
Abstract
During the twentieth century, there was an explosion in understanding of the malaria parasites infecting humans and wild primates. This was built on three main data sources: from detailed descriptive morphology, from observational histories of induced infections in captive primates, syphilis patients, prison inmates and volunteers, and from clinical and epidemiological studies in the field. All three were wholly dependent on parasitological information from blood-film microscopy, and The Primate Malarias” by Coatney and colleagues (1971) provides an overview of this knowledge available at that time. Here, 50 years on, a perspective from the third decade of the twenty-first century is presented on two pairs of primate malaria parasite species. Included is a near-exhaustive summary of the recent and current geographical distribution for each of these four species, and of the underlying molecular and genomic evidence for each. The important role of host transitions in the radiation of Plasmodium spp. is discussed, as are any implications for the desired elimination of all malaria species in human populations. Two important questions are posed, requiring further work on these often ignored taxa. Is Plasmodium brasilianum, circulating among wild simian hosts in the Americas, a distinct species from Plasmodium malariae? Can new insights into the genomic differences between Plasmodium ovale curtisi and Plasmodium ovale wallikeri be linked to any important differences in parasite morphology, cell biology or clinical and epidemiological features?
Similar content being viewed by others
Background
In The Primate Malarias (1971), by Coatney et al. [1], detailed species comparisons are presented based on descriptive morphology of both blood and mosquito stages, the geographic distribution of each parasite and certain features readily measurable in induced human infections, including the estimated duration of the liver-stage, time to symptoms and fever periodicity. Much of this work was performed in prison inmates in Georgia, USA. In this paper, fifty years since, the focus on the geographic, genomic and genetic characteristics of four primate malaria species—one currently regarded as zoonotic in South American monkeys, Plasmodium brasilianum, and three malaria parasites of Homo sapiens, namely Plasmodium malariae, Plasmodium ovale curtisi and Plasmodium ovale wallikeri. An exhaustive bibliography of reported identification of these species since 1890, across the globe and in different primate hosts, will also be presented.
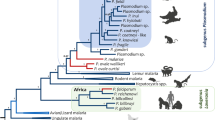
Over the last two decades, the analytical techniques of evolutionary biology and the task of reconstructing phylogenetic relationships within the genus have benefited greatly from the explosion in genomic data available for malaria parasites, and the now well-established practise of non-invasive faecal sampling of parasite genomic material from the faeces of wild primates [2]. This wealth of data provides new understanding of diversity both within and among the primate-infecting Plasmodium species, and points to the importance of transitions into new primate hosts. These transitions are gateways to the radiation of parasite species, but also act as genetic bottlenecks, as evidenced by reduced diversity among parasites in the new host [2, 3].
Among the homophilic species considered of clinical importance, a range of life history and transmission strategies are evident, and each of these strategies have their equivalent counterparts among the parasites of living simian hosts, and those of Pan and Gorilla. Thus, the majority of evolution leading to these diverse life histories occurred in the parasite lineages of non-human primates in the evolutionary past. However, as with Plasmodium knowlesi, the zoonotic potential of P. brasilianum shows that host transition can be a dynamic process operating over an extended time period, rather than a singular event, and understanding this in the present is essential to maintain effective malaria elimination strategies world-wide.
Plasmodium brasilianum
History & discovery
The first report of P. brasilianum is based on a finding in the blood of a bald uakari (Cacajao calvus) imported from the Brazil Amazonas region to Hamburg, Germany in 1908 [4]. Initial studies reported that P. brasilianum closely resembles P. malariae, and to be a relatively common parasite of New World monkeys in Panama and Brazil (reviewed in [1]).
Distribution and known non-human primate hosts
Historically, natural infections of P. brasilianum were reported in various primates in Central and Southern America—Panama, Colombia, Venezuela, Peru, and Brazil. The spectrum of primate hosts (incl. sequence confirmed reports) is given in Table 1 [5,6,7,8,9,10,11,12], indicating that P. brasilianum has promiscuous host-specificity compared to other malaria parasites. Moreover, natural infections in humans have been reported from Venezuela [13].
Genomic studies of Plasmodium brasilianum
Plasmodium brasilianum is a parasite thought to be closely related to P. malariae, and blood-stage infections of the two species present a morphologically identical picture, with discrimination determined by the host, monkey or human, respectively. The few molecular epidemiological studies reported so far have shown that P. brasilianum and P. malariae infections are almost indistinguishable genetically. Sequencing studies of the gene coding for the circumsporozoite protein (csp) appear not to differentiate the identity of the two parasites [14,15,16]. Similar, studies involving the merozoite surface protein-1 (msp1), the ssrRNA small subunit (18S) of ribosomes and the mitochondrial gene cytochrome b (cytb), have identified sequences that were 100% identical or that had only a few randomly distributed single nucleotide position differences [7, 13, 15,16,17,18]. Further, the close genetic resemblance of these parasites has been observed across studies in Brazil, Venezuela, Costa Rica, Peru, Colombia and French Guiana from infected humans, monkeys and mosquitoes [7,8,9, 11, 12, 15,16,17,18]. Under conditions of close contact, as shown in Yanomami people and monkeys species in the Venezuelan Amazon, both humans and non-human primates shared quartan parasites without any host specificity that are genetically identical in target candidate genes [13].
A small study using microsatellite genotyping showed that in 14 P. malariae isolates from infected individuals from the Brazilian Atlantic forest, all isolates had identical haplotypes, while in one mosquito sample from the same region a different haplotype was found [19]. In the same study, three P. brasilianum isolates from non-human primates sampled from a different region (Amazonia) were analysed, and diverse haplotypes were observed. Unfortunately, across all such studies to date only a small number of samples have been compared at only a few genetic loci. To understand the degree of similarity among P. brasilianum and P. malariae parasites, a comprehensive analysis of whole genome sequencing data is necessary, using many more parasites obtained from different hosts, across a range of geographic regions. Only one draft reference genome of P. brasilianum is available [20]. Similarly, only a few genomes are available for P. malariae, sourced from Africa and Asia, and none from South America [8, 20,21,22]. The apicoplast and mitochondrion genomes of P. brasilianum are indistinguishable from those of the P. malariae reference genome [20, 23], but further comparative analysis of nuclear genomes is needed to fully understand the status of these two species. This is made difficult by the scarcity of whole genome data, so it remains an open question whether these parasites are variants of a single species that is naturally adapted to both human and New World monkey hosts, and freely circulates between them. Related to this, it is also difficult to infer the direction of the cross-species transfer. Nevertheless, the similarity of these parasites suggests that monkeys can act as reservoirs of P. malariae / P. brasilianum, and this must be considered in control and eradication programmes.
Plasmodium malariae
History & discovery; epidemiology and disease
As Collins and Jeffery relate [24], P. malariae was named by Grassi and Feletti in 1890, following the observations of Golgi in 1886, who noted the existence of malaria parasites with either 48 h or 72 h cycles of fever, the latter subsequently being recognized as characteristic of P. malariae infections. This slow-growing species is widely distributed across the tropics and sub-tropics, with often asymptomatic infections characterized by low parasitaemia and a recognized ability to persist in a single host for years or decades [25, 26]. There is evidence that P. malariae can survive combination therapies used for treating acute P. falciparum malaria, and may present as a post-treatment recrudescence in P. falciparum patients [27,28,29]. Clinical malaria caused by P. malariae rarely progresses to severe, complicated or life-threatening illness, although the literature contains consistent reports of mortality due specifically to either glomerulonephritis or severe anaemia in small children with chronic infections [30].
Distribution and abundance
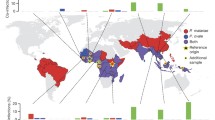
Plasmodium malariae is a cosmopolitan parasite distributed in sub-Saharan Africa, South-East Asia, western Pacific islands, and Central and South America [24]. Formerly this parasite was also present in the southern parts of the USA, Argentina, Bhutan, Brunei, South Korea, Morocco, Turkey, and parts of Europe where malaria was eradicated [31,32,33]. The distribution of this parasite is variable and patchy, and limited to particular mosquito vectors (sporogony needs a minimal temperature of 15 °C), yet autochthonous P. malariae cases have been documented from much of the tropics and sub-tropics (Fig. 1; Table 2) [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143].

Reported global distributions of P. malariae and P. ovale spp.
Assessment of the abundance of P. malariae is difficult because this parasite has been neglected by researchers, and studies differ (e.g. symptomatic patients vs. population studies; Table 2). Some epidemiological studies reported a high prevalence (15–30%) in Africa, Papua New Guinea, and the Western Pacific, in contrast to scanty observations (1–2%) from Asia, the Middle East, Central and Southern America [144]. However, with the advent of molecular diagnostic techniques this parasite species has been reported more frequently, being found in regions where it was not previously thought be present (e.g. Bangladesh), more commonly observed in mixed infections with P. falciparum [24], and identified as recrudescent infections in historical cases from areas such as Greece, formerly endemic for malariae malaria, but since having eliminated contemporary transmission of the disease [145].
Genomic studies of Plasmodium malariae
Large-scale genomic studies of the neglected malaria parasites and zoonotic species have been difficult to date, limited by infections having low parasite densities and being mixed with other Plasmodium species, thereby making it difficult to obtain sufficient parasite DNA to perform whole genome sequencing. For P. malariae, the first partial genome using next-generation sequencing was produced from CDC Uganda I strain DNA [22, 146]. A subsequent study generated a more complete reference using long-read sequencing technology from DNA of the P. malariae isolate PmUG01, from an Australian traveller infected in Uganda [22, 23]. Additional genomic data from short-read Illumina data of travellers’ isolates from Mali, Indonesia and Guinea, and one patient in Sabah, Malaysia, were also reported by Rutledge et al. Analysis of these genomes revealed that around 40% of the 33.6 Mbp genome (24% GC content), particularly in subtelomeric chromosome regions, is taken up by multigene families, as seen in P. ovale species [22, 25]. The P. malariae genome displays some unique characteristics, such as the presence of two large families, the fam-l and fam-m genes, with almost 700 members [22, 23]. Most of these genes encode proteins with a PEXEL export signal peptide and many encode proteins with structural homology to Rh5 of P. falciparum, the only known protein that is essential for P. falciparum red blood cell invasion [147]. These observations suggest that the fam-l and fam-m gene products may also have an important role in binding to host ligands. Other gene families, such as the Plasmodium interspersed repeat (pir) loci that are present in many species in the genus, including in Plasmodium vivax (~ 1500 vir genes), are present in the P. malariae genome. Of the 250 mir genes identified, half are possible pseudogenes. Products of the pir genes are predicted to be exported to the infected erythrocyte surface and may have a role in cell adhesion. Like pir genes, SURFIN proteins are also encoded in the P. malariae genome at around 125 loci, much greater than the number present in P. falciparum (ten) or P. vivax (two). Another unique feature of the P. malariae genome is the presence of 20 copies, in a single tandem array, of the P27/25 gene, a sexual-stage cytoplasmic protein with a possible role in maintaining cell integrity. P27/25 is encoded by a single copy gene in all other species evaluated to date [23, 25].
The sequences of an additional eighteen P. malariae genomes from Africa and Asia have recently been reported [21]. These were derived directly from patient isolates, using a selective whole genome DNA amplification (SWGA) approach to increase the relative abundance of parasite DNA sequence reads relative to host reads. A total of 868,476 genome-wide SNPs were identified, filtered to 104,583 SNPs after exclusion of the hypervariable subtelomeric regions. Phylogenetic analysis showed a clear separation of isolates sourced from Africa and Asia, similar to observations from the analysis of sequence data from the circumsporozoite (pmcsp) gene [148]. Many non-synonymous SNPs in orthologs of P. falciparum drug resistance-associated loci (pmdhfr, pmdhps and pmmdr1) were detected [21, 52], but their impact on drug efficacy remains unknown. Thus, to date, there are no validated molecular markers of drug resistance in P. malariae parasites although, as noted above, prophylaxis breakthrough, treatment failures and emergence following treatment for other species have been reported [26,27,28,29, 149].
In the wider Plasmodium species context, phylogenetic analysis has shown that P. malariae isolates group with malariae-like species that infect monkeys and non-human primates [2, 23]. Plasmodium malariae parasites also cluster closer to P. ovale spp., but in separate clades, and more generally in a clade with P. vivax, P. knowlesi and Plasmodium cynomolgi that is distant from the Laverania sub-genus exemplified by P. falciparum and Plasmodium reichenowi [2, 150]. Given the range of primate hosts that are infected by P. malariae, P. brasilianum and their close relatives, further genomic studies are needed to tease out the two main questions raised by the studies so far:
-
o
Should P. brasilianum, as is currently circulating in South America, and P. malariae be considered distinct, non-recombining species?
-
o
What is the extent of the radiation of P. malariae-like species in the great apes?
Plasmodium ovale curtisi and Plasmodium ovale wallikeri
History & discovery
First identified in Liverpool by Stephens in 1918, the index case of ovale malaria was a British army private, returning to the UK in 1918 following deployment in “East Africa”, and having reported an episode of symptomatic malaria in December, 1916 [151]. This soldier’s blood films were examined over several months, with no mention of any treatment being offered, during which time the presence of fimbriated, oval infected red cells was noted as a key feature, together with a 48 h fever periodicity. This “new parasite of man” (sic) was thus characterized as a benign tertian infection and named Plasmodium ovale in the primary paper, published in 1922. Some additional detailed description of the parasite and its presentation was published by Stephens and Owen in 1927 [152].
For much of the twentieth century, ovale malaria remained a minor entrant in parasitology textbooks, including Coatney et al. [1], until the advent of molecular diagnostic studies in the 1990s began to uncover evidence of genetic dimorphism [153], leading to a series of papers in the first decade of the twenty-first century examining the impact of this dimorphism on molecular and antigen-based diagnosis [154,155,156,157,158]. A multi-centre effort to gather 51 geographically diverse parasite isolates and generate sequencing data across seven genetic loci was then able to demonstrate that ovale malaria was the result of infection by either of two non-recombining, sympatric sibling parasite species, which were named P. ovale curtisi and P. ovale wallikeri [159]. In the decade that followed, various molecular tools were developed to distinguish the two ovale species, and there was an explosion of our understanding of the contribution of the newly recognized parasites to malaria burden across the tropics.
Distribution and abundance
Although the original identification of P. ovale sensu lato (s.l.) by Stephens was in a British soldier who contracted malaria in “East Africa”, the species was subsequently recognized as highly endemic in West Africa (especially Nigeria). Coatney et al. described the distribution of the species as extending to the East African Coast, and as far south as Mozambique [1]. Outside Africa, ovale malaria was sporadically reported from Papua New Guinea, Indonesian islands and some South-East Asian countries [144]. However, with the introduction of molecular diagnostic tools and recognition and widespread acceptance of the two sympatric species, P. o. curtisi (former “classic” type) and P. o. wallikeri (former “variant” type) [159], a much more complex understanding of these parasites has developed. Molecular diagnostics have greatly facilitated the confirmation of the presence of ovale malaria parasites in much of Africa and Asia, including countries where it was not previously known to be present (e.g. Bangladesh, Afghanistan, Angola) [35,36,37, 160,161,162], and in non-human primates [163]. However, it remains generally accepted that these parasites are not endemic in the Americas [159].
Infections with ovale malaria parasites are often asymptomatic and parasite densities low, leading to difficulties in accurate microscopic diagnosis and some uncertainties as to distribution in the recent past. Given the presence of intra-erythrocytic stippling on thin films, and the irregular shapes adopted by ovale-infected cells, there is some morphological similarity to P. vivax, which exacerbates diagnostic difficulties. This also influenced early phylogenetic thinking; Coatney and colleagues write that “from the vivax-like stem developed a morphologically similar species, P. ovale, that was capable of surviving in (African) hominids …” (1). Moreover, mixed infections with other human malaria parasites are very common. Double infections of P. ovale curtisi and P. ovale wallikeri in the same individual have also been reported (e.g. Angola, Bangladesh) [36, 161], confirming the lack of recombination between the two species. However, reported prevalence estimates vary widely among various studies, reflecting different study designs and blood sample collection strategies (e.g. asymptomatic vs. febrile patients). The known distribution of P. ovale spp., P. o. wallikeri and P. o. curtisi is presented in Fig. 2, and a detailed listing of reports identifying these species, including GenBank accession ID where relevant, is given in Table 3 [27, 36, 48, 58, 72, 76, 83, 90, 97, 102, 106, 116, 118, 137, 156, 159, 166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217].

Reported global distributions of P. ovale curtisi and P. ovale wallikeri. Poc Plasmodium ovale curtisi, Pow Plasmodium ovale wallikeri
Genomic studies of P. o. curtisi and P. o. wallikeri
In the period since the two genetically distinct forms of P. ovale spp. were recognized, there have been a limited number of studies that have explored the differences between them. A study in UK travellers with ovale malaria by Nolder and colleagues could not identify any robust features of morphology that can distinguish P. o. curtisi from P. o. wallikeri [168], but were able to provide evidence of a significant difference in the distribution of relapse periodicity: the former species displayed a geometric mean latency of 85.7 days (95% CI 66.1 to 111.1, N = 74), compared to the significantly shorter 40.6 days (95% CI 28.9 to 57.0, N = 60) of the latter. This contrasts with the earlier observation of Chin and Coatney, who conducted studies of experimentally infected volunteers whose initial infections (all with the same “West African strain”) were treated with quinine or chloroquine before extended follow-up for evidence of P. vivax-type relapse [218]. These authors concluded that “These results leave little doubt that ovale malaria is a relapsing disease, but there appears to be no definite relapse pattern…” Subsequent studies in European travellers, a group in which super-infection is absent as a potential confounder, have confirmed this difference in latency period between P. ovale curtisi and P. ovale wallikeri [168, 219, 220]. These studies were also consistent in finding that P. ovale wallikeri is associated with low platelet counts and thus more likely to elicit clinical thrombocytopenia, and more likely to be correctly identified by immunochromatographic lateral flow tests that detect the LDH antigen, which fail to identify > 90% of P. ovale curtisi infections, a reflection of differences in the amino acid sequence of LDH in the two species [158, 159].
Given the absence of distinguishing morphological characters, despite reliable differences in some clinical and diagnostic features, there has been increasing attention to characterisation of the genomic organisation of the two sibling species as a route to better understanding their divergence from each other, and to describe the level of within-species diversity. Initial efforts were based on direct sequencing of PCR-amplified loci, and gave a general picture of fixed differences in both synonymous and non-synonymous substitutions between the species in almost every coding region examined, but very little intra-species genetic diversity [159,160,161, 185, 210, 211]. This was also true of genes related to sexual stage development, which had been examined for evidence of a mating barrier between the two species [181]. Whole genome analysis would clearly be very informative, but very few draft genomes of either species are available due to the difficulty in obtaining parasite DNA from these typically very low parasitaemia infections. The first partial genomes to become available were assembled from Illumina short-read sequencing of two isolates of P. o. wallikeri from Chinese workers returning from West Africa, as well as one P. o. curtisi isolate also from a Chinese worker returning from West Africa and the genome of the chimpanzee-propagated Nigeria I strain [1, 22, 24]. Subsequently, three partial genomes of P. o. curtisi from two patients that tested positive for P. falciparum in Ghana and one mixed infection from Cameroon, together with two P. o. wallikeri genomes obtained from individual patients in Cameroon, were also assembled [23].
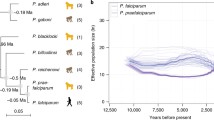
Analysis of the P. ovale spp. genomes published to date has estimated a total genome length for both species of ~ 35 Mbp (29% GC content), with 40% being subtelomeric [22, 23]. Differences in total length (maximum observed 38Mbp) were observed between isolates, primarily due to differences in the estimated size of expansion of the ocir/owir gene families. These species have considerably more pir genes (1500–2000), than other human plasmodium parasites (~ 300) [25]. A larger number of surfin genes have also been identified, with > 50 present in P. o. curtisi and > 125 in P. o. wallikeri. The variant protein isoforms expressed by members of these gene families may be important for interactions with multiple host ligands and, as they are likely to be antigenically variant, their expansion is thought to have been driven by host immune pressure. Expansion of reticulocyte binding-like proteins (RBP), involved in red blood cell invasion, has been observed in both ovale genomes (13–14 genes), gene copy numbers similar to P. vivax, while in other species only ~ 2–8 copies have been identified. An expansion of the Plasmodium ookinete surface protein P28 appears to be a specific feature of both P. ovale spp, as only one copy appears to exist in the genomes of other human-infecting species in the genus.
All the available data confirm that there is a close genetic relationship between the two species, supported by phylogenetic analysis that show P. o. curtisi and P. o. wallikeri grouping together in the same clade in all studies to date [2, 23, 159]. However, many differences between the two taxa have been observed when comparing surfin, pir and rbp genes, as isoforms with identical sequences have been observed between isolates of the same species, but these families are far more divergent in between-species comparisons of the few P. o. curtisi and P. o. wallikeri genomes assembled so far. Significant dimorphism has previously been reported in candidate genes across larger datasets from Asian and African isolates [159,160,161, 175, 185, 210, 211]. For example, specific analysis of nucleotide sequences of five protein-coding regions, likely involved in life cycle sexual stages and so potentially contributing to mating barriers, found that intra-species variation was minimal at each locus, but clear dimorphism were detected when comparing P. o. curtisi to P. o. wallikeri [181]. Similar results were observed across three vaccine candidate surface proteins in samples collected from Thailand and countries in Africa [185], and in multi-locus sequence analyses reported in a large study of both species in Bangladesh [161]. To better understand the intra- and inter -genetic diversity of these species, more complete reference genomes are needed, as well as a much greater number of isolates undergoing whole genome sequencing across geographic regions.
Likely origin of these two closely-related, sympatric and non-recombining species
The question as to how two non-recombining sibling species have ended up co-circulating in the same mammalian hosts, transmitted by the same arthropod vectors, has attracted some attention, as has the difficulty in estimating when the two lineages diverged, and in which primate hosts [2, 3, 23, 25, 159]. A thorough summary of the current thinking can be found therein, but the most parsimonious explanation for the current co-circulation of P. o. curtisi and P. o. wallikeri, in what appears to be perfect sympatry, can be paraphrased from reference 26: pre-ovale parasites in an unknown non-human primate host underwent an initial host transition into hominids some millions of years before the present. This new lineage thus began from a single event, representing an extreme genetic bottleneck, and developed apart from the progenitor stock. Substantial genetic drift occurred, while the two parasite lineages were partitioned in different hosts, a form of allopatry. When a second transition into hominid hosts occurred, again through an extreme genetic bottleneck, both lineages now shared the same hosts, but there was insufficient genetic similarity for fertilisation, meiotic pairing and recombination to occur. However, as the two new species shared almost all features of biology and life history, they together flourished in settings where conditions were favourable and appropriate vectors abundant, and both perished where conditions were harsh. This provides a plausible scenario to explain the contemporary observation that P. o. curtisi and P. o. wallikeri are now always found co-circulating in the same host and vector populations. Considering these observations, and the irrefutable evidence assembled since 2010 that the ovale parasites represent two distinct sibling species, it is clear that the trinomial nomenclature currently in use is not fit for purpose. Some of the arguments around this can be found in Box 2 of reference 26; to resolve this situation, the current authors and collaborators have developed a proposed solution in which two new binomials are utilized in place of the current nomenclature (manuscript in preparation). In the meantime, correspondence on this topic is most welcome.
As to the evolutionary origins of the ovale parasites, despite twentieth century phylogenetic analyses in general favouring kinship with P. vivax [1, 221], genomic sequencing and elucidation of nuclear protein-coding, ribosomal RNA-coding, and mitochondrial genes have more recently placed these species distant from the vivax clade, which includes P. cynomolgi, P. knowlesi and other SE Asian parasites of simian hosts. Rather a position closer to P. malariae [159], Lemuroidea [222], or perhaps the rodent parasite clade [23], have also been put forward. As more genomic information becomes available for P. o. curtisi and P. o. wallikeri the kinship of these species, and therefore identification of their closest contemporary relatives, should become clearer.
Concluding remarks
Multi-population genomic studies of the neglected malaria parasites considered here are essential to provide insights into the biology underlying mechanisms of infection, disease progression and adaptation to different hosts. Many questions, for these and other Plasmodium species, remain answered, including the ability of some species to form dormant stages in the liver (hypnozoites) as observed for P. vivax and P. ovale species, and suggested as also possible for P. malariae [26], and the regulation of the blood stage cycles that can differ among species (e.g., P. malariae has a quartan cycle, a quotidian cycle is observed for P. knowlesi, while the other primate species all follow a tertian cycle).
Although genomics studies of these parasites have been difficult, the development of new assays such as SWGA allow the whole genome sequencing of parasite DNA from clinical samples [21], and have therefore opened up new opportunities to understand genomic diversity. Sequencing developments, such as real-time selective sequencing using Nanopore technology, will favour the selection of parasite DNA molecules for sequencing while excluding human molecules [223]. Phenotypic studies of important characters such as drug susceptibility are challenging for these species [224], but the recently developed strategy of “orthologue exchange” now permits detailed in vitro studies of gene function for every species, using transgenic lines with P. falciparum or P. knowlesi as the recipient parasite cell. These and future advances can support the large-scale and cost-effective genomic studies of neglected malaria that are now needed. The resulting gains in knowledge will greatly assist the design of species-specific diagnostics, treatments, and surveillance tools, thereby supporting malaria elimination goals.
References
Coatney GR, Collins WE, Warren M, Contacos PG. The primate malarias. 1st edn. Bethesda, MD: US National Institute of Allergy and Infectious Diseases; 1971. Digital Version 1.0. Atlanta, GA: CDC; 2003.
Sharp PM, Plenderleith LJ, Hahn BH. Ape origins of human malaria. Annu Rev Microbiol. 2020;74:39–63.
Sutherland CJ, Polley SD. Genomic insights into the past, current, and future evolution of human parasites of the genus Plasmodium. In: Tibayrenc M, editor. Genetics and Evolution of Infectious Diseases (Second Edition). Elsevier; 2017. p. 487-507. https://doi.org/10.1016/B978-0-12-799942-5.00021-4
Gonder R, Berenberg-Gossler von HV. Untersuchungen uber Malaria-Plasmodien der Affen. Malaria-Intern. Arch. Leipzig. 1908:47–56.
Carrillo-Bilbao G, Martin-Solano S, Saegerman C. Zoonotic blood-borne pathogens in non-human primates in the Neotropical Region: a systematic review. Pathogens. 2021;10:1009.
Abreu FVS, Santos ED, Mello ARL, Gomes LR, Alvarenga DAM, Gomes MQ, et al. Howler monkeys are the reservoir of malarial parasites causing zoonotic infections in the Atlantic forest of Rio de Janeiro. PLoS Negl Trop Dis. 2019;13:e0007906.
Fandeur T, Volney B, Peneau C, de Thoisy B. Monkeys of the rainforest in French Guiana are natural reservoirs for P. brasilianum/P. malariae malaria. Parasitology. 2000;120:11–21.
Rondón S, León C, Link A, González C. Prevalence of Plasmodium parasites in non-human primates and mosquitoes in areas with different degrees of fragmentation in Colombia. Malar J. 2019;18:276.
Araújo MS, Messias MR, Figueiró MR, Gil LH, Probst CM, Vidal NM, et al. Natural Plasmodium infection in monkeys in the state of Rondônia (Brazilian Western Amazon). Malar J. 2013;12:180.
Alvarenga DA, Pina-Costa A, Bianco C Jr, Moreira SB, Brasil P, Pissinatti A, et al. New potential Plasmodium brasilianum hosts: tamarin and marmoset monkeys (family Callitrichidae). Malar J. 2017;16:71.
Erkenswick GA, Watsa M, Pacheco MA, Escalante AA, Parker PG. Chronic Plasmodium brasilianum infections in wild Peruvian tamarins. PLoS ONE. 2017;12:e0184504.
Guimarães LO, Wunderlich G, Alves JM, Bueno MG, Röhe F, Catão-Dias JL, et al. Merozoite surface protein-1 genetic diversity in Plasmodium malariae and Plasmodium brasilianum from Brazil. BMC Infect Dis. 2015;15:529.
Lalremruata A, Magris M, Vivas-Martínez S, Koehler M, Esen M, Kempaiah P, et al. Natural infection of Plasmodium brasilianum in humans: Man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine. 2015;2:1186–92.
Escalante AA, Barrio E, Ayala FJ. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol. 1995;12:616–26.
Lal AA, de la Cruz VF, Collins WE, Campbell GH, Procell PM, McCutchan TF. Circumsporozoite protein gene from Plasmodium brasilianum Animal reservoirs for human malaria parasites? J Biol Chem. 1988;263:5495–8.
Tazi L, Ayala FJ. Unresolved direction of host transfer of Plasmodium vivax v. P. simium and P. malariae v. P. brasilianum. Infect Genet Evol. 2011;11:209–21.
de Castro Duarte AM, Malafronte Rdos S, Cerutti C Jr, Curado I, de Paiva BR, Maeda AY, et al. Natural Plasmodium infections in Brazilian wild monkeys: reservoirs for human infections? Acta Trop. 2008;107:179–85.
Fuentes-Ramírez A, Jiménez-Soto M, Castro R, Romero-Zuñiga JJ, Dolz G. Molecular detection of Plasmodium malariae/Plasmodium brasilianum in non-human primates in captivity in Costa Rica. PLoS ONE. 2017;12:e0170704.
Guimarães LO, Bajay MM, Wunderlich G, Bueno MG, Röhe F, Catão-Dias JL, et al. The genetic diversity of Plasmodium malariae and Plasmodium brasilianum from human, simian and mosquito hosts in Brazil. Acta Trop. 2012;124:27–32.
Talundzic E, Ravishankar S, Nayak V, Patel DS, Olsen C, Sheth M, et al. First full draft genome sequence of Plasmodium brasilianum. Genome Announc. 2017;5:e01566-e1616.
Ibrahim A, Diez Benavente E, Nolder D, Proux S, Higgins M, Muwanguzi J, et al. Selective whole genome amplification of Plasmodium malariae DNA from clinical samples reveals insights into population structure. Sci Rep. 2020;10:10832.
Ansari HR, Templeton TJ, Subudhi AK, Ramaprasad A, Tang J, Lu F, et al. Genome-scale comparison of expanded gene families in Plasmodium ovale wallikeri and Plasmodium ovale curtisi with Plasmodium malariae and with other Plasmodium species. Int J Parasitol. 2016;46:685–96.
Rutledge GG, Böhme U, Sanders M, Reid AJ, Cotton JA, Maiga-Ascofare O, et al. Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature. 2017;542:101–4.
Collins WE, Jeffery GM. Plasmodium malariae: parasite and disease. Clin Microbiol Rev. 2007;20:579–92.
Sutherland CJ. Persistent parasitism: the adaptive biology of malariae and ovale malaria. Trends Parasitol. 2016;32:808–19.
Teo BH, Lansdell P, Smith V, Blaze M, Nolder D, Beshir KB, et al. Delayed onset of symptoms and atovaquone-proguanil chemoprophylaxis breakthrough by Plasmodium malariae in the absence of mutation at codon 268 of pmcytb. PLoS Negl Trop Dis. 2015;9:e0004068.
Dinko B, Oguike MC, Larbi JA, Bousema T, Sutherland CJ. Persistent detection of Plasmodium falciparum, P. malariae, P. ovale curtisi and P. ovale wallikeri after ACT treatment of asymptomatic Ghanaian school children. Int J Parasitol Drugs Drug Resist. 2013;3:45–50.
Betson M, Sousa-Figueiredo JC, Atuhaire A, Arinaitwe M, Adriko M, Mwesigwa G, et al. Detection of persistent Plasmodium spp. infections in Ugandan children after artemether-lumefantrine treatment. Parasitology. 2014;141:1880–90.
Lubis IND, Wijaya H, Lubis M, Lubis CP, Beshir KB, Staedke SG, et al. Recurrence of Plasmodium malariae and P. falciparum following treatment of uncomplicated malaria in North Sumatera with dihydroartemisinin-piperaquine or artemether-lumefantrine. Open Forum Infect Dis. 2020. https://doi.org/10.1093/ofid/ofaa116.
Langford S, Douglas NM, Lampah DA, Simpson JA, Kenangalem E, Sugiarto P, et al. Plasmodium malariae infection associated with a high burden of anemia: a hospital-based surveillance study. PLoS Negl Trop Dis. 2015;9:e0004195.
Bardach A, Ciapponi A, Rey-Ares L, Rojas JI, Mazzoni A, Glujovsky D, et al. Epidemiology of malaria in Latin America and the Caribbean from 1990 to 2009: systematic review and meta-analysis. Value Health Reg Issues. 2015;8:69–79.
Hong YJ, Yang SY, Lee K, Kim TS, Kim HB, Park KU, et al. A case of imported Plasmodium malariae malaria. Ann Lab Med. 2012;32:229–33.
Özbilgin A, Topluoglu S, Es S, Islek E, Mollahaliloglu S, Erkoc Y. Malaria in Turkey: successful control and strategies for achieving elimination. Acta Trop. 2011;120:15–23.
Mikhail AF, Leslie TJ, Mayan MI, Zekria R, Mohammad N, Hasanzai MA, et al. Field trial of three different Plasmodium vivax-detecting rapid diagnostic tests with and without evaporative cool box storage in Afghanistan. Malar J. 2011;10:169.
Ramachandra T. Malaria control using indoor residual sprays in the Eastern Province of Afghanistan. Bull World Health Organ. 1951;3:639–61.
Fançony C, Gamboa D, Sebastião Y, Hallett R, Sutherland C, Sousa-Figueiredo JC, et al. Various pfcrt and pfmdr1 genotypes of Plasmodium falciparum cocirculate with P. malariae, P. ovale spp, and P. vivax in northern Angola. Antimicrob Agents Chemother. 2012;56:5271–7.
Pembele G, Rivero L, Fraga J. Detection and species identification of malaria parasites by nested-pcr: comparison with light microscopy and with SD BIOLINE malaria Ag test in Luanda, Angola. Int J Trop Dis Health. 2015;10:1–3.
Fuehrer HP, Swoboda P, Harl J, Starzengruber P, Habler VE, Bloeschl I, et al. High prevalence and genetic diversity of Plasmodium malariae and no evidence of Plasmodium knowlesi in Bangladesh. Parasitol Res. 2014;113:1537–43.
Doderer-Lang C, Atchade PS, Meckert L, Haar E, Perrotey S, Filisetti D, et al. The ears of the African elephant: unexpected high seroprevalence of Plasmodium ovale and Plasmodium malariae in healthy populations in Western Africa. Malar J. 2014;13:240.
Motshoge T, Ababio GK, Aleksenko L, Read J, Peloewetse E, Loeto M, et al. Molecular evidence of high rates of asymptomatic P. vivax infection and very low P. falciparum malaria in Botswana. BMC Infect Dis. 2016;16:520.
Scopel KK, Fontes CJ, Nunes AC, Horta MF, Braga EM. Low sensitivity of nested PCR using Plasmodium DNA extracted from stained thick blood smears: an epidemiological retrospective study among subjects with low parasitaemia in an endemic area of the Brazilian Amazon region. Malar J. 2004;3:8.
Cunha MG, Santos CS, Raiol M, Costa SPT, Ventura AMR, Póvoa MM, et al. Mixed Plasmodium malariae infections were underdetected in a malaria endemic area in the Amazon region, Brazil. Am J Trop Med Hyg. 2021;105:1184–6.
de Alencar FEC, Malafronte RDS, Cerutti Junior C, Natal Fernandes L, Buery JC, Fux B, et al. Assessment of asymptomatic Plasmodium spp. infection by detection of parasite DNA in residents of an extra-Amazonian region of Brazil. Malar J. 2018;17:113.
Williams J, Njie F, Cairns M, Bojang K, Coulibaly SO, Kayentao K, et al. Non-falciparum malaria infections in pregnant women in West Africa. Malar J. 2016;15:53.
Geiger C, Agustar HK, Compaoré G, Coulibaly B, Sié A, Becher H, et al. Declining malaria parasite prevalence and trends of asymptomatic parasitaemia in a seasonal transmission setting in North-Western Burkina Faso between 2000 and 2009–2012. Malar J. 2013;12:27.
Culleton RL, Mita T, Ndounga M, Unger H, Cravo PV, Paganotti GM, et al. Failure to detect Plasmodium vivax in West and Central Africa by PCR species typing. Malar J. 2008;7:174.
Gnémé A, Guelbéogo WM, Riehle MM, Tiono AB, Diarra A, Kabré GB, et al. Plasmodium species occurrence, temporal distribution and interaction in a child-aged population in rural Burkina Faso. Malar J. 2013;2013(12):67.
Li P, Zhao Z, Xing H, Li W, Zhu X, Cao Y, et al. Plasmodium malariae and Plasmodium ovale infections in the China-Myanmar border area. Malar J. 2016;15:557.
Wang RB, Zhang J, Zhang QF. Malaria baseline survey in four special regions of northern Myanmar near China: a cross-sectional study. Malar J. 2014;13:302.
Protopopoff N, Van Bortel W, Marcotty T, Van Herp M, Maes P, Baza D, et al. Spatial targeted vector control is able to reduce malaria prevalence in the highlands of Burundi. Am J Trop Med Hyg. 2008;79:12–8.
Nimpaye H, Nisubire D, Nyandwi J. Plasmodium falciparum and P. malariae: infection rates in the population of Northern Imbo plain Burundi. East Afr Health Res J. 2020;4:189–93.
Khim N, Kim S, Bouchier C, Tichit M, Ariey F, Fandeur T, et al. Reduced impact of pyrimethamine drug pressure on Plasmodium malariae dihydrofolate reductase gene. Antimicrob Agents Chemother. 2012;56:863–8.
Durnez L, Pareyn M, Mean V, Kim S, Khim N, Menard D, et al. Identification and characterization of areas of high and low risk for asymptomatic malaria infections at sub-village level in Ratanakiri. Cambodia Malar J. 2018;17:27.
Lek D, Popovici J, Ariey F, Vinjamuri SB, Meek S, Bruce J, et al. National malaria prevalence in Cambodia: microscopy versus polymerase chain reaction estimates. Am J Trop Med Hyg. 2016;95:588–94.
Tahar R, Ringwald P, Basco LK. Heterogeneity in the circumsporozoite protein gene of Plasmodium malariae isolates from sub-Saharan Africa. Mol Biochem Parasitol. 1998;92:71–8.
Feufack-Donfack LB, Sarah-Matio EM, Abate LM, Bouopda Tuedom AG, Ngano Bayibéki A, Maffo Ngou C, et al. Epidemiological and entomological studies of malaria transmission in Tibati, Adamawa region of Cameroon 6 years following the introduction of long-lasting insecticide nets. Parasit Vectors. 2021;14:247.
Roman DNR, Rosalie NNA, Kumar A, Luther KMM, Singh V, Albert MS. Asymptomatic Plasmodium malariae infections in children from suburban areas of Yaoundé Cameroon. Parasitol Int. 2018;67:29–33.
Mapua MI, Fuehrer HP, Petrželková KJ, Todd A, Noedl H, Qablan MA, et al. Plasmodium ovale wallikeri in western lowland gorillas and humans Central African Republic. Emerg Infect Dis. 2018;24:1581–3.
Bylicka-Szczepanowska E, Korzeniewski K, Lass A. Prevalence of Plasmodium spp. in symptomatic BaAka Pygmies inhabiting the rural Dzanga Sangha region of the Central African Republic. Ann Agric Environ Med. 2021;28:483–90.
Terveer EM, Brienen EA, Erkens MA, van Lieshout L. Late manifestation of a mixed Plasmodium falciparum and Plasmodium malariae infection in a non-immune toddler after traveling to Chad. Travel Med Infect Dis. 2016;14:533–4.
Niño CH, Cubides JR, Camargo-Ayala PA, Rodríguez-Celis CA, Quiñones T, Cortés-Castillo MT, et al. Plasmodium malariae in the Colombian Amazon region: you don’t diagnose what you don’t suspect. Malar J. 2016;15:576.
Camargo M, Soto-De León SC, Del Río-Ospina L, Páez AC, González Z, González E, et al. Micro-epidemiology of mixed-species malaria infections in a rural population living in the Colombian Amazon region. Sci Rep. 2018;8:5543.
Papa Mze N, Ahouidi AD, Diedhiou CK, Silai R, Diallo M, Ndiaye D, et al. Distribution of Plasmodium species on the island of Grande Comore on the basis of DNA extracted from rapid diagnostic tests. Parasite. 2016;23:34.
Nundu SS, Culleton R, Simpson SV, Arima H, Muyembe JJ, Mita T, et al. Malaria parasite species composition of Plasmodium infections among asymptomatic and symptomatic school-age children in rural and urban areas of Kinshasa Democratic Republic of Congo. Malar J. 2021;20:389.
Kiyonga Aimeé K, Lengu TB, Nsibu CN, Umesumbu SE, Ngoyi DM, Chen T. Molecular detection and species identification of Plasmodium spp. infection in adults in the Democratic Republic of Congo: a population-based study. PLoS ONE. 2020;15:e0242713.
Podgorski RM, Goff KA, Penney TP, Maness NJ, Keating J, Yukich JO, et al. DNA analysis reveals non-falciparum malaria in the Democratic Republic of the Congo. Acta Trop. 2020;212:105557.
Kavunga-Membo H, Ilombe G, Masumu J, Matangila J, Imponge J, Manzambi E, et al. Molecular identification of Plasmodium species in symptomatic children of Democratic Republic of Congo. Malar J. 2018;17:334.
Calvo N, Morera J, Solórzano-Morales A, Herrero MV, Dolz G. Re-emergence of Plasmodium malariae in Costa Rica. Sci Postprint. 2015;1:e00049.
Ehounoud BCH, Boumbanda Koyo CS, Doua Bongue L, Cortaredona S, N’Douba Kakou A, Konan DB, et al. Assessment of the burden of malaria and bacteraemia by retrospective molecular diagnosis in febrile illnesses and first-line anti-infectives in Côte d’Ivoire. Travel Med Infect Dis. 2021;43:102105.
Guerra-Neira A, Rubio JM, Royo JR, Ortega JC, Auñón AS, Diaz PB, et al. Plasmodium diversity in non-malaria individuals from the Bioko Island in Equatorial Guinea (West Central-Africa). Int J Health Geogr. 2006;5:27.
Schindler T, Robaina T, Sax J, Bieri JR, Mpina M, Gondwe L, et al. Molecular monitoring of the diversity of human pathogenic malaria species in blood donations on Bioko Island Equatorial Guinea. Malar J. 2019;18:9.
Schlagenhauf P, Grobusch MP, Hamer DH, Asgeirsson H, Jensenius M, Eperon G, et al. Area of exposure and treatment challenges of malaria in Eritrean migrants: a GeoSentinel analysis. Malar J. 2018;17:443.
Mekonnen SK, Aseffa A, Medhin G, Berhe N, Velavan TP. Re-evaluation of microscopy confirmed Plasmodium falciparum and Plasmodium vivax malaria by nested PCR detection in southern Ethiopia. Malar J. 2014;13:48.
Getnet G, Getie S, Srivastava M, Birhan W, Fola AA, Noedl H. Diagnostic performance of rapid diagnostic tests for the diagnosis of malaria at public health facilities in north-west Ethiopia. Trop Med Int Health. 2015;20:1564–8.
Maghendji-Nzondo S, Nzoughe H, Lemamy GJ, Kouna LC, Pegha-Moukandja I, Lekoulou F, et al. Prevalence of malaria, prevention measures, and main clinical features in febrile children admitted to the Franceville Regional Hospital Gabon. Parasite. 2016;23:32.
Woldearegai TG, Lalremruata A, Nguyen TT, Gmeiner M, Veletzky L, Tazemda-Kuitsouc GB, et al. Characterization of Plasmodium infections among inhabitants of rural areas in Gabon. Sci Rep. 2019;9:9784.
Owusu EDA, Brown CA, Grobusch MP, Mens P. Prevalence of Plasmodium falciparum and non-P. falciparum infections in a highland district in Ghana, and the influence of HIV and sickle cell disease. Malar J. 2017;16:167.
Ceesay SJ, Koivogui L, Nahum A, Taal MA, Okebe J, Affara M, et al. Malaria prevalence among young infants in different transmission settings. Africa Emerg Infect Dis. 2015;21:1114–21.
Tanomsing N, Imwong M, Pukrittayakamee S, Chotivanich K, Looareesuwan S, Mayxay M, et al. Genetic analysis of the dihydrofolate reductase-thymidylate synthase gene from geographically diverse isolates of Plasmodium malariae. Antimicrob Agents Chemother. 2007;51:3523–30.
Arez AP, Pinto J, Pålsson K, Snounou G, Jaenson TG, Rosário VE do. Transmission of mixed Plasmodium species and Plasmodium falciparum genotypes. Am J Trop Med Hyg. 2003;68:161–8.
Baird JK, Tiwari T, Martin GJ, Tamminga CL, Prout TM, Tjaden J, et al. Chloroquine for the treatment of uncomplicated malaria in Guyana. Ann Trop Med Parasitol. 2002;96:339–48.
Lindo JF, Bryce JH, Ducasse MB, Howitt C, Barrett DM, Lorenzo Morales J, et al. Plasmodium malariae in Haitian refugees, Jamaica. Emerg Infect Dis. 2007;13:931–3.
Chaturvedi N, Bhandari S, Bharti PK, Basak SK, Singh MP, Singh N. Sympatric distribution of Plasmodium ovale curtisi and P. ovale wallikeri in India: implication for the diagnosis of malaria and its control. Trans R Soc Trop Med Hyg. 2015;109:352–4.
Pati P, Rana RK, Khuntia HK, Bal MS, Ranjit MR. The prevalence of P. malariae in Odisha, India. Trop Biomed. 2017;34:607–14.
Kaisar MM, Supali T, Wiria AE, Hamid F, Wammes LJ, Sartono E, et al. Epidemiology of Plasmodium infections in Flores Island Indonesia using real-time PCR. Malar J. 2013;12:169.
Adel E, Asghar F. The risk of re-emergence of Plasmodium malariae in South-East of Iran as detected by nested polymerase chain reaction. Asian J Epidemiol. 2008;1:47–52.
Lo E, Nguyen K, Nguyen J, Hemming-Schroeder E, Xu J, et al. Plasmodium malariae prevalence and csp gene diversity, Kenya, 2014 and 2015. Emerg Infect Dis. 2017;23:601–10.
Lover AA, Dantzer E, Hongvanthong B, Chindavongsa K, Welty S, Reza T, et al. Prevalence and risk factors for asymptomatic malaria and genotyping of glucose 6-phosphate (G6PD) deficiencies in a vivax-predominant setting, Lao PDR: implications for sub-national elimination goals. Malar J. 2018;17:218.
Björkman A, Hedman P, Brohult J, Willcox M, Diamant I, Pehrsson PO, et al. Different malaria control activities in an area of Liberia—effects on malariometric parameters. Ann Trop Med Parasitol. 1985;79:239–46.
Cao Y, Wang W, Liu Y, Cotter C, Zhou H, Zhu G, et al. The increasing importance of Plasmodium ovale and Plasmodium malariae in a malaria elimination setting: an observational study of imported cases in Jiangsu Province, China, 2011–2014. Malar J. 2016;15:459.
Mehlotra RK, Howes RE, Cramer EY, Tedrow RE, Rakotomanga TA, Ramboarina S, et al. Plasmodium falciparum parasitemia and band sensitivity of the SD Bioline Malaria Ag P.f/Pan rapid diagnostic test in Madagascar. Am J Trop Med Hyg. 2019;100:1196–201.
Bruce MC, Macheso A, McConnachie A, Molyneux ME. Comparative population structure of Plasmodium malariae and Plasmodium falciparum under different transmission settings in Malawi. Malar J. 2011;10:38.
Lee KS, Cox-Singh J, Brooke G, Matusop A, Singh B. Plasmodium knowlesi from archival blood films: further evidence that human infections are widely distributed and not newly emergent in Malaysian Borneo. Int J Parasitol. 2009;39:1125–8.
William T, Jelip J, Menon J, Anderios F, Mohammad R, Awang Mohammad TA, et al. Changing epidemiology of malaria in Sabah, Malaysia: increasing incidence of Plasmodium knowlesi. Malar J. 2014;13:390.
Vythilingam I, Noorazian YM, Huat TC, Jiram AI, Yusri YM, Azahari AH, et al. Plasmodium knowlesi in humans, macaques and mosquitoes in peninsular Malaysia. Parasit Vectors. 2008;1:26.
Koita OA, Sangaré L, Sango HA, Dao S, Keita N, Maiga M, et al. Effect of seasonality and ecological factors on the prevalence of the four malaria parasite species in northern Mali. J Trop Med. 2012;2012:367160.
Ouldabdallahi Moukah M, Ba O, Ba H, Ould Khairy ML, Faye O, Bogreau H, et al. Malaria in three epidemiological strata in Mauritania. Malar J. 2016;15:204.
Ould Ahmedou Salem MS, Basco LK, Ouldabdallahi M, Mint Lekweiry K, Konaté L, Faye O, et al. Malaria-associated morbidity during the rainy season in Saharan and Sahelian zones in Mauritania. Acta Trop. 2015;152:1–7.
Maillard O, Lernout T, Olivier S, Achirafi A, Aubert L, Lepère JF, et al. Major decrease in malaria transmission on Mayotte Island. Malar J. 2015;14:323.
Marques PX, Saúte F, Pinto VV, Cardoso S, Pinto J, Alonso PL, et al. Plasmodium species mixed infections in two areas of Manhiça district, Mozambique. Int J Biol Sci. 2005;1:96–102.
Noor AM, Uusiku P, Kamwi RN, Katokele S, Ntomwa B, Alegana VA, et al. The receptive versus current risks of Plasmodium falciparum transmission in northern Namibia: implications for elimination. BMC Infect Dis. 2013;13:184.
Doudou MH, Mahamadou A, Ouba I, Lazoumar R, Boubacar B, Arzika I, et al. A refined estimate of the malaria burden in Niger. Malar J. 2012;11:89.
May J, Mockenhaupt FP, Ademowo OG, Falusi AG, Olumese PE, Bienzle U, et al. High rate of mixed and subpatent malarial infections in southwest Nigeria. Am J Trop Med Hyg. 1999;61:339–43.
Nnoso AC, Chike DC, Agore VM, Afam FN. A study of Plasmodium species DNA in urine sample of HIV positive individuals using PCR amplification method. African J Malar Trop Dis. 2015;3:193–9.
Oyedeji SI, Awobode HO, Bassi PU. Molecular investigation of sub-microscopic and mixed Plasmodium species infection in North-Central Nigeria. Asian Pacific J Trop Dis. 2017;7:220–4.
Abdulraheem MA, Ernest M, Ugwuanyi I, Abkallo HM, Nishikawa S, Adeleke M, et al. High prevalence of Plasmodium malariae and Plasmodium ovale in co-infections with Plasmodium falciparum in asymptomatic malaria parasite carriers in southwestern Nigeria. Int J Parasitol. 2021;52:23–33.
Beg MA, Sani N, Mehraj V, Jafri W, Khan MA, Malik A, et al. Comparative features and outcomes of malaria at a tertiary care hospital in Karachi. Pakistan Int J Infect Dis. 2008;12:37–42.
Hurtado L, Cumbrera A, Rigg C, Perea M, Santamaría AM, Chaves LF, et al. Long-term transmission patterns and public health policies leading to malaria elimination in Panamá. Malar J. 2020;19:265.
Mehlotra RK, Lorry K, Kastens W, Miller SM, Alpers MP, Bockarie M, et al. Random distribution of mixed species malaria infections in Papua New Guinea. Am J Trop Med Hyg. 2000;62:225–31.
Hetzel MW, Morris H, Tarongka N, Barnadas C, Pulford J, Makita L, et al. Prevalence of malaria across Papua New Guinea after initial roll-out of insecticide-treated mosquito nets. Trop Med Int Health. 2015;20:1745–55.
Sulzer AJ, Cantella R, Colichon A, Gleason NN, Walls KW. A focus of hyperendemic Plasmodium malariae-P. vivax with no P. falciparum in a primitive population in the Peruvian Amazon jungle. Bull World Health Organ. 1975;52:273–8.
Oberst R, Schultz G, Laughlin L, Sy N, Santos M, Casimiro C. Epidemiological study of malaria in Palawan. Phil J Microbiol Infect Dis. 1988;17:41–8.
Dacuma MGB, Dimalibot JC, Baril JA, Allian F, Bahidjan DK, Mori V, et al. Subpatent Plasmodium with mutant pfmdr1, pfcrt, and pvmdr1 alleles from endemic provinces in Mindanao, the Philippines: implications for local malaria elimination. Int J Infect Dis. 2021;110:45–53.
Lee PW, Liu CT, Rampao HS, Rosario VE do, Shaio MF. Pre-elimination of malaria on the island of Príncipe. Malar J. 2010;9:26.
Amer OS, Waly MI, Burhan IW, Al-Malki ES, Smida A, Al-Benasy KS. Epidemiological trends of malaria in the Western regions of Saudi Arabia: a cross sectional study. J Infect Dev Ctries. 2020;14:1332–7.
Badiane AS, Ndiaye T, Thiaw AB, Binta DA, Diallo MA, Seck MC, et al. High prevalence of asymptomatic Plasmodium infection in Bandafassi South-East Senegal. Malar J. 2021;20:218.
Gbakima AA. Inland valley swamp rice development: malaria, schistosomiasis, onchocerciasis in south central Sierra Leone. Public Health. 1994;108:149–57.
Leski TA, Taitt CR, Swaray AG, Bangura U, Reynolds ND, Holtz A, et al. Use of real-time multiplex PCR, malaria rapid diagnostic test and microscopy to investigate the prevalence of Plasmodium species among febrile hospital patients in Sierra Leone. Malar J. 2020;19:84.
Oldfield EC 3rd, Rodier GR, Gray GC. The endemic infectious diseases of Somalia. Clin Infect Dis. 1993. https://doi.org/10.1093/clinids/16.Supplement_3.S132.
Newton JA Jr, Schnepf GA, Wallace MR, Lobel HO, Kennedy CA, Oldfield EC 3rd. Malaria in US Marines returning from Somalia. JAMA. 1994;272:397–9.
Omer AH. Species prevalence of malaria in northern and southern Sudan, and control by mass chemoprophylaxis. Am J Trop Med Hyg. 1978;27:858–63.
Imirzalioglu C, Soydan N, Schaller M, Bretzel RG, Chakraborty T, Domann E. Diagnosis of mixed Plasmodium malariae and P. vivax infection in a development aid volunteer by examination of bone-marrow specimens by real-time PCR. J Clin Microbiol. 2006;44:2307–10.
Ageep A. Diagnosis of malaria in red sea state. Sudan Ann Trop Med Public Health. 2013;6:232–5.
Peek R, Van Gool T, Panchoe D, Greve S, Bus E, Resida L. Drug resistance and genetic diversity of Plasmodium falciparum parasites from Suriname. Am J Trop Med Hyg. 2005;73:833–8.
Hsiang MS, Hwang J, Kunene S, Drakeley C, Kandula D, Novotny J, et al. Surveillance for malaria elimination in Swaziland: a national cross-sectional study using pooled PCR and serology. PLoS ONE. 2012;7:e29550.
Xu W, Morris U, Aydin-Schmidt B, Msellem MI, Shakely D, Petzold M. SYBR Green real-time PCR-RFLP assay targeting the Plasmodium cytochrome B gene–a highly sensitive molecular tool for malaria parasite detection and species determination. PLoS ONE. 2015;10:e0120210.
Baltzell KA, Shakely D, Hsiang M, Kemere J, Ali AS, Björkman A, et al. Prevalence of PCR detectable malaria infection among febrile patients with a negative Plasmodium falciparum specific rapid diagnostic test in Zanzibar. Am J Trop Med Hyg. 2013;88:289–91.
Yman V, Wandell G, Mutemi DD, Miglar A, Asghar M, Hammar U, et al. Persistent transmission of Plasmodium malariae and Plasmodium ovale species in an area of declining Plasmodium falciparum transmission in eastern Tanzania. PLoS Negl Trop Dis. 2019;13:e0007414.
Yorsaeng R, Saeseu T, Chotivanich K, Felger I, Wampfler R, Cui L, et al. Indigenous Plasmodium malariae infection in an endemic population at the Thai-Myanmar border. Am J Trop Med Hyg. 2019;100:1164–9.
Bragonier R, Nasveld P, Auliffe A. Plasmodium malariae in East Timor. Southeast Asian J Trop Med Public Health. 2002;33:689–90.
Elmes NJ. Malaria notifications in the Australian Defence Force from 1998 to 2007. Int Health. 2010;2:130–5.
Dorkenoo AM, Yehadji D, Agbo YM, Layibo Y, Agbeko F, Adjeloh P, et al. Therapeutic efficacy trial of artemisinin-based combination therapy for the treatment of uncomplicated malaria and investigation of mutations in k13 propeller domain in Togo, 2012–2013. Malar J. 2016;15:331.
Hayakawa T, Culleton R, Otani H, Horii T, Tanabe K. Big bang in the evolution of extant malaria parasites. Mol Biol Evol. 2008;25:2233–9.
Murphy KJ, Conroy AL, Ddungu H, Shrestha R, Kyeyune-Byabazaire D, Petersen MR, et al. Malaria parasitemia among blood donors in Uganda. Transfusion. 2020;60:955–64.
Maguire JD, Bangs MJ, Brennan L, Rieckmann K, Taleo G. Cross-sectional characterization of malaria in Sanma and Shefa Provinces, Republic of Vanuatu: malaria control implications. P N G Med J. 2006;49:22–31.
Maeno Y, Culleton R, Quang NT, Kawai S, Marchand RP, Nakazawa S. Plasmodium knowlesi and human malaria parasites in Khan Phu, Vietnam: gametocyte production in humans and frequent co-infection of mosquitoes. Parasitology. 2017;144:527–35.
Nguyen HV, Eede PVD, van Overmeir C, Thang ND, Hung LX, D’Alessandro U, et al. Marked age-dependent prevalence of symptomatic and patent infections and complexity of distribution of human Plasmodium species in central Vietnam. Am J Trop Med Hyg. 2012;87:989–95.
Al-Eryani SM, Kelly-Hope L, Harbach RE, Briscoe AG, Barnish G, Azazy A, et al. Entomological aspects and the role of human behaviour in malaria transmission in a highland region of the Republic of Yemen. Malar J. 2016;15:130.
Al-Mekhlafi AM, Al-Mekhlafi HM, Mahdy MA, Azazy AA, Fong MY. Human malaria in the highlands of Yemen. Ann Trop Med Parasitol. 2011;105:187–95.
Nambozi M, Malunga P, Mulenga M, Van Geertruyden JP, D’Alessandro U. Defining the malaria burden in Nchelenge District, northern Zambia using the World Health Organization malaria indicators survey. Malar J. 2014;13:220.
Sitali L, Miller JM, Mwenda MC, Bridges DJ, Hawela MB, Hamainza B, et al. Distribution of Plasmodium species and assessment of performance of diagnostic tools used during a malaria survey in Southern and Western Provinces of Zambia. Malar J. 2019;18:130.
Laban NM, Kobayashi T, Hamapumbu H, Sullivan D, Mharakurwa S, Thuma PE, et al. Comparison of a PfHRP2-based rapid diagnostic test and PCR for malaria in a low prevalence setting in rural southern Zambia: implications for elimination. Malar J. 2015;14:25.
Taylor P, Mutambu SL. A review of the malaria situation in Zimbabwe with special reference to the period 1972–1981. Trans R Soc Trop Med Hyg. 1986;80:12–9.
Mueller I, Zimmerman PA, Reeder JC. Plasmodium malariae and Plasmodium ovale–the “bashful” malaria parasites. Trends Parasitol. 2007;23:278–83.
Vinetz JM, Li J, McCutchan TF, Kaslow DC. Plasmodium malariae infection in an asymptomatic 74-year-old Greek woman with splenomegaly. N Engl J Med. 1998;338:367–71.
Collins WE, Skinner JC, Broderson JR, Pappaioanou M, Filipski V, Sutton BB, et al. The Uganda I/CDC strain of Plasmodium malariae in Aotus lemurinus griseimembra monkeys. J Parasitol. 1989;75:61–5.
Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011;480:534–7.
Saralamba N, Mayxay M, Newton PN, Smithuis F, Nosten F, Archasuksan L, et al. Genetic polymorphisms in the circumsporozoite protein of Plasmodium malariae show a geographical bias. Malar J. 2018;17:269.
Rutledge GG, Marr I, Huang GKL, Auburn S, Marfurt J, Sanders M, et al. Genomic characterization of recrudescent Plasmodium malariae after treatment with artemether/lumefantrine. Emerg Infect Dis. 2017;23:1300–7.
Arisue N, Hashimoto T, Mitsui H, Palacpac NM, Kaneko A, Kawai S, et al. The Plasmodium apicoplast genome: conserved structure and close relationship of P. ovale to rodent malaria parasites. Mol Biol Evol. 2012;29:2095–9.
Stephens JWW. A new malaria parasite of man. Ann Trop Med Parasitol. 1922;16:383–6.
Stephens JWW, Owen DU. Plasmodium ovale. Ann Trop Med Parasitol. 1927;21:293–302.
Li J, Wirtz RA, McConkey GA, Sattabongkot J, Waters AP, Rogers MJ, et al. Plasmodium: genus-conserved primers for species identification and quantitation. Exp Parasitol. 1995;81:182–90.
Faye FB, Spiegel A, Tall A, Sokhna C, Fontenille D, Rogier C, et al. Diagnostic criteria and risk factors for Plasmodium ovale malaria. J Infect Dis. 2002;186:690–5.
Tachibana M, Tsuboi T, Kaneko O, Khuntirat B, Torii M. Two types of Plasmodium ovale defined by SSU rRNA have distinct sequences for ookinete surface proteins. Mol Biochem Parasitol. 2002;122:223–6.
Win TT, Jalloh A, Tantular IS, Tsuboi T, Ferreira MU, Kimura M, et al. Molecular analysis of Plasmodium ovale variants. Emerg Infect Dis. 2004;10:1235–40.
Calderaro A, Piccolo G, Perandin F, Gorrini C, Peruzzi S, Zuelli C, et al. Genetic polymorphisms influence Plasmodium ovale PCR detection accuracy. J Clin Microbiol. 2007;45:1624–7.
Talman AM, Duval L, Legrand E, Hubert V, Yen S, Bell D, et al. Evaluation of the intra- and inter-specific genetic variability of Plasmodium lactate dehydrogenase. Malar J. 2007;6:140.
Sutherland CJ, Tanomsing N, Nolder D, Oguike M, Jennison C, Pukrittayakamee S, et al. Two nonrecombining sympatric forms of the human malaria parasite Plasmodium ovale occur globally. J Infect Dis. 2010;201:1544–50.
Fuehrer HP, Starzengruber P, Swoboda P, Khan WA, Matt J, Ley B, et al. Indigenous Plasmodium ovale malaria in Bangladesh. Am J Trop Med Hyg. 2010;83:75–8.
Fuehrer HP, Habler VE, Fally MA, Harl J, Starzengruber P, Swoboda P, et al. Plasmodium ovale in Bangladesh: genetic diversity and the first known evidence of the sympatric distribution of Plasmodium ovale curtisi and Plasmodium ovale wallikeri in southern Asia. Int J Parasitol. 2012;42:693–9.
Nguyen HTT, Romano F, Wampfler R, Mühlethaler K, Tannich E, Oberli A. Case Report: Diagnostic challenges in the detection of a mixed Plasmodium vivax/ovale infection in a non-endemic setting. Am J Trop Med Hyg. 2020;103:1085–7.
Duval L, Nerrienet E, Rousset D, Sadeuh Mba SA, Houze S, Fourment M, et al. Chimpanzee malaria parasites related to Plasmodium ovale in Africa. PLoS ONE. 2009;4:e5520.
Bauffe F, Desplans J, Fraisier C, Parzy D. Real-time PCR assay for discrimination of Plasmodium ovale curtisi and Plasmodium ovale wallikeri in the Ivory Coast and in the Comoros Islands. Malar J. 2012;11:307.
Motshoge T, Haiyambo DH, Ayanful-Torgby R, Aleksenko L, Ntebela D, Malleret B, et al. Recent molecular assessment of Plasmodium vivax and Plasmodium falciparum asymptomatic infections in Botswana. Am J Trop Med Hyg. 2021;104:2159–64.
Calderaro A, Piccolo G, Gorrini C, Montecchini S, Rossi S, Medici MC, et al. A new real-time PCR for the detection of Plasmodium ovale wallikeri. PLoS ONE. 2012;7:e48033.
Frickmann H, Wegner C, Ruben S, Loderstädt U, Tannich E. A comparison of two PCR protocols for the differentiation of Plasmodium ovale species and implications for clinical management in travellers returning to Germany: a 10-year cross-sectional study. Malar J. 2019;18:272.
Nolder D, Oguike MC, Maxwell-Scott H, Niyazi HA, Smith V, Chiodini PL, et al. An observational study of malaria in British travellers: Plasmodium ovale wallikeri and Plasmodium ovale curtisi differ significantly in the duration of latency. BMJ Open. 2013;3:e002711.
Incardona S, Chy S, Chiv L, Nhem S, Sem R, Hewitt S, et al. Large sequence heterogeneity of the small subunit ribosomal RNA gene of Plasmodium ovale in Cambodia. Am J Trop Med Hyg. 2005;72:719–24.
Chavatte JM, Tan SB, Snounou G, Lin RT. Molecular characterization of misidentified Plasmodium ovale imported cases in Singapore. Malar J. 2015;14:454.
Kojom Foko LP, Kouemo Motse FD, Kamgain Mawabo L, Pande V, Singh V. First evidence of local circulation of Plasmodium ovale curtisi and reliability of a malaria rapid diagnostic test among symptomatic outpatients in Douala, Cameroon. Infect Genet Evol. 2021;91:104797.
Zhou R, Li S, Zhao Y, Yang C, Liu Y, Qian D, et al. Characterization of Plasmodium ovale spp. imported from Africa to Henan Province. China Sci Rep. 2019;9:2191.
Gabrielli S, Bellina L, Milardi GL, Katende BK, Totino V, Fullin V, et al. Malaria in children of Tshimbulu (Western Kasai, Democratic Republic of the Congo): epidemiological data and accuracy of diagnostic assays applied in a limited resource setting. Malar J. 2016;15:81.
Chen M, Dong Y, Deng Y, Xu Y, Liu Y, Zhang C, et al. Polymorphism analysis of propeller domain of k13 gene in Plasmodium ovale curtisi and Plasmodium ovale wallikeri isolates original infection from Myanmar and Africa in Yunnan Province. China Malar J. 2020;19:246.
Oguike MC, Betson M, Burke M, Nolder D, Stothard JR, Kleinschmidt I, et al. Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in African communities. Int J Parasitol. 2011;41:677–83.
de Santi VP, Khaireh BA, Chiniard T, Pradines B, Taudon N, Larréché S, et al. Role of Anopheles stephensi mosquitoes in malaria outbreak, Djibouti, 2019. Emerg Infect Dis. 2021;27:1697–700.
Gundelfinger BF. Observations on malaria in Indonesian Timor. Am J Trop Med Hyg. 1975;24:393–6.
Roggelin L, Tappe D, Noack B, Addo MM, Tannich E, Rothe C. Sharp increase of imported Plasmodium vivax malaria seen in migrants from Eritrea in Hamburg Germany. Malar J. 2016;15:325.
Alemu A, Fuehrer HP, Getnet G, Tessema B, Noedl H. Plasmodium ovale curtisi and Plasmodium ovale wallikeri in North-West Ethiopia. Malar J. 2013;12:346.
Groger M, Veletzky L, Lalremruata A, Cattaneo C, Mischlinger J, Manego Zoleko R, et al. Prospective clinical and molecular evaluation of potential Plasmodium ovale curtisi and wallikeri relapses in a high-transmission setting. Clin Infect Dis. 2019;69:2119–26.
Oguike MC, Sutherland CJ. Dimorphism in genes encoding sexual-stage proteins of Plasmodium ovale curtisi and Plasmodium ovale wallikeri. Int J Parasitol. 2015;45:449–54.
Heinemann M, Phillips RO, Vinnemeier CD, Rolling CC, Tannich E, et al. High prevalence of asymptomatic malaria infections in adults, Ashanti Region, Ghana, 2018. Malar J. 2020;19:366.
Joste V, Bailly J, Hubert V, Pauc C, Gendrot M, Guillochon E, et al. Plasmodium ovale wallikeri and P. ovale curtisi infections and diagnostic approaches to imported malaria, France, 013–018. Emerg Infect Dis. 2021;27:372–84.
Zhou R, Liu Y, Li S, Zhao Y, Huang F, Yang C, et al. Polymorphisms analysis of the Plasmodium ovale tryptophan-rich antigen gene (potra) from imported malaria cases in Henan Province. Malar J. 2018;17:127.
Saralamba N, Nosten F, Sutherland CJ, Arez AP, Snounou G, White NJ, et al. Genetic dissociation of three antigenic genes in Plasmodium ovale curtisi and Plasmodium ovale wallikeri. PLoS ONE. 2019;14:e0217795.
Krishna S, Bhandari S, Bharti PK, Basak S, Singh N. A rare case of quadruple malaria infection from the highly malaria-endemic area of Bastar, Chhattisgarh, India. PLoS Negl Trop Dis. 2017;11:e0005558.
Miller RH, Obuya CO, Wanja EW, Ogutu B, Waitumbi J, Luckhart S, et al. Characterization of Plasmodium ovale curtisi and P. ovale wallikeri in Western Kenya utilizing a novel species-specific real-time PCR assay. PLoS Negl Trop Dis. 2015;9:e0003469.
Toma H, Kobayashi J, Vannachone B, Arakawa T, Sato Y, Nambanya S, et al. Plasmodium ovale infections detected by PCR assay in Lao PDR. Southeast Asian J Trop Med Public Health. 1999;30:620–2.
Iwagami M, Nakatsu M, Khattignavong P, Soundala P, Keomalaphet S, Lorpachan L, et al. Heterogeneous distribution of k13 mutations in Plasmodium falciparum in Laos. Malar J. 2018;17:483.
Randriamiarinjatovo DNAL. Mise en évidence de la coexistence de Plasmodium ovale curtisi et Plasmodium ovale wallikeri à Saharevo (Moramanga, Madagascar). 2015. http://www.recherches.gov.mg/spip.php?page=detail_article&id_article=3272. Accessed 26 Oct 2021.
Noordin NR, Lee PY, Mohd Bukhari FD, Fong MY, Abdul Hamid MH, Jelip J, et al. Prevalence of asymptomatic and/or low-density malaria infection among high-risk groups in peninsular Malaysia. Am J Trop Med Hyg. 2020;103:1107–10.
Yusof R, Lau YL, Mahmud R, Fong MY, Jelip J, Ngian HU, et al. High proportion of knowlesi malaria in recent malaria cases in Malaysia. Malar J. 2014;13:68.
Rojo-Marcos G, Rubio-Muñoz JM, Ramírez-Olivencia G, García-Bujalance S, Elcuaz-Romano R, Díaz-Menéndez M, et al. Comparison of imported Plasmodium ovale curtisi and P. ovale wallikeri infections among patients in Spain, 2005–2011. Emerg Infect Dis. 2014;20:409–16.
Haiyambo DH, Uusiku P, Mumbengegwi D, Pernica JM, Bock R, Malleret B, et al. Molecular detection of P. vivax and P. ovale foci of infection in asymptomatic and symptomatic children in Northern Namibia. PLoS Negl Trop Dis. 2019;13:e0007290.
Oyedeji SI, Awobode HO, Ojurongbe O, Anumudu C, Bassi PU. Molecular identification and characterization of Plasmodium ovale curtisi in field isolates from symptomatic children in North-Central Nigeria. Acta Parasitol. 2021;66:915–24.
Mehlotra RK, Kasehagen LJ, Baisor M, Lorry K, Kazura JW, Bockarie MJ, et al. Malaria infections are randomly distributed in diverse holoendemic areas of Papua New Guinea. Am J Trop Med Hyg. 2002;67:555–62.
Robinson LJ, Wampfler R, Betuela I, Karl S, White MT, et al. Strategies for understanding and reducing the Plasmodium vivax and Plasmodium ovale hypnozoite reservoir in Papua New Guinean children: a randomised placebo-controlled trial and mathematical model. PLoS Med. 2015;12:e1001891.
Cabrera BD, Arambulo PV 3rd. Malaria in the Republic of the Philippines. A review. Acta Trop. 1977;34:265–79.
Reyes RA, Fornace KM, Macalinao MLM, Boncayao BL, De La Fuente ES, Sabanal HM, et al. Enhanced health facility surveys to support malaria control and elimination across different transmission settings in the Philippines. Am J Trop Med Hyg. 2021;104:968–78.
Sifft KC, Geus D, Mukampunga C, Mugisha JC, Habarugira F, Fraundorfer K, et al. Asymptomatic only at first sight: malaria infection among schoolchildren in highland Rwanda. Malar J. 2016;15:553.
Pinto J, Sousa CA, Gil V, Ferreira C, Gonçalves L, Lopes D, et al. Malaria in São Tomé and Príncipe: parasite prevalences and vector densities. Acta Trop. 2000;76:185–93.
Echeverry DF, Deason NA, Davidson J, Makuru V, Xiao H, Niedbalski J, et al. Human malaria diagnosis using a single-step direct-PCR based on the Plasmodium cytochrome oxidase III gene. Malar J. 2016;2020(15):128.
Echeverry DF, Deason NA, Makuru V, Davidson J, Xiao H, Niedbalski J, et al. Fast and robust single PCR for Plasmodium sporozoite detection in mosquitoes using the cytochrome oxidase I gene. Malar J. 2017;16:230.
Russell TL, Grignard L, Apairamo A, Kama N, Bobogare A, Drakeley C, et al. Getting to zero: micro-foci of malaria in the Solomon Islands requires stratified control. Malar J. 2021;20:248.
Centers for Disease Control and Prevention (CDC). Malaria among U.S. military personnel returning from Somalia 993. Morb Mortal Wkly Rep. 1993;42:524–6.
Himeidan YE, Elbashir MI, El-Rayah el-A, Adam I. Epidemiology of malaria in New Halfa, an irrigated area in eastern Sudan. East Mediterr Health J. 2005;11:499–504.
El Sayed BB, Arnot DE, Mukhtar MM, Baraka OZ, Dafalla AA, Elnaiem DE, et al. A study of the urban malaria transmission problem in Khartoum. Acta Trop. 2000;75:163–71.
Calderaro A, Piccolo G, Gorrini C, Rossi S, Montecchini S, Dell’Anna ML, et al. Accurate identification of the six human Plasmodium spp. causing imported malaria, including Plasmodium ovale wallikeri and Plasmodium knowlesi. Malar J. 2013;12:321.
Cook J, Xu W, Msellem M, Vonk M, Bergström B, Gosling R, et al. Mass screening and treatment on the basis of results of a Plasmodium falciparum-specific rapid diagnostic test did not reduce malaria incidence in Zanzibar. J Infect Dis. 2015;211:1476–83.
Putaporntip C, Hughes AL, Jongwutiwes S. Low level of sequence diversity at merozoite surface protein-1 locus of Plasmodium ovale curtisi and P. ovale wallikeri from Thai isolates. PLoS ONE. 2013;8:e58962.
Tanomsing N, Imwong M, Sutherland CJ, Dolecek C, Hien TT, Nosten F, et al. Genetic marker suitable for identification and genotyping of Plasmodium ovale curtisi and Plasmodium ovale wallikeri. J Clin Microbiol. 2013;51:4213–6.
Baum E, Sattabongkot J, Sirichaisinthop J, Kiattibutr K, Jain A, Taghavian O, et al. Common asymptomatic and submicroscopic malaria infections in Western Thailand revealed in longitudinal molecular and serological studies: a challenge to malaria elimination. Malar J. 2016;15:333.
Gbary AR. Emergence du paludisme chloroquinorésistant en Afrique de l’Ouest : cas de Sokode (Togo). Trop Med Parasitol. 1988;39:142–4.
Ministère de la Santé et de la Protection Sociale (MSPS) et ICF. Enquête sur les Indicateurs du Paludisme au Togo 2017. Rockville, Maryland, USA, 2017 ; MSPS et ICF.
Al-Maktari MT, Bassiouny HK. Ovale malaria: a case report from the Republic of Yemen. Revue Santé Méditerr Orient. 1999;5:826–8.
Hayashida K, Kajino K, Simukoko H, Simuunza M, Ndebe J, Chota A, et al. Direct detection of falciparum and non-falciparum malaria DNA from a drop of blood with high sensitivity by the dried-LAMP system. Parasit Vectors. 2017;10:26.
Taylor P. The malaria problem in Zimbabwe epidemiology. Central African J Med. 1985;31:163–6.
Chin W, Coatney GR. Relapse activity in sporozoite-induced infections with a West African strain of Plasmodium ovale. Am J Trop Med Hyg. 1971;20:825–7.
Nabarro LEB, Nolder D, Broderick C, Nadjm B, Smith V, Blaze M, et al. Geographical and temporal trends and seasonal relapse in Plasmodium ovale spp. and Plasmodium malariae infections imported to the UK between 1987 and 2015. BMC Med. 2018;16:218.
Rojo-Marcos G, Rubio-Muñoz JM, Angheben A, Jaureguiberry S, García-Bujalance S, Tomasoni LR, et al. Prospective comparative multi-centre study on imported Plasmodium ovale wallikeri and Plasmodium ovale curtisi infections. Malar J. 2018;17:399.
Qari SH, Shi YP, Pieniazek NJ, Collins WE, Lal AA. Phylogenetic relationship among the malaria parasites based on small subunit rRNA gene sequences: monophyletic nature of the human malaria parasite Plasmodium falciparum. Mol Phylogenet Evol. 1996;6:157–65.
Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, et al. Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011;11:299.
Loose M, Malla S, Stout M. Real-time selective sequencing using nanopore technology. Nat Methods. 2016;13:751.
van Schalkwyk DA, Moon RW, Duffey M, Leroy D, Sutherland CJ. Ex vivo susceptibility to new antimalarial agents differs among human-infecting Plasmodium species. Int J Parasitol Drugs Drug Resist. 2018;17:5–11.
Acknowledgements
The authors thank the ERASMUS Plus programme for funding a visit by H-PF to London in 2017. CJS is supported by the UK Health Security Agency, the EDCTP WANECAM II Project and the Medical Research Council. SC is supported by Research England Bloomsbury SET Project Grant CCF17-7779.
Funding
CJS is supported by the UK Health Security Agency, the EDCTP WANECAM II Project and the UK Medical Research Council. SC is supported by Research England Bloomsbury SET Project Grant CCF17-7779.
Author information
Authors and Affiliations
Contributions
All three authors together wrote the first draft and edited the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fuehrer, HP., Campino, S. & Sutherland, C.J. The primate malaria parasites Plasmodium malariae, Plasmodium brasilianum and Plasmodium ovale spp.: genomic insights into distribution, dispersal and host transitions. Malar J 21, 138 (2022). https://doi.org/10.1186/s12936-022-04151-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-022-04151-4