.org.)
KRAS is a GTPase, which interacts with a set of activators and effectors. KRAS is activated with the binding of GTP and begins transmitting signals. KRAS is inactivated when GTP is converted to GDP, and stops signaling. (19) When mutated, KRAS becomes constitutively active, which results in continuous cellular proliferation independent from its upstream effector epidermal growth factor receptor (EGFR) (5,19) (Figure 2).
KRAS genotypes in NSCLC. In addition to lung cancer, KRAS mutations are frequently found in colon and pancreatic cancers, which are among the most aggressive, deadliest forms of cancer. (1,3,10) KRAS mutations most often arise in smokers or former smokers. However, they are not exclusive to smokers. Recent studies document substantial KRAS mutation cases in patients who have never smoked. (7)
The prognostic value of KRAS mutations in NSCLC has been controversial. (20) However, there are accumulating studies reporting poor prognosis and worse OS of patients with KRAS mutations compared with other NSCLC patients. A recent metaanalysis carried out by Meng et al. showed that KRAS mutations have an inverse association with OS in adenocarcinoma patients. (21) In addition, these mutations are negatively associated with response to standard chemotherapy, as well as certain available targeted therapies such as EGFR TKIs. (22)
Specific KRAS genotypes include those in the 12th and 13th codons. Mutations are mainly missense mutations with a single nucleotide change. The most commonly found genotypes are G12C, G12D and G12V (Table 1). KRAS mutations are generally mutually exclusive of other driver mutations and genetic anomalies such as BRAF and EGFR mutations and ALK and ROS1 rearrangements. (1) However, there are exceptional cases showing co-mutations. EGFR and KRAS coexistence as well as PIK3CA and KRAS coexistence have recently been reported. (23)
Targeting KRAS
KRAS is a mandatory target for NSCLC as one of the most frequently mutated genes. KRAS targeting strategies could include targeting upstream and/or downstream molecules, as well as direct targeting. Currently, a direct target molecule for KRAS is not available in the clinic. Earlier efforts proved farnesyltransferase inhibitors as direct targets to be of minimal use. (9)
Most of the signaling cascades downstream of KRAS have been described as shown in Figure 2. Targeting KRAS indirectly has been inefficient. (24) The focus was mainly on inhibiting the activities of RAF-MAPK and PI3K-AKT pathways. Yet many of the attempts to target these cascades did not provide major improvement in treatment. Figure 2 shows KRAS-related pathways and available targeted treatment agents for NSCLC driver mutations. It should be noted that inhibitors specifically designed for a certain gene are also being clinically investigated to target other mutations.
A novel approach being sought is to inhibit multiple agents in the pathways KRAS utilizes, with the main idea being parallel inhibition of different agents linked in different pathways. (24) In this respect, the linkage of EGFR-RAS-RAF-MAPK cascades is critical due to their function in cell proliferation, especially when their frequent mutations in cancers are considered.
Direct Targeting of KRAS
Recently, direct targeting of the KRAS G12C genotype with irreversible inhibitors has been reported to show activity in preclinical models. Ostrem et al. developed a small molecule that irreversibly binds to mutated KRAS (G12C) and does not have any effect on wild-type KRAS. When these inhibitors are bound to KRAS (G12C), GDP is preferred over GTP, inhibiting RAF binding. These cysteine-reactive small molecules selectively bind to mutant KRAS, providing promising results in vitro. (25)
Hunter et al. developed a molecule called SML-8-73-1, which irreversibly inhibits KRAS, specifically the G12C driver mutation. It was observed that SML-8-73-1 binds to G12C-mutated KRAS irreversibly and selectively, even under circumstances where GDP and GTP were found in high levels. The significance of SML-8-73-1 is that it is the first GTP-competitive inhibitor discovered so far. (26) The main challenge in targeting KRAS directly has been its constant activation; it binds to GTP as long as there is no other agent to take GTP’s place. SML-8-73-1 provides inactivation of KRAS by competing with GTP and binding to KRAS irreversibly. It was shown that SML-8-73-1 is highly selective for the KRAS G12C genotype, regardless of the GTP concentration. (26)
On the other hand, previous findings by Sunaga et al. in a knockdown study suggested that targeting KRAS directly and alone does not provide successful results for treatment. The authors concluded that direct targeting combined with other strategies could potentially provide better results. (27)
Targeting Upstream of KRAS
KRAS functions downstream of receptor tyrosine kinases (RTKs), some of which are growth factors, which provide targeted therapy options in NSCLC (Figure 2). In this section, some of the upstream molecules of KRAS implicated in NSCLC are detailed.
Anaplastic lymphoma kinase. Anaplastic lymphoma kinase (ALK) is an RTK that regulates multiple cellular processes. (1) ALK gene rearrangements cause fusion with the echinoderm microtubule-associated protein-like 4 (EML4) gene, which leads to its activation in cancer cells. About 2% to 5% of NSCLC cases, mostly young adenocarcinoma patients with no former smoking history, carry these rearrangements (Figure 1). ALK mutations are generally mutually exclusive of EGFR and KRAS mutations. (28) Several inhibitors for ALK mutations in NSCLC are in use, such as alectinib, ceritinib and crizotinib (Figure 2). ALK-positive NSCLC patients have high response rates of about 60% for crizotinib. However, most patients with ALK mutations develop drug resistance and undergo relapse after a few years. Based on these limitations about crizotinib, several other ALK inhibitors are under development. (29)
Epidermal growth factor receptor. Epidermal growth factor receptor (EGFR) is an RTK, activated by ligand binding, which in turn activates PI3K and MAPK/ERK pathways. (1) EGFR is frequently mutated in NSCLC (Figure 1) and is being clinically targeted with TKIs (Figure 2). Mutations in EGFR increase its activity, causing a chain hyperactivation of the RAS pathway downstream of it. KRAS mutations decrease the responsivity of the tumor cell to EGFR TKIs. (11) On the other hand, EGFR and KRAS co-mutations are reported to be rare, and such tumors are also unlikely to be responsive to EGFR TKIs. (23,30) The EGFR TKIs afatinib, erlotinib and gefitinib are being clinically used for NSCLC patients harboring specific EGFR mutations, either combined with chemotherapy drugs or alone. EGFR inhibitors decrease tumor formation for several months, but eventually patients stop responding most of the time. (3)
Fibroblast growth factor receptor 1. FGFR1 is an RTK in the fibroblast growth factor receptor (FGFR) family (Figure 2). When activated, FGFR1 affects the dowstream effectors, influencing cell differentiation and mitogenesis. (11) It is mainly involved in cell proliferation, differentiation and migration and embryonic development regulation. It is involved in the activation of MAPK/ERK pathways. In different cancer types, mutated FGFRs are shown to be deregulated by translocation, amplification and point mutation. Mutated FGFR1 is more frequently involved in SCCs (20%)than with adenocarcinomas (5%). Clinical trials for inhibitors of FGFRs are currently ongoing. (11)
Human epidermal growth factor receptor 2. Human epidermal growth factor receptor 2 (HER2) belongs to the ERBB RTK family (Figure 2). HER2 amplifications are found in 2% to 4% of NSCLCs (Figure 1). Adenocarcinomas harbor HER2 mutations more prevalently compared with other types. HER2 amplifications have negative prognostic value, and several HER2 targeted therapies are available in clinic and in trial. (11,31) An inhibitor developed for EGFR, afatinib, has also shown activity against HER2 mutations. (32) A combination of an irreversible pan-HER inhibitor, neratinib, and an mTOR inhibitor, temsirolimus, provided clinical activity in patients with HER2 mutations. (33) Initial clinical trials using available therapeutic agents, such as trastuzumab, together with chemotherapy to target HER2 in NSCLC were unsuccesful, especially with amplified HER2. Some inhibitors under investigation to target HER2 are afatinib, dacomitinib and neratinib. (31) In addition, some HER2-directed antibodies such as pertuzumab or trastuzumab are being studied. (11)
Mesenchymal-epidermal transition. Mesenchymal-epidermal transition (MET) is a proto-oncogene RTK controlling tyrosine kinase activities. MET activates several signaling pathways such as RAS-RAF-MAPK and PI3K-AKT-mTOR by going through homodimerization and autophosphorylation (Figure 2). In NSCLC, specifically in adenocarcinomas, MET amplifications are found in around 2% to 5% of cases (11,34) (Figure 1). Crizotinib, an ALK/ROS1 inhibitor, was first developed for inhibiting MET and demonstrates clinical activity in tumors with MET amplification. (29) Erlotinib and tivantinib are being clinically evaluated for MET inhibition. (35) Other TKIs are still under clinical investigation for MET inhibition, such as cabozantinib and forenitib. (11,36)
Targeting Downstream Effectors
In some NSCLC tumors, downstream effectors of KRAS are mutated, although not as frequently as KRAS itself (Figure 1). These effectors are potential target molecules for inhibition of constitutive KRAS activation (Figure 2). Many of the downstream effectors are kinases, presenting a targetable opportunity via kinase inhibitors. (24) It has also been shown that some downstream effectors are frequently co-mutated with KRAS. For example, Wang et al. recently reported that PIK3CA and KRAS mutations co-occur in high frequencies. (37) In general, with co-mutations, targeted therapy becomes even more challenging and thus a more negative prognosis is expected. (37,38) In this section, some of the downstream effectors of KRAS implicated in NSCLC are detailed.
v-Raf murine sarcoma viral oncogene homolog B. RAF proteins are downstream effectors of the RAS protein cascade. They belong to a kinase family that transmit signals from growth factor receptors outside the cell to the nucleus. (11) v-Raf murine sarcoma viral oncogene homolog B (BRAF), found downstream of KRAS, is upstream of MEK-ERK and functions as a serine threonine kinase, which affects cell division, differentiation and secretion (Figure 2). BRAF mutations are involved in several cancer types. (1,6) Up to 5% of NSCLC adenocarcinomas contain BRAF mutations, specifically the V600E genotype (11,39) (Figure 1). Studies show that when mutated, BRAF can signal independent from its upstream effectors. Due to BRAF’s constant activation, downstream molecules MEK and ERK become overactivated, leading to uncontrolled and excess cell growth independent from the growth factors, and resistance against apoptosis can arise. In preclinical studies, BRAF mutations were reported to be sensitive to MEK inhibitors. Moreover, combining BRAF and MEK inhibitors showed activity as well. (11)
MEK. MEK proteins are a family of enzymes upstream of mitogen-activated protein kinases (MAPK), with mutations in about 1% of NSCLC cases (Figure 1). Together with conventional chemotherapy, inhibition of MEK, a downstream effector of KRAS, appears to be a promising approach to some extent (Figure 2). In a clinical trial, NSCLC patients with KRAS mutation were given the MEK inhibitor selumetinib in addition to the chemotherapy drug docetaxel. They showed increased overall response rate and improved median progression-free survival rates, along with improvement in OS. Yet the combination of docetaxel and MEK inhibitor also gave rise to side effects such as neutropenia and penumonia. (40) Another MEK inhibitor, trametinib, was reported to show activity in patients with KRAS mutation when given together with chemotherapy drugs, but failed to improve the therapy outcome when applied alone, in comparison with chemotherapy alone. (41)
Phosphatidylinositol 3-kinase. Phosphatidylinositol 3-kinase (PI3K) is a pathway involved in cell growth and survival (Figure 2). The PI3K-AKT-mTOR pathway is a very complicated signaling cascade and is frequently deregulated in several cancer types. (1,37,42) PI3K is involved in the phosphorylation of AKT when activated. Phosphorylated AKT affects the downstream signaling cascades, which have roles in cell proliferation, survival, motility and invasion. About 1% to 3% of all NSCLC cases harbor somatic PIK3CA mutations (1) (Figure 1). PIK3CA mutations frequently coexist with other driver mutations. This indicates that PIK3CA mutations are generally not driver mutations in NSCLC. These mutations also confer an acquired resistance against EGFR TKIs. (37,43,44) PIK3CA mutations provide prognostic value for EGFR/KRAS wild-type patients, therefore determining the PIK3CA mutation status could prove useful for personal therapeutic strategies. (37) Tumors with PIK3CA mutations are reported to be highly susceptible to PI3K inhibitors, as the preclinical data indicates. (42,43)
RET. RET is an RTK and an oncogene involved in several cancer types with gain of function mutations. RET translocations are found in 1.5% of NSCLC cases, specifically in adenocarcinomas (Figure 1). Translocations in RET do not coexist with other driver mutations. In NSCLC, alectinib, an inhibitor developed for ALK, has shown activity against RET alterations. Other inhibitors for RET, such as sorafenib, sunitinib and vandetanib, caused loss of cell viability. In a precilinical model, cabozantinib, a multityrosine kinase inhibitor, showed partial response. In addition, vandetanib has shown activity in 2 clinical trials of adenocarcinoma patients harboring RET translocations. Clinical trials on cabozantinib, vandetanib and other potential inhibitors to evaluate activity against RET are still ongoing.(11,47)
C-ros oncogene 1. C-ros oncogene 1 (ROS1) belongs to the insulin receptor family and is an RTK (Figure 2). ROS1 rearrangements are found in 0.7% to 1.7% of NSCLC cases (Figure 1), mainly in adenocarcinomas and mostly in light smokers or never-smoking young patients. Crizotinib, which was developed as a MET inhibitor, has also displayed activity against ROS1 rearrangements. However, some novel mutations in ROS1 showed resistance against crizotinib. (11) There is also a novel approach under investigation for patients with ALK or ROS1 translocations to overcome resistance against crizotinib, using a combination of ALK/ROS1 and heat shock protein 90 inhibitors. (45) Other inhibitors under investigation include ceritinib, foretinib, AP26113 and PF-06463922. As an inhibitor of MET/RET/VEGFR pathways, cabozantinib revealed activity in alterations resistant to ROS1 inhibitor in a preclinical model. (36,46)
Discussion
There are several factors challenging treatment of NSCLC. One is the histological and molecular subtypes of NSCLC. (48) A 2015 article suggests that an EGFR-mutant lung adenocarcinoma can transform into SCC by formation of a secondary mutation, which presents a challenge for treatment with EGFR inhibitors. (49) Another recently published article accentuates a new potential subtype as adenosquamous carcinoma (ADSQ), which can also be a transition state between 2 predominant histological subtypes, SCC and adenocarcinoma. ADSQ accounts for 2% to 4% of NSCLC subtypes. It is not yet clear if ADSQ is a hybrid or a genuine type, owing to its morphological pattern consisting of both SCC and adenocarcinoma characteristics. Yet ADSQ is found to be more aggressive compared with these subtypes. Patients with ADSQ could benefit from targeted therapy, due to infrequent KRAS mutation compared with EGFR mutation. (50) Even though adenocarcinoma is less aggressive than ADSQ, treatment remains challenging due to the highly frequent KRAS mutation, once again emphasizing the difficulty of KRAS mutation presence in targeted therapies.
Another challenge stems from cancer evolution. Cancer is a an evolving, multistep process, where tumor heterogeneity is a fact. (14) KRAS mutations occur early in cancer progression, thus targeting KRAS alone might not be a sufficient therapeutic approach, especially in advanced NSCLC. (51) Furthermore, sequencing cancer tissue is generally done only once. For improvement in molecular profiling, when possible, sequencing could be done several times during the course of treatment, to guide treatment and combat drug resistance.
Co-mutations present another challenge. As long as KRAS co-occurs with other driver mutations, treatment will not be as effective and will require inhibition of multiple agents (Figures 1 and 2). Resistance to current therapy can be a result of co-occuring driver mutations and the tumor can relapse. This was documented in a clinical trial, where knocking down of KRAS did not prevent uncontrolled cell proliferation, due to abnormalities in associated molecules and pathways. (27)
KRAS mutations also co-occur with mutations in STK11 and TP53 tumor suppressor genes. (52) Coexistence of these mutations has a critical effect on KRAS-driven lung cancers in the sense of tumor formation, immune surveillance and proliferative response. TP53 and STK11 can promote tumor progression with different mechanisms. TP53 mutations have a role in increased cell proliferation, where STK11 mutations were understood to be related to supression of immune response to the tumor. Mutated STK11 is reported to increase KRAS signal transduction, both independent and dependent of the coexisting mutated KRAS. (52)
Recently, Zhu et al. suggested that multitargeted therapy is required to combat NSCLC. Combining a group of inhibitors in a novel fashion could provide better results. (24) However, in combination therapies, drug-drug interactions are a challenge and can cause resistance. As an alternative, a combination of therapies targeting KRAS directly and inhibiting other factors, either ones that have already provided considerable results or novel agents, in the cascades where KRAS is involved seems to be more promising. When putting these appraoches into clinical use, it should be noted that combining drugs with different mechanisms can result in either overlapping or nonoverlapping toxicities, negatively affecting the flow of the treatment. (24)
It should also be noted that recent developments in cancer immunotherapy could be promising in NSCLC, although these are not discussed here, as they are outside of this review’s scope. Briefly, in 2015, 2 new immunotherapy agents, nivolumab and pembrolizumab, were approved for lung cancer treatment after standard chemotherapy has stopped working. These immune checkpoint inhibitor antibodies function by interfering with the programmed death protein 1 (PD-1) signaling, with the aim of improving immunity against tumor. (53)
Conclusion
The majority of NSCLC cases are not yet addressed by any available targeted therapy. Specifically, as outlined in this review, KRAS genotypes, covering the majority of driver mutations, do not have a target match in clinic and are not responsive to standard chemotherapy. NSCLC consists of a large number of genomic anomalies, and the documented aberrations are increasing, making NSCLC harder to cope with.
The combined use of multiple signaling inhibitors targeting more than one pathway is conceptually appealing. Although clinical experience with this approach is limited, to date such combination therapies have not shown great promise. The reason for this lack of success is not clear, but it is thought to be due to cumulative toxicity of the combined agents and/or limited suppression of signaling by the inhibitors at the doses used. Despite the promise, it is possible that the extensive subclonal tumor heterogeneity of KRAS-driven NSCLC may make even the inhibition of multiple downstream effectors insufficient for long-term tumor control and prolonged survival.
Concentrated research efforts should be made in exploring and developing inhibitors to target KRAS as well as potential molecules in the associated signaling pathways. Combining the efforts of collaborative international groups and studying international patient data could facilitate and hasten discovery. More funding in this respect, as well as increasing the numbers and availability of international clinical studies, could prove useful. New efforts to ensure that even negative results of clinical trials are published will also help focus future efforts in new, potentially more successful directions.
Dedication
Dedicated to the loving memory of an amazing father, A. Ertuğ Taneri.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
My Cancer Genome online. c2010–2016. Nashville (TN): Vanderbilt-Ingram Cancer Center; [accessed 2016 Jan 8]. http://www.mycancergenome.org.
Cancer Research UK online. c2013. London (UK): British Journal of Cancer, National Cancer Research Institute, Cancer Research Technology; [accessed 2016 Feb 9]. http://www.cancerresearchuk.org.
American Cancer Society online. c2016. New York (NY): National Health Council, Health On The Net, Better Business Bureau; [accessed 2016 Feb 5]. http://www.cancer.org.
World Health Organization online. c2016. Geneva (Switzerland): Worldwide Collaboration Centers; [accessed 2016 Feb 9]. http://www.who.int.
Riely GJ, Marks J, Pao W. (2009) KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 6(2):201–05.
Brose MS et al. (2002) BRAF and RAS mutations in human lung cancer and melanoma. Cancer. Res. 62(23):6997–7000.
Riely, GJ et al. (2008) Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res. 14(18):5731–34.
Sun JM, Hwang DW, Ahn JS, Ahn MJ, Park K. (2013) Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One. 8(5):e64816.
Sequist LV, Neil JW. (2015) Personalized, genotype directed therapy for advanced non-small cell lung cancer; [Accessed 2016 Jan 13]. Available at: http://www.uptodate.com/contents/personalized-genotype-directed-therapy-for-advanced-non-small-cell-lung-cancer.
Globocan online. c2012. Lyon (France): International Agency for Research on Cancer; [accessed 2016 Apr 14]. http://globocan.iarc.fr.
Rothschild SI. (2015) Targeted Therapies in Non-Small Cell Lung Cancer: Beyond EGFR and ALK. Cancers. 7(2):930–49.
Li W, Zhao K, Kirberger M, Liao W, Yan Y. (2015) Next generation sequencing technologies in cancer diagnostics and therapeutics. Cell Mol. Biol. 61(5):91–102.
Meldrum C, Doyle MA, Tothill RW. (2011) Next-Generation Sequencing for Cancer Diagnostics: a Practical Perspective. Clin. Biochem. Rev. 32(4):177–95.
Majem M, Remon J. (2013) Tumor heterogeneity: evolution through space and time in EGFR mutant non-small cell lung cancer patients. Transl. Lung Cancer Res. 2(3):226–37.
Alsdorf WH, et al. (2013) Intratumoral heterogeneity of KRAS mutation is rare in non-small-cell lung cancer. Exp. Mol. Pathol. 94(1):155–59.
Mount DW, Pandey R. (2005) Using bioinformatics and genome analysis for new therapeutic interventions. Mol. Cancer Ther. 4:1636.
Balbin OA, et al. (2013) Reconstructing targetable pathways in lung cancer by integrating diverse omics data. Nat. Commun. 4:2617.
The Clinical Lung Cancer Genome Project, Network Genomic Medicine. (2013) A genomics-based classification of human lung tumors. Sci. Transl. Med. 5(209):209ra153.
Karachaliou N, et al. (2013) KRAS mutations in lung cancer. Clin. Lung Cancer. 14(3):205–14.
Califano R, Landi L, Cappuzzo F. (2012) Prognostic and predictive value of K-RAS mutations in non-small cell lung cancer. Drugs. Suppl. 1:28–36.
Meng D, et al. (2013) Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: a systematic review with meta-analysis. Lung Cancer. 81(1):1–10.
Hames ML, et al. (2016) Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer. Lung Cancer. 92:29–34.
Li S, et al. (2014) Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer. 110(11):2812–20.
Zhu Z, Golay HG, Barbie DA. (2014) Targeting pathways downstream of KRAS in lung adenocarcinoma. Pharmacogenomics. 15(11):1507–18.
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. (2013) K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503(7477):548–51.
Hunter JC, et al. (2014) In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. U.S.A. 111(24):8895–900.
Sunaga N, et al. (2011) Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol. Cancer Ther. 10(2):336–46.
Gridelli C, et al. (2014) ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat. Rev. 2:300–06.
Awad MM, Shaw AT. (2014) ALK inhibitors in non-small cell lung cancer: crizotinib and beyond. Clin. Adv. Hematol. Oncol. 7:429–39.
Benesova L, Minarik M, Jancarikova D, Belsanova B, Pesek M. (2010) Multiplicity of EGFR and KRAS mutations in non-small cell lung cancer (NSCLC) patients treated with tyrosine kinase inhibitors. Anticancer Res. 30(5):1667–71.
Garrido-Castro AC, Felip E. (2013) HER2 driven non-small cell lung cancer (NSCLC): potential therapeutic approaches. Transl. Lung Cancer Res. 2(2):122–27.
Yonesaka K, et al. (2015) The pan-HER family tyrosine kinase inhibitor afatinib overcomes HER3 ligand heregulin-mediated resistance to EGFR inhibitors in non-small cell lung cancer. Oncotarget. 32:33602–11.
Gandhi L, et al. (2014) Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J. Clin. Oncol. 2:68–75.
Park S, et al. (2015) MET amplification, protein expression, and mutations in pulmonary adenocarcinoma. Lung Cancer. 3:381–87.
Sharma N, Adjei AA. (2011) In the clinic: ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther. Adv. Med. Oncol. (1 Suppl):S37–50.
Katayama R, et al. (2015) Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin. Cancer Res. 1:166–74.
Wang L, et al. (2014) PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One. 9(2):e88291.
Richer AL, Friel JM, Carson VM, Inge LJ, Whitsett TG. (2015) Genomic profiling toward precision medicine in non-small cell lung cancer: getting beyond EGFR. Pharmgenomics Pers. Med. 8:63–79.
Naoki K, Chen TH, Richards WG, Sugarbaker DJ, Meyerson M. (2002) Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 62(23):7001–03.
Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. (2013) MEK and the inhibitors: from bench to bedside. J. Hematol. Oncol. 12:6–27.
Mas C, et al. (2015) Antitumour efficacy of the selumetinib and trametinib MEK inhibitors in a combined human airway-tumour-stroma lung cancer model. J. Biotechnol. 205:111–19.
Rodon J, Dienstmann R, Serra V, Tabernero J. (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 3:143–53.
Chaft JE, et al. (2012) Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol. Cancer Ther. 2:485–91.
Ludovini V, et al. (2012) Optimization of patient selection for EGFR-TKIs in advanced non-small cell lung cancer by combined analysis of KRAS, PIK3CA, MET, and non-sensitizing EGFR mutations. Cancer Chemother. Pharmacol. 5:1289–99.
Kohno T, et al. (2015) Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 4(2):156–64.
Zou HY, et al. (2015) PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc. Natl. Acad. Sci. U.S.A. 11:3493–98.
Kodama T, et al. (2014) Alectinib shows potent antitumor activity against RET-rearranged non-small cell lung cancer. Mol. Cancer Ther. 12:2910–18.
Cancer Genome Atlas Research Network. (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature. 511(7511):543–50.
Jukna A, et al. (2015) Squamous Cell Carcinoma “Transformation” Concurrent with Secondary T790M Mutation in Resistant EGFR-Mutated Adenocarcinomas. J. Thorac. Oncol.(15):00213–20.
Vassella E, et al. (2015) Molecular profiling of lung adenosquamous carcinoma: hybrid or genuine type? Oncotarget. 15;6(27):23905–16.
Baines AT, Xu D, Der CJ. (2011) Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3(14):1787–1808.
Schabath MB, et al. (2015) Differential association of STK11 and TP53 with KRAS mutationassociated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 35(24):3209–16.
Langer CJ. (2015) Emerging immunotherapies in the treatment of non-small cell lung cancer (NSCLC): the role of immune checkpoint inhibitors. Am. J. Clin. Oncol. 4:422–30.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Kilgoz, H.O., Bender, G., Scandura, J.M. et al. KRAS and the Reality of Personalized Medicine in Non-Small Cell Lung Cancer. Mol Med 22, 380–387 (2016). https://doi.org/10.2119/molmed.2016.00151
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2016.00151