
Abstract
The KRAS mutation remains the most common driver mutation in patients with non-small cell lung cancer (NSCLC) and confers a poor prognosis. Thus far, efforts to target this mutation over the last two decades have been unsuccessful. Over the past 5 years, many efforts to develop drugs that target the RAS-RAF-MEK-ERK (MAPK) pathway have resulted in enhanced understanding of the KRAS mutant NSCLC and have provided optimism that this disease can be targeted.
Similar content being viewed by others
Introduction
Lung cancer is the leading cause of cancer mortality in the United States [1]. Currently, the 5-year survival remains an abysmal 17 %. In 2015, in the US alone, an estimated 158,040 patients are expected to die of lung cancer accounting for 27 % of all cancer deaths and accounts for more deaths than prostate, colorectal and breast cancers combined [1]. The advances made in the last decade have revealed that non-small cell lung cancer (NSCLC) is not a single cancer, but rather a collection of molecularly defined neoplasms with distinct biology and clinical outcomes. This principle is supported by the seminal finding that sensitizing epidermal growth factor receptor (EGFR) mutations are present in about 17 % of lung cancers and are targetable with the FDA approved EGFR tyrosine kinase inhibitors (TKIs) (erlotinib, gefitinib and afatinib) [2, 3]. Additionally, translocations involving the anaplastic lymphoma kinase (ALK) are found in 7 % of adenocarcinomas of the lung and can be targeted by several ALK TKIs (crizotinib and ceritinib) [2, 3]. However, currently there are no FDA approved drugs that target the most common driver oncogenic driver, mutant KRAS [4].
The KRAS mutation is present in approximately 25 % of patients with NSCLC (mostly adenocarcinoma) and was first discovered more than 3 decades ago [5]. This mutation confers a poor prognosis in the metastatic setting, and a high risk of cancer recurrence as seen in several studies [6–9]. It is mutated in one-third of all cancers including colon cancer and pancreatic cancer. In addition, the three human RAS genes (NRAS, HRAS, KRAS) have been identified and mutations in these three isoforms have been seen throughout human cancers [5].
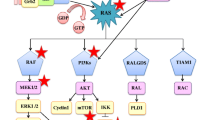
In lung cancer, KRAS mutations occur frequently at codons 12 and 13 and less frequently at codon 61 [10]. The most frequently observed mutation in lung cancer is G12C and is associated with exposure to tobacco [11]. The mutation G12C accounts for 40 % of total mutations, followed by G12V (22 %) and G12D (16 %) [12, 13]. Interestingly, KRAS mutations at G12C and G12V have a worse clinical outcome possibly due to their ability to engage in multiple downstream effectors including the RAL pathway [14]. Conversely, the G12D mutant protein predominantly activates the RAF/MAPK and PI3K pathways [14] (Fig. 1). Finally, codon 61 mutant are more severely deficient in intrinsic GTPase activity and may therefore have increased activity compared to alterations at codons 12 and 13 [15].

The RAS-RAF-MEK-ERK Signaling Cascade in NSCLC. In a normal cell, the activation of the KRAS protein through binding of GTP and translocation to the plasma membrane is a tightly controlled process. However, in NSCLC, the KRAS protein is often mutated at codons 12, 13, and 61 leading to the inactivation of its intrinsic GTPase activity resulting in constitutive activation of KRAS. Mutant KRAS can then promote tumorigenesis through multiple downstream signaling pathways
Regardless of the site of the mutation, these mutations result in loss of GTPase activity making this oncoprotein constitutively active and leading to activation of a series of downstream pathways including the RAF-MEK-ERK (MAPK) signaling pathway and AKT-PI3K-MTOR pathway (Fig. 1). Thus far, efforts to inhibit KRAS have focused primarily on targeting the RAS-RAF-MEK-ERK (MAPK) signaling pathway.
RAS-RAF-MEK-ERK pathway
In the RAS signaling cascade, the binding of either GTP or GDP to RAS serves as the “on” or “off” switch for RAS signaling respectively. In the normal cell, RAS is GDP bound and is inactive unless an extracellular stimuli causes formation of an active GTP-bound molecule. RAS is subsequently inactivated through hydrolysis of its GTP to GDP primarily through the function of GTPase-activating proteins (GAPs). Upon mutation, its intrinsic GTPase activity is lost and GAPs are unable to bind RAS resulting in RAS primarily bound to GTP and therefore constitutively activated [16]. KRAS mutations in lung cancer occur primarily at codon 12 or 13, making the protein GAP insensitive and constitutively GTP bound leading to the activation of downstream effectors. It then drives oncogenesis through a multitude of effectors and downstream signaling pathways to promote tumor growth. These signaling pathways include RAF (MAP kinase pathway), PI3K (AKT/MTOR pathway), ERK, RLIP and RALGDS as seen in Fig. 1. The unregulated signaling of RAS in these pathways thereby leads to increased proliferation, decreased apoptosis, disrupted cellular metabolism, and increased angiogenesis which in turn leads to tumor cell proliferation [16].
The RAF, MEK, ERK, PI3K, AKT, MTOR, and RALGDS pathways are targets for drug development. However, an understanding of the nature of these pathways is paramount before designing therapeutic strategies. For example, activated RAF phosphorylates and activates the kinase MEK, which then phosphorylates and activates the ERK kinase. Upon activation, ERK phosphorylates a number of substrates including kinases and transcription factors that mediate entry and progression through the cell cycle, inhibition of differentiation, protein translation and suppression of apoptosis [16]. Despite understanding the underlying cascade for the RAF/MEK/ERK pathway, it is still unclear what node is the most efficacious to target clinically. Therefore, not only is an understanding of the critical signaling pathways downstream of KRAS required but also the knowledge of which node to target within in these essential pathways. Finally, it is clear that an understanding of the critical pathways for each KRAS mutant codon [14] and possibly mutational subset (KRAS/STK11 or KRAS/TP53) [17, 18] will be required to target KRAS mutant NSCLC.
Over the last two decades, a variety of strategies have been developed and tested to target oncogenic KRAS signaling. These include the development of direct inhibitors of the KRAS protein, use of RNA interference strategies, development of inhibitors which prevent localization of RAS to the plasma membrane, and pharmacologic targeting of its downstream effectors. It remains to be seen whether any of these strategies will produce a significant clinical benefit. Both the past and present early clinical trial efforts to test these clinical trial strategies are described.
Previous efforts to target KRAS mutant NSCLC in the clinic
Cytotoxic chemotherapy
Although platinum based chemotherapy remains the standard of care in the first line adjuvant and metastatic setting for mutational unselected patients, the benefit of cytotoxic therapy in KRAS mutant NSCLC has been an active area of research. A retrospective analysis conducted by Tsao and colleagues [19] looked at the prognostic effects of p53 mutations or RAS mutations in a set of 450 patients with NSCLC who were randomized to observation or a platinum doublet in the adjuvant setting as part of the JBR10 study. In this group of patients, 117 patients harbored a RAS gene mutation (26 %). This study did not find that KRAS mutational status was a prognostic determinant of disease free survival. However, patients with RAS mutations who were given adjuvant chemotherapy did not appear significantly benefit from adjuvant therapy (HR of 0.95, 95 % CI 0.53–1.71, p = 0.87) compared to observation. In contrast, the wild-type RAS group, adjuvant chemotherapy did appear prolong survival significantly when compared to observation (HR of 0.69, 95 % CI 0.49–0.97, p = 0.03). An important caveat is that in this retrospective analysis, a test for interaction failed to find a significant interaction between chemotherapy and RAS mutational status (p = 0.29). Therefore, the authors concluded that KRAS mutation status was not a predictive factor for chemotherapy [19].
In order to obtain a more definitive answer, the Lung Adjuvant Cisplatin Evaluation Biomarker (LACE-Bio) collaborative group undertook a pooled analysis of KRAS mutant vs. wild type patients in four different trials to ascertain if KRAS mutational status had an effect on patients who received adjuvant chemotherapy. Data from 1543 patients (763 observation patients and 780 patients who received adjuvant chemotherapy) was compiled in a blinded fashion. In this analysis, 300 patients had KRAS mutations (19 %) including 275 codon 12 mutations, 24 codon 13 mutations and only 1 codon 14 mutation. The median follow-up time was 5.5 years and there were 754 deaths (49 %) among all patients [20].
This trial’s pooled results found that KRAS mutational status was weakly prognostic and did not show a significant predictive benefit from adjuvant chemotherapy. Between these studies, results were not significantly different with respect to disease free survival. The hazards ratio when comparing adjuvant chemotherapy with observation in the KRAS wild type patients was 0.89 (95 % CI, 0.76–1.05, p = 0.15). In comparison, the hazard ratio when comparing adjuvant chemotherapy with observation in the patients with KRAS mutations at codon 12 was 0.95 (95 % CI, 0.77–1.44, p = 0.77). Interestingly, there did appear to be small subset of KRAS mutant patients who failed to benefit from chemotherapy. Patients with codon 13 mutations seemed to have a detrimental effect from adjuvant chemotherapy (HR of 5.78, CI 2.06–16.22, p = 0.001). However, this was limited by the small sample size, as there were only 24 patients with codon 13 mutations but was statistically significant [20].
In the metastatic setting, several studies have looked at the predictive role of KRAS mutations in patients treated with chemotherapy. To date, the conclusions of these studies have been mixed with some studies finding that a KRAS mutation predicts for worse response and outcome while others have found no difference based on KRAS mutation status [21–24]. One of the first studies to examine this question retrospectively analyzed the prognostic and predictive value of KRAS mutations among 484 patients with KRAS and EGFR mutation information available. Of these patients, 8 % (39 patients) had KRAS mutations and 38 % (182) had EGFR mutations. In the multi-variate analysis, a KRAS mutation was a poor prognostic factor (hazard ratio = 2.6, 95 % CI 1.8–3.7). Patients with the KRAS mutation tended to do worse after gemcitabine-based or pemetrexed-based chemotherapy (RR 14 % mutation vs. 28 % in wild type) [21]. A second larger study looking at 204 patients with advanced non-squamous NSCLC that included 77 patients with KRAS mutant phenotype who had a significant inferior outcome with respect to disease response rate (0.04), disease control rate (0.05) and progression-free survival (p = 0.05) in patients being treated with first-line platinum-based chemotherapy regimens when compared to patients conferring KRAS wild-type and EGFR wild-type mutations [22]. However, a recent large study in 505 Caucasian patients that included 167 KRAS mutant patients failed to find a difference in either PFS or OS [24]. In summary, the KRAS mutation has been found to be predictive in some but not all studies and all these analyses are limited by their retrospective nature and diverse ethnic populations examined.
Targeted approaches
Preclinical studies have demonstrated that localization of KRAS to the plasma membrane is essential for its function. One of the first efforts to block RAS signaling was with farnesyl transferase inhibitors. These drugs were designed to block farnesylation of RAS and therefore prevent its localization to the membrane and subsequent signaling. A phase II trial conducted examined the efficacy and pharmacodynamics of a farnesyl transferase inhibitor (R1155777) in 44 patients with NSCLC (mutation status unknown). Patients were pre-treated patients with Stage IIIB to Stage IV disease and were given R1155777 at 300 mg twice daily for 21 days out of a 28-day cycle. Unfortunately, there was no objective response seen in this study as seen in Table 1. Median survival was 7.7 months (95 % CI 6.5–10.5) and 7 patients had disease stabilization for 6 months (16 %, 95 % CI 6.5–10.5). Interestingly, 83 % of the patients had FTI inhibition in vivo although this did not correlate with objective response. Grade 4 toxicities observed in this study included neutropenia, anemia, and anorexia [25]. Although it is likely that a fraction of these patients would be KRAS mutant positive, it is unclear whether those patients with disease stabilization had KRAS mutant positive tumors. This class of drugs was never specifically tested in a defined KRAS mutant NSCLC population, in part due to the recognition that KRAS could be undergo an alternative modification which would allow it to localize to the membrane even in the absence of farnesylation. The failure of this agent dampened interest in further developed of any targeted agents for RAS for almost a decade [26].
A second potential RAS inhibitor was developed again with the goal of inhibiting localization to the plasma membrane. Salirasib (s-trans, trans-farnyesilthiosalicylic acid) was developed as a RAS farnesylcysteine mimetic which prevents attachment to the RAS membrane [27]. FTS successfully outcompetes RAS for binding to its escort protein that possesses putative farnesyl-binding domains that are necessary for its localization to the membrane. Preclinical data in vitro and in vivo demonstrated promising efficacy in KRAS mutant driven malignancies and therefore, Salirasib was tested in KRAS mutant NSCLC in the clinic [28].
In a phase I trial salirasib’s safety profile was evaluated in patients with advanced cancers and neurofibromatosis. Common side effects included diarrhea (occurred in 79 % of patients), as well as fatigue, nausea and vomiting. Of note, there was not a single patient in this trial with KRAS mutant advanced lung cancer [29]. Subsequently, Riely et al. [28] conducted a Phase II trial to determine the activity of salirasib in patients with advanced KRAS mutant lung cancer (Stage III–IV). A total of 33 patients were enrolled in the trial of which 30 had KRAS mutant NSCLC. The primary end point was rate of non-progression at 10 weeks and the secondary endpoints included RECIST response rate, duration of response, time to progression, and overall survival (OS).
The results from this trial were disappointing. No responses were observed. In patients who were previously treated with chemotherapy, 7 out of the 23 had stable disease at 10 weeks (30 %, 95 % CI 15–51 %). In the patients not previously treated with chemotherapy, 4 out of the 10 had stable disease at 10 weeks (40 %, 95 % CI 17–69 %). Of these 11 patients who had stable disease, the median time of stable disease was about 7 months. Overall, the median survival was 15 months in the pre-treated patients and was not reached (>9 months) in the untreated patients as seen in Table 1. This was at the cost of moderate toxicity of diarrhea, nausea and fatigue [28]. Although this trial’s results and toxicities were disappointing, it is significant that it was the first trial to prospectively examine a targeted therapy specifically in KRAS mutant NSCLC.
Kim et al. [30] subsequently published a trial in 2011 that was quite unique in that it is a prospective trial that mandated biopsies in patients with pre-treated lung cancer. This trial entitled biomarker-integrated approaches of targeted therapy for lung cancer elimination (BATTLE) enrolled 255 patients with pre-treated non-small cell lung cancer who were randomized into one of four arms based on biomarker analysis: erlotinib, vandetanib, erlotinib plus bexarotene, or sorafenib. Subsequently, patients were randomized in an adaptive manner into four predefined biomarker subgroups including EGFR mutation/copy number, KRAS/BRAF mutation, VEG/VEGFR-2 expression, RXRs/Cyclin D1 expression and CCND1 copy number. In this study population, about 20 % had a KRAS mutation and another 15 % had EGFR mutations.
The results of this trial show an impressive benefit to patients with KRAS mutant genotype who were treated with sorafenib. Those with KRAS/BRAF marker group who were treated with sorafenib had a 79 % disease control rate at 8 weeks compared to 14 % in those just treated with erlotinib (p = 0.016). However, sorafenib produced the most drug toxicity causing 21 % treated dose reductions and causing 19 % discontinuation of the drug. The median overall survival was 8.8 months (95 % CI 6.3–10.6) and the median patient follow-up was 10.3 months. The 1-year survival was 35 %. There were no complete responses and only 9 partial responses. These findings are currently being followed up in the BATTLE-2 trial, which hopes to look at pre-specified biomarkers and then conduct prospective testing for biomarker signatures [30].
Despite the provocative findings above, it is unclear whether sorafenib will be an effective therapeutic strategy for KRAS mutant NSCLC. Another study conducted at the National Cancer Institute looked at sorafenib in mutationally defined populations as well. Thirty-seven patients with relapsed or recurrent NSCLC were enrolled in this trial and given sorafenib at 400 mg by mouth twice a day for a 28-day cycle. The median duration of treatment was 3 months. KRAS mutations occurred in 32 % of patients (11/34). However, KRAS or EGFR status showed no correlation with response (progression free survival (PFS) or OS). The disease control rate in the KRAS mutant group was 60 % and was 71 % in the KRAS wild-type group (p = 0.69). The disease control rate in the EGFR mutant group was 40 % and in the wild-type group was 69 % (p = 0.33). The conclusion made from this trial is that sorafenib inhibits progression of disease independent of KRAS mutational status with no statistical differences when looking at either PFS or OS [31]. Similar results were found in patients with EGFR mutations as well. This study is limited primarily by the small size but did have a varied population that is representative of the scope of patients seen in lung cancer clinics nationwide.
Current therapeutic strategies in clinical trials for targeting KRAS mutant NSCLC
MEK inhibitors
Two MEK inhibitors (MEKi), selumetinib and trametinib, have been explored as either monotherapy or in combination with cytotoxic agents to target KRAS mutant NSCLC. Selumetinib, was developed by AstraZeneca is an oral, selective non-ATP competitive inhibitor of MEK1/MEK2, which are key mediators of KRAS signaling (Fig. 1) [32]. Early phase studies showed that selumetinib monotherapy resulted in tumor response in patients with advanced cancer [33]. However, phase II studies with selumetinib monotherapy showed little clinical activity in patients with previously treated NSCLC including patients that had KRAS mutations [34]. Although monotherapy did not appear to be effective in the clinic, in vivo models looking at docetaxel in combination with selumetinib found that there was a synergistic effect that resulted in tumor growth inhibition and regression [17]. A subsequent phase I study in the setting of advanced cancers showed that the side effects of docetaxel and selumetinib were manageable [35].
Therefore a phase II trial was conducted to look at the combination of selumetinib and docetaxel in patients with KRAS mutant NSCLC. Participants in this study had stage III-IV NSCLC with KRAS mutant disease and had failure of treatment after first-line chemotherapy. The study accrued 87 patients who were randomized 1:1 to receive selumetinib and docetaxel or to receive placebo and docetaxel in a double blind fashion. Patients received docetaxel at 75 mg/m2 on day 1 every 21-day cycle and were expected to receive a total of 6 cycles. Patients also received either selumetinib capsules 75 mg twice a day or placebo capsules twice a day until disease progression or toxic effects of the drug. The primary end point of this study was overall survival and secondary endpoints were PFS, proportion of patients with objective response, duration of response, change in tumor size, 6-month PFS and safety [36].
Although the trend in overall survival was encouraging, the study failed to meet its primary endpoint. Median overall survival was 9.4 months (95 % CI 6.8–13.6) in the selumetinib group and 5.2 months (3.8–not calculable [NC]) in the placebo group (HR for death with selumetinib, 0.80, 80 % CI 0.56–1.14; one-sided p = 0.21) and hazards ratio were non-proportional (HR for death with selumetinib 0.8, 80 % CI 0.56-1.14, one sided p = 0.21). Median progression free survival was 5.3 months (95 % CI 4.6–6.4) in the selumetinib group and 2.1 months in the placebo group (95 % CI 1.4–3.7) with a HR for progression with selumetinib of 0.58 (80 % CI 0.42–0.79, one-sided p = 0.014) as seen in Table 1. No complete responses were observed. However, 16 patients in the selumetinib arm had partial response versus no patients in the placebo arm had a partial response thus a 37 % objective response for selumetinib [36]. Unfortunately, the utility of this regimen was limited by the higher frequency of adverse events in the selumetinib group. Febrile neutropenia occurred in 14 % of the selumetinib group and did not occur in the placebo group. Additionally, pneumonia occurred in 9 % of the selumetinib group and did not occur in the placebo group. [36].
This was the first prospective study to examine the efficacy of docetaxel in combination with selumetinib, a MEK1/2 inhibitor, in patients with pre-treated KRAS mutant Stage III-IV NSCLC Overall survival did not significantly improve however there were statistically significant improvements in progression-free survival and response rate. One major drawback of the study was the study size. Although more than 300 patients were screened for the study, only 87 were included in the study. The success of these drugs in combination was at the cost of adverse events including febrile neutropenia, pneumonia and neutropenia. Currently an ongoing phase III trial with mandated growth factor support is underway assessing the efficacy and safety of selumetinib and docetaxel in advanced or metastatic KRAS mutant NSCLC in the second line and beyond (NCT01933932).
A second MEKi, trametinib which is a selective allosteric inhibitor of MEK1/MEK2, has also been examined as a single agent in KRAS mutant NSCLC. The first trial of trametinib monotherapy in KRAS-mutant NSCLC patients showed 2/30 (2 %) partial responses and 16/30 (53 %) stable disease response [37]. In a subsequent phase II trial conducted by Blumenschein and colleagues [38], patients with KRAS mutant NSCLC who had previously been treated with a platinum regimen were randomized in a 2:1 fashion with trametinib (2 mg once daily) or docetaxel (75 mg/m2 IV every 3 weeks). There were one hundred and twenty-nine patients enrolled in this study in which 86 patients received trametinib and 43 received docetaxel. The primary end point analyzed was progression free survival. Progression free survival was 12 weeks in the trametinib group and 11 weeks in the docetaxel group (HR of 1.14; 95 % CI 0.75–1.75; p = 0.5197). This was not statistically significant or clinically meaningful. There were 10 (12 %) partial responses in the trametinib arm and 5 (12 %) in the docetaxel arm (p = 1.00). No patients had a complete response but 3 patients in the trametinib arm had tumor reduction of greater than 80 %. The overall survival was 8 months in the trametinib arm and was not reached in the docetaxel arm. The overall response rate was 12 % in the trametinib arm and 12 % in the docetaxel arm. Unfortunately, the trial ended prematurely due to interim analysis results that showed that the trial crossed the futility boundary after the 92 PFS events were analyzed. In the trametinib arm, all patients had some type of adverse event (mostly grade 1 or 2). The most common adverse effects were rash, diarrhea, hypertension, nausea, dyspnea and fatigue and 5 fatal events were reported. In the docetaxel group, the most common adverse event was neutropenia and no fatal events were noted. In this study, trametinib did not show superiority to docetaxel as a single agent. However, the authors suggested that similarly to selumetinib, there is strong rationale to trametinib with other chemotherapy agents, biologics or radiation as a potent therapy.
Focal adhesion kinase (FAK) inhibitors
Other efforts to target KRAS mutant NSCLC have focused on inhibition of the focal adhesion kinase (FAK). The extracellular signal-regulated kinase ERK/RHOA/(FAK) network is unregulated in patients with lung cancer. Furthermore, the KRAS-RHOA-FAK signaling pathway appears to be critical for KRAS mutant tumorigenesis in the subset of lung cancers that have lost either TP53 or the CDKN2A loci that encodes p16 and ARF proteins. Furthermore, inhibition of FAK causes tumor regression in mice with mutant KRAS and Cdkn2a deficiency [39]. These studies served as the preclinical rationale for testing the FAK inhibitor, Defactinib (VS-6063) in KRAS mutant NSCLC. To test the efficacy of FAK inhibition in KRAS mutant NSCLC with distinct mutation cohorts (A: TP53, CDKN2A wild type, B: TP53 wild type, CDKN2A altered, C: TP53 mutant, CDKN2A wild type, and D: TP53 mutant, CDKN2A altered), a phase II multi-center study is currently underway in patients with KRAS mutant NSCLC who have already received one platinum doublet regimen. The trial has completed accrual and the data analysis is underway [40] as seen in Table 2. This trial represents the first effort to test the efficacy of a targeted therapy in a molecular defined subset of KRAS mutant NSCLC and may serve as a model for future trials.
Cyclin-dependent kinase (CDK) inhibitors
Another potential set of targets that may be successfully targeted in KRAS mutant NSCLC are the cyclin-dependent kinases (CDKs) which are critical regulatory enzymes that drive the cell cycle. Many of the key concepts of the biology of CDK were learned greater than 20 years ago through the study of yeast. Proliferation requires the activation and formation of Cyclin/CDK4 or CDK6 complexes. Active Cyclin/CDK4/6 complexes phosphorylate the critical tumor suppressor, RB1 (Retinoblastoma 1) leading to gene expression and progression through the cell cycle. Additionally, CDK4 and CDK6 lead to increase levels of expression and stability of E-type and A-type cyclins and activation of CDK2 leading to entry into S phase and DNA replication [41]. Interestingly, preclinical data both in vitro and in vivo suggest that CDK function is critical for KRAS tumorigenesis and that inhibition of CDK in KRAS mutant NSCLC leads to potent synthetic lethality [42].
Translating the knowledge that the cyclin dependent kinase pathway is a potential therapeutic target into a successful clinical drug has been challenging in various cancer subtypes. Flavopiridol was the first drug CDK inhibitor that was extensively investigated with over 60 clinical trials between 1998 and 2014. Unfortunately, flavopiridol had low levels of activity seen in Phase II clinical trials in several solid tumor types. Roscovitine is another CDK inhibitor that was evaluated in patients with NSCLC. The APPRAISE trial is a Phase II that randomized NSCLC patients to roscovitine versus best supportive care. The trial terminated after 187 patients were enrolled. The results were not published but were disappointing as there was no impact on progression free survival [41].
Recently, a novel CDK inhibitor, palbociclib has been demonstrated to have significant clinical activity in estrogen receptor positive (ER+) breast cancer patients which is likely due to the key role the CDK4/6-RB1-E2F plays in this breast cancer subtype. The PALOMA-1/TRIO-18 was a Phase II clinical study of 165 women with advanced ER+ breast cancer. This study compared the aromatase inhibitor letrozole alone versus the combination of letrozole plus the CDK4/6i, palbociclib and showed that the group that received the combination therapy had a significant improvement in median progression-free survival when compared with letrozole alone (20.2 months compared with 10.2 months; HR of 0.448; 95 % CI 0.319–0.748; p = 0.0004). Additionally, the overall survival analysis showed that there was a trend of favoring the letrozole plus palbociclib combination (37.5 vs. 33.3 months respectively, HR of 0.813, p = 0.2105) [43]. This lead to the FDA granting accelerated approval to palbociclib for use in combination with letrozole for the treatment of postmenopausal women in the first line metastatic setting in ER +, human epidermal growth factor receptor 2 (HER2)-negative breast cancer [44] and increased interest in examining this agent in other solid tumors including lung.
Efforts to use palbociclib in patients with NSCLC are currently in clinical trials, however, palbociclib has not been specifically examined in KRAS mutant NSCLC. In a Phase II study by Gopolan et al. [45], 19 previously treated patients with recurrent or metastatic NSCLC and negative for p16 expression by immunohistochemistry were given palbociclib at 125 mg daily with the primary endpoint of response rate. Of the 16 evaluable patients who received at least 1 month of therapy there were no responses. Eight patients had stable disease and the remaining eight patients had progressive disease within 8 weeks of treatment. The median progression free survival was 12.5 weeks. Grade 3 toxicities included thrombocytopenia and neutropenia that was seen in three patients. Another patient experienced rhabdomyolysis and transaminitis when taken together with high dose simvastatin. The drug was otherwise well tolerated.
Another CDK inhibitor LY2835219 has shown great activity specifically in KRAS mutant xenografts and is currently being tested in KRAS mutant NSCLC. An initial Phase I trial was conducted in a 49 patient cohort of progressive or metastatic NSCLC patients who had had an average of 4 lines of treatment. In this cohort of patients, 26 were KRAS mutant and 19 KRAS wild type. Patients were given an oral pill of LY2835219 for a 28-day cycle. The drug was relatively well tolerated with 41 % of patients reaching at least 4 cycles with only 2 % of the study cohort suffering from grade 3 toxicities including diarrhea and nausea, and 2 % suffering from fatigue, vomiting and anemia. Consistent with preclinical studies, the study drug was more effective in patients with KRAS mutant NSCLC. The disease control rate was 54 % in KRAS mutant patients [46]. Given these promising findings, a phase III trial is being conducted using Abemaciclib (LY2835219) plus best supportive care versus erlotinib plus best supportive care in patients with KRAS mutant pretreated NSCLC. This study is currently underway and recruiting patients. (NCT02152631).
Heat shock protein 90 (Hsp90) inhibitors
Hsp90 is part of the class of molecular chaperone proteins that plays a central role in the assembly of multi-protein chaperone complexes and regulates folding, stability and function of many client proteins that are oncogenic drivers of lung adenocarcinoma subsets such as mutant EGFR, wild-type CRAF, mutant BRAF, wild-type and mutant HER2, and EML4-ALK fusion protein positive NSCLC. The inhibition of Hsp90 depletes these kinases from cancer cells and disrupts signaling pathways that are critical for proliferation and survival, and thus are an exciting therapeutic target [47].
Since Hsp90 inhibitors are able to inhibit multiple downstream signaling pathways of mutant KRAS, there has been significant interest in testing Hsp90i in KRAS mutant NSCLC. In a trial by Socinski and colleagues in 2013 [48], ganetespib was given to 99 patients with a median of 2 prior systemic therapies. Patients were divided into three cohorts, including mutant EGFR, mutant KRAS and wild type for EGFR and KRAS. Patients were given 200 mg/m2 ganetespib intravenously once weekly for 3 weeks until disease progression. At 16 weeks, the EGFR mutant patients had a 13.3 % progression free survival, the KRAS mutant patients had a 5.9 % progression free survival and those that were KRAS/EGFR wild type had a 19.7 % progression free survival. Interestingly, four patients who harbored the anaplastic lymphoma kinase (ALK) gene rearrangement had a partial response and there appears to be significant single agent activity in this patient population. Eight patients (8.1 %) experienced treatment related serious advents of which 2 resulted in death (cardiac arrest and renal failure). The most common adverse events included diarrhea, fatigue, nausea and anorexia. In summary, single agent Hsp90i have failed to demonstrate significant activity in KRAS mutant NSCLC.
Since preclinical studies demonstrated a synergistic effect of taxanes in combination with ganetespib, this combination has been examined in a completed Phase II where PFS in KRAS mutant patients was a co-primary endpoint and an ongoing Phase III trial. The GALAXY-1 trial looked at ganetespib in combination with docetaxel in pretreated patients with NSCLC. Patients were given docetaxel (at 75 mg/m2 on day 1) alone or in combination with ganetespib (at 150 mg/m2 on days 1 and 15) of a 21-day cycle. Patients were randomized in a 1:1 fashion between docetaxel and docetaxel and ganetespib therapy. In this trial, 385 patients were initially enrolled but it was found early on that the combination therapy resulted in hemoptysis and lack of efficacy in those that did not have adenocarcinoma. Subsequently, only patients with adenocarcinoma were enrolled in this trial. There was a trend in favor of the combination therapy in the adenocarcinoma treated arm with respect to progression free survival (n = 253, HR of 0.82, p = 0.0784) and overall survival (HR of 0.84, p = 0.1139). The most common grade 3 adverse events in the combination arm included neutropenia, leukopenia, anemia and neutropenic fever. Grade 1 or 2 diarrhea was common in the combination arm about 48 h after the infusion. For the patients with KRAS mutations (89 patients), the combination therapy did not result in improved progression free survival (combination median 3.9 months vs. control of 3.0 months, HR of 1.1) or overall survival (combination median 7.6 months versus control of 6.4 months with HR of 1.23). One explanation regarding the lack of efficacy in the KRAS mutant population is that every 2 weeks dosing of ganetespib was not sufficient to suppress the KRAS pathway. Additionally, it was observed that those with advanced disease defined as advanced disease greater than 6 months before study entry seemed to do better with the ganetespib and docetaxel combination therapy with regards to progression free survival (n = 177, adjusted HR of 0.74, p = 0.0417) and overall survival (adjusted HR = 0.69, p = 0.0191) [49]. Currently, the next phase of the study called GALAXY-2 is currently enrolling patients and will definitely determine if there is any benefit in the KRAS mutant NSCLC patient population. This Phase III study is looking at patients with pretreated advanced NSCLC (diagnosis greater than 6 months prior) and randomizing in a 1:1 fashion with docetaxel and the ganetespib and docetaxel combination (NCT01798485). Finally, preclinical studies have suggested that KRAS mutant NSCLC may be more sensitive combinations of Hsp90i and other pathway inhibitors such as MTOR inhibitors [50, 51]. Results of phase Ib/II trial looking at the combination of the HSP90i, Retaspimycin (IPI-504) and TORC1 inhibitor, Everolimus is still pending as seen in Table 2.
Future directions
In addition to the current strategies being tested in the clinic to target KRAS mutant NSCLC, several potential promising preclinical agents are in development. One class of promising agents is the direct RAS inhibitors including those that directly target the KRAS mutant protein. As oncogenic mutations inhibit GTP hydrolysis and therefore drives the activation of RAS; direct inhibition of KRAS proteins would have great clinical significance. Ostrem, Peters and their colleagues [52] reported the development of a small molecule that irreversibly binds to the KRAS mutant molecule (G12C). The molecule targets the mutant cysteine amino acid which is only present in KRAS-mutant proteins and therefore does not interfere with wild-type KRAS proteins. Binding of these inhibitors to KRAS at the switch-I and switch-II regions of the molecule shifts the preference to GDP rather than GTP impairing RAF binding. This study suggests that it may be possible to directly inhibit RAS proteins and target mutant proteins with no affect on wild-type proteins. Currently, these inhibitors are pre-clinical compounds; however, there is significant increase in the development of second generation compounds that could be tested in a phase I or II trials. Others groups are actively pursuing strategies to develop compounds that bind either RAS-GDP or RAS-GTP isoforms and prevent critical intra-molecular interactions with key RAS signaling partners [53]. In addition, there has been a reemergence of interest in preventing RAS localization to cellular membranes [54]. Furthermore, multiple preclinical studies have focused on identifying synthetic lethal interactions with KRAS mutations; however, the majority of these synthetic lethal interactions have not been reproducible [55]. This may be in part to the context dependence nature of these interactions. These studies lead in part to the realization that responses to agents may depend on presence or/absence of genetic modifiers such as TP53 mutations, CDKN2A alterations or STK11/LKB1 alterations [17, 39]. As discussed above, there is already one example of a phase II trial of a FAK inhibitor which presorted KRAS mutant patients into four cohorts based on the co-occurrence of TP53 mutations or CDKN2A mutations [40]. This is likely the first of many trials that will go beyond just selecting patients based solely on the presence of a KRAS mutation and hopefully lead to more effective treatments for genetically defined subsets of KRAS mutant NSCLC.
Conclusions
In conclusion, the KRAS mutation is the most common oncogene driver mutation in patients with NSCLC and confers a poor prognosis in the metastatic setting making it an important target for drug development. This mutation that was discovered more than 3 decades ago result in the loss of GTPase activity making the onco-protein constitutively active and leading to activation of the RAF-MEK-ERK (MAPK) signaling pathway. This pathway is quite complex and multiple strategies have been proposed to target this mutation. To date, an effective agent to KRAS mutant NSCLC remains elusive.
In the last 5 years, there has been an impressive amount of drug development that has resulted in better understanding of the pathway. But unlike ALK and EGFR mutations, there is still no targeted therapy available for patients with KRAS mutations. Currently, there are promising strategies in clinical trials. We feel that the most promising of these agents are the MEKi agents (Trametinib and Selumetinib in combination with chemotherapy), CDK inhibitors, and hopefully direct KRAS inhibitors. We hope that these strategies result in successful drug development and provide an era of personalized medicine for this common mutation.
Abbreviations
- NSCLC:
-
non small cell lung cancer
- KRAS mutation:
-
Kristen Rat Sarcoma virus mutation
- EGFR mutation:
-
epidermal growth factor receptor (EGFR) mutation
- ALK mutation:
-
anaplastic lymphoma kinase (ALK) mutation
- FAK inhibitor:
-
focal adhesion kinase inhibitor
- CDK inhibitor:
-
cyclin dependent kinase inhibitor
- Hsp90 inhibitor:
-
heat shock protein 90 inhibitor
References
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65(1):5–29
Somasundaram A, Socinski MA, Burns TF (2014) Personalized treatment of EGFR mutant and ALK-positive patients in NSCLC. Expert Opin Pharmacother 15(18):2693–2708
Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II et al (2014) Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311(19):1998–2006
Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA (2010) Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol 28(31):4769–4777
Karnoub AE, Weinberg RA (2008) Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9(7):517–531
Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M et al (2005) The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer 92(1):131–139
Graziano SLGG, Newman NB et al (1999) Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol 17:668–675
Schiller JHAS, Feins RH et al (2001) Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small-cell lung cancer on E4592: a laboratory ancillary study on an Eastern Cooperative Oncology Group prospective randomized trial of postoperative adjuvant therapy. J Clin Oncol 19:448–457
Villaruz LC, Socinski MA, Cunningham DE, Chiosea SI, Burns TF, Siegfried JM et al (2013) The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer 119(12):2268–2274
Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA et al (1982) Mechanism of activation of a human oncogene. Nature 300:143–149
Prior IA, Lewis PD, Mattos C (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res 72(10):2457–2467
Forbes SABN, Bamford S et al (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39:D945–D950
Garassino MCMM, Rusconi P et al (2011) Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol 22(1):235–237
Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR et al (2012) Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 104(3):228–239
Stephen AG, Esposito D, Bagni RK, McCormick F (2014) Dragging ras back in the ring. Cancer Cell. 25(3):272–281
Vasan N, Boyer JL, Herbst RS (2014) A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res 20(15):3921–3930
Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T et al (2012) A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483(7391):613–617
Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M et al (2013) LKB1 Inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 23(2):143–158
Tsao MS, Aviel-Ronen S, Ding K, Lau D, Liu N, Sakurada A et al (2007) Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol 25(33):5240–5247
Shepherd FA, Domerg C, Hainaut P, Janne PA, Pignon JP, Graziano S et al (2013) Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol 31(17):2173–2181
Sun JM, Hwang DW, Ahn JS, Ahn MJ, Park K (2013) Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One 8(5):e64816
Metro G, Chiari R, Bennati C, Cenci M, Ricciuti B, Puma F et al (2014) Clinical outcome with platinum-based chemotherapy in patients with advanced nonsquamous EGFR wild-type non-small-cell lung cancer segregated according to KRAS mutation status. Clin Lung Cancer 15(1):86–92
Macerelli M, Caramella C, Faivre L, Besse B, Planchard D, Polo V et al (2014) Does KRAS mutational status predict chemoresistance in advanced non-small cell lung cancer (NSCLC)? Lung Cancer (Amsterdam, Netherlands) 83(3):383–388
Cserepes M, Ostoros G, Lohinai Z, Raso E, Barbai T, Timar J et al (2014) Subtype-specific KRAS mutations in advanced lung adenocarcinoma: a retrospective study of patients treated with platinum-based chemotherapy. Eur J Cancer (Oxford, England: 1990) 50(10):1819–1828
Adjei AA, Mauer A, Bruzek L, Marks RS, Hillman S, Geyer S et al (2003) Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J Clin Oncol 21(9):1760–1766
Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ et al (1997) K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 272(22):14459–14464
Gana-Weisz M, Haklai R, Marciano D, Egozi Y, Ben-Baruch G, Kloog Y (1997) The Ras antagonist S-farnesylthiosalicylic acid induces inhibition of MAPK activation. Biochem Biophys Res Commun 239(3):900–904
Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG et al (2011) A Phase II Trial of Salirasib in Patients with Lung Adenocarcinomas with KRAS Mutations. J Thorac Oncol 6(8):1435–1437
Tsimberidou A, Rudek M, Hong D, Ng C, Blair J, Goldsweig H et al (2010) Phase 1 first-in-human clinical study of S-trans, trans-farnesylthiosalicylic acid (salirasib) in patients with solid tumors. Cancer Chemother Pharmacol 65(2):235–241
Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR Jr, Tsao A et al (2011) The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 1(1):44–53
Kelly RJ, Rajan A, Force J, Lopez-Chavez A, Keen C, Cao L et al (2011) Evaluation of KRAS mutations, angiogenic biomarkers, and DCE-MRI in patients with advanced non-small-cell lung cancer receiving sorafenib. Clin Cancer Res 17(5):1190–1199
Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ et al (2007) Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 13(5):1576–1583
Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB et al (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16(5):1613–1623
Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, Ganchev H, Pover G, Morris C, Tzekova V (2010) A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol 5(10):1630–1636. doi:10.1097/JTO.0b013e3181e8b3a3
Kim K, Infante J, Cohen R, Burris H, Emeribe U, Curt G et al (2011) Abstract B225: a phase I dose-escalation study of selumetinib in combination with docetaxel in patients with advanced solid tumors. Mol Cancer Ther 10(11 Supplement):B225
Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C et al (2013) Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 14(1):38–47
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C et al (2012) Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 13(8):773–781
Blumenschein GR, Smit EF, Planchard D, Kim D-W, Cadranel J, De Pas T et al (2015) A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann Oncol 26(5):894–901
Konstantinidou G, Ramadori G, Torti F, Kangasniemi K, Ramirez RE, Cai Y et al (2013) RHOA-FAK is a required signaling axis for the maintenance of KRAS-driven lung adenocarcinomas. Cancer Discov 3(4):444–457
Gerber D (2014) A phase 2 study of defactinib (VS-6063), a cancer stem cell inhibitor that acts through inhibition of focal adhesion kinase (FAK), in patients with KRAS-mutant non-small cell lung cancer. J Clin Oncol 32:5s (suppl; abstr TPS8126)
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14(2):130–146
Puyol M (2010) A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 18:63–73
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO et al (2015) The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16(1):25–35
FDA. Palbociclib. FDA. 2015
Gopalan PK (2014) A phase II clinical trial of the CDK 4/6 inhibitor palbociclib (PD 0332991) in previously treated, advanced non-small cell lung cancer (NSCLC) patients with inactivated CDKN2A. J Clin Oncol 32:5s (suppl; abstr 8077)
Goldman JW (2014) Clinical activity of LY2835219, a novel cell cycle inhibitor selective for CDK4 and CDK6, in patients with non-small cell lung cancer. J Clin Oncol 32:5s (suppl; abstr 8026)
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18(1):64–76
Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L et al (2013) A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res 19(11):3068–3077
Ramalingam S, Goss G, Rosell R, Schmid-Bindert G, Zaric B, Andric Z, et al (2015) A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1). Ann Oncol 26:1741–1748
Acquaviva J, Smith DL, Sang J, Friedland JC, He S, Sequeira M, Zhang C, Wada Y, Proia DA (2012) Targeting KRAS-Mutant Non-Small Cell Lung Cancer with the Hsp90 Inhibitor Ganetespib. Mol Cancer Ther 11(12):2633–2643. doi:10.1158/1535-7163.MCT-12-0615. (Epub 2012 Sep 25)
De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF et al (2011) Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 20(3):400–413
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503(7477):548–551
Marcus K, Mattos C (2015) Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res 21(8):1810–1818
Cox AD, Der CJ, Philips MR (2015) Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res 21(8):1819–1827
Downward J (2015) RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res 21(8):1802–1809
Authors’ contributions
SB, MAS and TFB: all authors contributed to the primary writing, revision and editing of the manuscript. In addition, all authors provided intellectual input into the topics discussed. All authors read and approved the final manuscript.
Acknowledgements
T.F.B. receives funding from the UPCI LUNG SPORE (P50 CA9045440), the LUNGevity Foundation, the V Foundation, the Sidney Kimmel Foundation for Cancer Research, and the Doris Duke Charitable Foundation to support his work on KRAS mutant NSCLC.
Authors’ information
S.B. is a Hematology Oncology fellow at the University of Pittsburgh Medical Center. MAS is a professor of Medicine and Cardiothoracic Surgery, Director, Lung Cancer Section, Division of Hematology/Oncology, Co-Director, UPMC Lung Cancer Center of Excellence, Clinical Associate Director, Lung SPORE and Co-Leader, UPCI Lung Cancer Program. T.F.B. is an assistant professor of Medicine, Division of Hematology/Oncology, member of the UPCI SPORE and Lung Cancer Program.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bhattacharya, S., Socinski, M.A. & Burns, T.F. KRAS mutant lung cancer: progress thus far on an elusive therapeutic target. Clin Trans Med 4, 35 (2015). https://doi.org/10.1186/s40169-015-0075-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40169-015-0075-0