
Abstract
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer deaths worldwide. Besides common therapeutic approaches, such as surgery, chemotherapy, and radiotherapy, novel therapeutic approaches, including immunotherapy, have been an advent in CRC treatment. The immunotherapy approaches try to elicit patients` immune responses against tumor cells to eradicate the tumor. Monoclonal antibodies (mAbs) and chimeric antigen receptor (CAR) T cells are two branches of cancer immunotherapy. MAbs demonstrate the great ability to completely recognize cancer cell-surface receptors and blockade proliferative or inhibitory pathways. On the other hand, T cell activation by genetically engineered CAR receptor via the TCR/CD3 and costimulatory domains can induce potent immune responses against specific tumor-associated antigens (TAAs). Both of these approaches have beneficial anti-tumor effects on CRC. Herein, we review the different mAbs against various pathways and their applications in clinical trials, the different types of CAR-T cells, various specific CAR-T cells against TAAs, and their clinical use in CRC treatment.
Similar content being viewed by others


Background
Colorectal cancer (CRC) is the second leading cause of cancer death (about 881,000 deaths) and the third most common malignancy (about 1.8 million new cases) in the world [1, 2]. CRC is a multifactorial disease resulting from genetic alterations [3], diet, lifestyle [4], exposure to environmental xenobiotics [5], and intestinal microbiota [6]. Despite enormous improvements in the strategies of CRC treatment, including chemotherapy, radiotherapy, surgery, and nutritional-supplement therapy [7], the prognosis of patients with metastatic CRC remains poor, with a median overall survival (OS) of about 30 months [8]. For this reason, the development of novel and more effective therapeutic approaches are necessary for metastatic CRC. Owing to the suitable tumor microenvironment (TME) in CRC for expressing molecular markers and receptors such as VEGF (vascular endothelial growth factor) [9], EGFR (epidermal growth factor receptor) [10], IGF-1R (insulin-like growth factor 1 receptor) [11], HER2/neu (human epidermal growth factor receptor-2) [12], αVβ3 integrin [13], MUC5AC (mucin 5AC) [14], DR5 (death receptor) [15], CTLA-4 (cytotoxic T-lymphocyte associated protein 4), and PD-L1 (programmed death-ligand 1) [16], compared with healthy cells, immunotherapy strategies using monoclonal antibodies (mAbs) and chimeric antigen receptor (CAR) T cells are promising therapeutic candidates in CRC treatment. MAbs display important ability to completely identify and classify cancer cell-surface receptors, while CAR T-cells are activated upon antigen engagement to counteract physiologic response suppression and enhancement the therapeutic efficacy against cancers. In this paper, we review the development of two novel therapeutic approaches, mAbs and CAR T-cells, in CRC treatment. We also summarize the available data with the use mAbs and CAR T-cells as well as their challenges and hopes.
Immune cells and responses in the CRC
The immune system has a multi-faceted and complex role in CRC, affecting its all aspect from tumorigenesis to treatment. Immune cells can act as tumor promotors by inducing tumor cell proliferation and metastasis, as well as tumor suppressors by inhibiting tumor initiation and progression. Various immune cells within the TME, including macrophages, dendritic cells (DCs), neutrophils, mast cells, natural killer (NK) cells, myeloid-derived suppressor cells (MDSCs), and B and T lymphocytes, interact with tumor cells (directly or through cytokine and chemokine signals), shaping the tumor cells behavior and response to treatment [17]. An algorithmic study revealed 22 immune cell types in the TME of CRC patients, which were heterogenic in different tumor stages [18].
It has been shown that infiltration of M1 macrophages, T follicular helper (T-fh) cells, Th1 cells, NK cells, and DCs are associated with a good prognosis in CRC patients, whereas a poor outcome is associated with MDSCs, B cells, Th17, and M2 macrophages [19]. Immunosuppressive cells inhibit immune responses against tumor cells, leading to tumor progression and development. For example, MDSCs can suppress anti-tumor activities of NK cells and T cells. Furthermore, they establish pre-metastasis niches, promote angiogenesis, and recruit other immunosuppressive cells, such as regulatory T cells [20]. Hu et al. found that CD39+ γδT cells, as immunosuppressive T cells, significantly increased in the CRC tissues and highly expressed immunosuppression-related molecules, including CD25, CD161, FOXP3, programmed cell death protein 1 PD-1, CTLA-4, PD-L1, whereas they expressed markedly lower levels of immunostimulatory factors [21]. Also, M2 macrophages can mediate resistance to chemotherapy, tumor cell migration and invasion, and angiogenesis [22,23,24]. Moreover, it has been shown that the gut microbiome can affect the immune responses in CRC patients. For instance, Fusobacterium nucleatum inhibits T cell proliferation and increases T cell apoptosis by expanding MDSCs [6, 25]. Thus, understanding immune cells in TME and their interaction with tumor cells allows scientists to identify, develop, and individualize novel immune-based therapeutic agents in CRC patients.
Monoclonal antibodies
Overview of monoclonal antibodies
Whilst antibodies (Abs) secreted by B-cells in response to and neutralizing an antigen are usually polyclonal, Kohler and Milstein produced murine mAbs from hybridomas in 1975 [26]. Although murine mAbs were developed for clinical application, allergic reactions, the induction of anti-drug antibodies (ADAs), and short half-life in humans shifted the technology toward chimeric mouse-human and then humanized Abs [27]. In the chimeric mouse-human Abs, the entire variable regions of a mice Ab is fused with the constant regions of a human Ab to reduce immunogenicity and extend the half-life, but the tendency of chimeric mAbs to induce ADAs is still substantial [28]. The next generation of mAbs was humanized ones in which only the hypervariable regions (complementarity determining regions/CDRs) of the mAb are originated from mice [29]. Fully human mAbs are state of the art in the construction of mAbs which are produced in transgenic mice carrying the human immunoglobulin locus [30]. The structure of different types of mAbs is presented in Fig. 1.

The structure of different types of mAbs. All the components of mouse mAbs are derived from mice. In chimeric mAbs, the variable regions of a mice Ab is fused with the constant regions of a human Ab. In humanized mAbs, only the hypervariable regions (CDRs) of the mAb are originated from mice. All the components of human mAbs are derived from humans
Over the past decades, the significance of therapeutic mAbs has been dramatically increased due to their efficacy in the treatment of different diseases. Muromonab-CD3, as the first therapeutic mAb, was approved against T cell-expressed CD3 for the prevention of acute transplant rejection in 1986 [31]. Until the late 1990s, the growth of approved mAbs was slow, but the advent of chimeric, humanized, and fully human mAbs increased the rate of approval and sales of mAb products. Until 2019, the US FDA has approved 79 therapeutic mAbs, including 30 mAbs for cancer treatment [32]. Besides recombinant proteins, mAbs are the foremost leaders of the biopharmaceutical market [33]. Sales of mAbs showed a 90% increase between 2008 and 2013 from $39 billion to $75 billion [31], and according to forecasts, Ab-based medicine will occupy 20% of the pharmaceutical market in 2022, with sales of $172.8 billion [33].
Monoclonal antibodies for CRC therapy
It has been shown that molecularly targeted agents are more efficient for improving OS and progression-free survival (PFS) of metastatic CRC [34]. For this reason, several molecularly targeted approaches are developed for first- and second-line treatment in combination with chemotherapy regimens (FOLFOX, FOLFIRI, and FOLFOXIRI) [8, 35]. For example, bevacizumab (anti-VEGF), cetuximab, and panitumumab (anti-EGFR) are approved for both first- and second-line treatment [8]. In this section, we reviewed the developed mAbs in the (pre)-clinical studies according to their targets. Moreover, Table 1 summarizes the application of mAbs in the treatment of CRC in clinal trials.
Vascular endothelial growth factor pathway
VEGF family, as the prominent factor in angiogenesis, consists of six members, including VEGF-A, B, C, D, E, and placental growth factors (PlGF) [36]. VEGF families activate several intracellular signaling by binding to their receptors, including VEGFR1, VEGFR2, and VEGFR3 [37]. During solid tumor growth, cells become hypoxic and need new blood vessels to provide oxygen and nutrients for surviving [38]. It has been shown that the expression of VEGF is increased in several malignancies, including pancreatic, liver, gastric, and colorectal cancer [39]. There is a positive correlation between the expression of VEGF and stage of CRC; VEGF expression is higher in patients with stage IV than patients with stage II and III. Moreover, the expression of VEGF is significantly higher in dead patients compared with those who survived for 10 years [40].
Bevacizumab (Avastin®)
Bevacizumab is a humanized mAb that binds to VEGF-A and prevents its interaction with VEGFR, leading to inhibition of VEGF signaling pathways and angiogenesis [41]. Although bevacizumab was first approved for the treatment of metastatic CRC in the combination of chemotherapy in 2004 [42], it is widely recommended for the treatment of various cancers, including breast, ovarian, and lung cancers [43,44,45]. Several randomized trials confirmed the efficacy of bevacizumab in combination with chemotherapy agents in multiple lines of treatment of patients with CRC [46,47,48]. Recently, a meta-analysis study was conducted on seven randomized trials to evaluate the effect of bevacizumab in combination with first-line chemotherapy agents in metastatic CRC. The study revealed that the addition of bevacizumab to only the fluoropyrimidine monotherapy regimen could prolong PFS and OS [49].
Ramucirumab (Cyramza®)
Ramucirumab, a fully human mAb against the extracellular domain of VEGFR2, was approved by the US FDA for the treatment of metastatic CRC in combination with 5-FU, leucovorin, and irinotecan (FOLFIRI) in the second-line setting [50]. According to two phase I clinical trials using ramucirumab, the recommended doses of the mAb for phase II were established as 8 mg/kg every 2 weeks or 10 mg/kg every 3 weeks [51, 52]. In a phase II study, intravenous administration of ramucirumab in combination with the mFOLFOX-6 regimen prolonged PFS and OS, but some adverse events were observed, including neutropenia, neuropathy, and hypertension [53]. A randomized, double-blind phase III RAISE trial demonstrated that ramucirumab versus placebo in combination with FOLFIRI in patients with metastatic CRC significantly improved OS [54].
Aflibercept (Zaltrap®)
Aflibercept, a fully humanized recombinant fusion protein, consists of extracellular domains of VEGFR1 and VEGFR2 fused to the Fc portion of human immunoglobulin G1 which binds to VEGF-A, -B, and PlGF [55]. It has been shown that aflibercept has strong anti-angiogenesis effects compared with bevacizumab owing to longer half-life, higher affinity to soluble VEGFs, and added affinity to PlGF [56]. Multiple phase I trials have demonstrated the safety of aflibercept in combination with chemotherapy regimens in various cancers [57,58,59]. A phase II clinical trial using aflibercept in combination with FOLFIRI in Japanese patients with CRC showed beneficial effects as a second-line treatment [60]. In a multicenter phase III study, aflibercept combined with FOLFIRI regimen improved survival and response rates in CRC patients treated with prior oxaliplatin-based therapy [61].
Tanibirumab
Tanibirumab is a fully human mAb against VEGFR2 which blocks angiogenesis and thereby inhibits tumor growth. It has been shown that tanibirumab has strong anti-tumor effects as a single agent in glioblastoma, breast, and colorectal tumor models [62]. A phase I trial of tanibirumab in patients with refractory solid tumors revealed modest clinical efficacy and tolerable toxicity profile [63].
Vanucizumab
Vanucizumab is a bispecific mAb targeting angiopoietin-2 (Ang-2), an angiogenesis growth factor, and VEGF-A [64]. It has been found that vanucizumab inhibits tumor growth, metastasis, and angiogenesis more effectively than monotherapy using mAbs against Ang-2 or VEGF-A in a murine cancer model [64, 65]. A phase I study using biweekly administration of vanucizumab in patients with solid tumors confirmed its safety and anti-angiogenesis activities [66]. Recently, the results of a phase II clinical trial, McCAVE trial, in patients with previously untreated metastatic CRC revealed that angiogenesis inhibition by vanucizumab does not provide additional benefits over bevacizumab for first‐line treatment of metastatic CRC [67].
Epidermal growth factor receptor pathway
Epidermal growth factor receptor (EGFR) family, transmembrane proteins involved in the cell growth, differentiation, and survival, includes EGFR (HER1), HER2, HER3, and HER4 [68]. Binding of the EGFRs to their ligands, such as EGF, induces homo-/hetero-dimerization of EGFR that triggers the phosphorylation of tyrosine kinases in the intracellular domain of the receptor. Subsequently, the complex signaling network activated via EGFR, such as the PI3K-Akt pathway and RAS/RAF/MEK/MAPK pathway, plays vital roles in several cellular processes, including inhibition of apoptosis, tumor growth, metastasis, and angiogenesis [69, 70]. It has been demonstrated that EGFR is overexpressed in CRC and it can be used as a prognostic biomarker in metastatic CRC [71, 72]. For this reason, mAbs have been designed to target EGFR for the treatment of CRC.
Cetuximab
Cetuximab, a chimeric mAb with approximately 152 kDa molecular weight, has a higher affinity to EGFR compared to its natural ligands, thereby inhibits the tumorigenesis effects of EGFR activation [73, 74]. Cetuximab, combined with chemotherapy, is the standard practice for first-line treatment of RAS wild-type metastatic CRC patients [75]. A phase II trial study revealed that once-weekly intravenous administration of cetuximab has modest anti-tumor activity and is well-tolerated in patients with chemotherapy-refractory CRC [76]. Although weekly administration of cetuximab is the standard protocol in patients with metastatic CRC, the results of a meta-analysis study indicated that biweekly administration of cetuximab instead of weekly administration shows equivalent efficacy and safety [77].
Panitumumab
Panitumumab, a fully human mAb against EGFR, was approved for the treatment of metastatic CRC in combination FOLFIRI and FOLFOX in the first- and second-line setting [78]. Binding of panitumumab to EGFR leads to internalization of the receptor, induction of apoptosis, inhibition of cell growth, and decrease in the production of VEGF and interleukin 8 [79, 80]. A randomized phase III study revealed that using panitumumab combined with FOLFOX4 significantly improved OS in patients with wild-type KRAS metastatic CRC [81]. It has been shown that testing KRAS mutation is critical in the administration of panitumumab to CRC patients, whereas KRAS mutations act as a predictor of resistance to therapy with panitumumab [82].
Necitumumab
High affinity of necitumumab, a fully humanized mAb, for EGFR inhibits phosphorylation of the EGFR and subsequent downstream signaling, leading to inhibition of tumor cell proliferation and tumor growth [83]. A phase II trial demonstrated that using 800 mg necitumumab every 2 weeks combined with chemotherapy (modified FOLFOX6) was active with manageable toxicity in metastatic CRC patients [84].
Immune Checkpoint Inhibitors
Naïve T-cells require two signals to respond to tumor antigens and activation: The first one comes from the binding of T-cell receptors (TCRs) to complexes of an antigen presented on a major histocompatibility complex (MHC) class I molecules on the surface of a antigen presenting cell (APC)/tumor cell which is insufficient for activation of T-cell. The second signal that completes T-cell activation and proliferation occurs by binding of co-stimulatory receptor (CD28) on T-cell to B7 proteins on the APC/tumor cell [85]. Alongside the co-stimulatory receptors, T-cells also express co-inhibitory receptors, such as programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), which suppress T-cell function following binding to B7 and PD-1 ligand 1 or 2 (PD-L1 or PD-L2), respectively [86].
Immune checkpoint inhibitors (ICIs) which target co-inhibitory receptors or their ligands have revolutionized cancer therapy in several cancers. Anti-PD-1 and anti-PD-L1 antibodies directly block the PD-1/PD-L1 axis as well as the direct immune rejection of tumors through antibody-dependent cellular cytotoxicity (ADCC), whereas anti-CTLA-4 inhibitors allow free ligation of B7 to CD28 and enhance co-stimulatory signals of T-cells by directly blocking the receptor [87].
Pembrolizumab
Pembrolizumab, a humanized mAb against PD-1, was approved by the FDA in 2014 for preventing interaction between PD-1 and PD-L1/2 from restoring the immune responses [88]. A phase Ib study conducted by O’Neil et al. revealed that 24% of patients with CRC express PD-L1 in which treatment with pembrolizumab showed a manageable safety profile [89]. A phase 2 clinical trial demonstrated that 10 mg/kg intravenously administration of pembrolizumab is more efficient in mismatch repair–deficient (dMMR) CRC patients compared with mismatch repair–proficient CRC patients [90]. In another study, Dung et al. reported that once every 3 weeks administration of pembrolizumab to patients with dMMR/microsatellite instability-high (MSI-H) CRC provides durable anti-tumor activity and reasonable safety [91]. It has been shown that mismatch repair-deficient tumors strongly express immune checkpoint ligands, indicating that their microenvironment is resistant to tumor elimination [92].
Nivolumab
Nivolumab, a fully human mAb with a molecular weight of 146 kDa, targets PD-1 to block its interaction with PD-L1 and PD-L2 and subsequently avoiding PD-1 mediated inhibition of anti-tumor responses [93]. According to a phase I clinical trial by Yamamoto et al. in patients with solid tumors, nivolumab safety was reasonable at doses of up to 20 mg/kg [94]. Overman et al. conducted a multicentric phase II trial using nivolumab monotherapy and the combination of nivolumab with ipilimumab for the treatment of metastatic CRC patients with dMMR/MCI-H. They reported that the combination regimen improved outcomes, including disease control rate for 12 weeks or longer (80%), ORR (55%), and 12-month OS (85%). They also demonstrated that the treatment-related adverse effects in CRC patients were manageable [95].
Atezolizumab
Atezolizumab, a fully humanized mAb, targets PD-L1 and blocks its interactions with PD-1 and B7.1 (CD80) to restore the anti-tumor immune responses [96]. Antoniotti et al. designed a phase II clinical trial to assess the efficacy of FOLFOXIRI /bevacizumab regimen in combination with atezolizumab for the treatment of metastatic CRC. They indicated that this combination regimen is safe for patients without grade 4 adverse events [97]. A phase III randomized clinical trial, IMblaze 370, was conducted on metastatic CRC patients using atezolizumab (1200 mg), regorafenib (160 mg), and atezolizumab (840 mg) plus cobimetinib (60 mg). The results showed that the median OS in the atezolizumab, regorafenib, and atezolizumab plus cobimetinib groups was 7.10, 8.5, and 8.87 months, respectively. Moreover, the combination therapy showed more adverse events than monotherapy with atezolizumab. Overall, this trial did not demonstrate significant improvements and safety in patients with microsatellite-stable metastatic CRC receiving the combination therapy [98].
Ipilimumab
Ipilimumab, a fully humanized mAb against CTLA-4, was approved by FDA for the treatment of melanoma in 2011 [99]. The positive results of ipilimumab plus nivolumab was demonstrated in the treatment of renal cell carcinoma, melanoma, and metastatic CRC [100]. A multicentric phase II study, CheckMate 142, is designed to assess the safety and efficacy of nivolumab in combination with low-dose ipilimumab in MSI-H/dMMR CRC patients. The results of the study revealed that the combinational regimen is safe and the adverse events were generally low grade. However, the occurrence of adverse events did not affect the efficacy of the treatment with nivolumab in combination with low-dose ipilimumab. Thus, combinational therapy targeting both immune checkpoints (PD-1 and CTLA-4) provides promising [101].
Tremelimumab
Tremelimumab is a fully human mAb against CTLA-4, which blocks inhibitory signals of CTLA-4, leading to immune activation [102]. Chung et al. evaluated the efficacy of tremelimumab in patients with refractory CRC. They reported that intravenously administration of tremelimumab (15 mg/kg) every 90 days, as a monotherapy, had not clinically benefit. However, the manageable toxicity and mechanism of action suggest that a combination of tremelimumab with other therapeutic agents may be effective [103].
Other targets
In addition to the mentioned targets, other mAbs also have been designed to target different pathways in CRC. For example, tigatuzumab, a fully human mAb targeting Apo2L/TRAIL death receptor DR5, was applied in combination with mFOLFOX6 and bevacizumab in patients with metastatic CRC. This combinational regimen showed evidence anti-tumor activities with no dose-limiting toxicities [104]. A phase II study using robatumumab, a fully human anti- insulin-like growth factor 1 receptor (IGF-1R), revealed that patients with refractory CRC might show transient response to robatumumab [105]. Other mAbs also were tested for the treatment of CRC patients, including nimotuzumab, drozitumab, i-huA33, MNRP1685A, KRN330, tigatuzumab, and RG7212 [106].
Challenges of mAbs
Some factors are determinants in comparing different clinical trial results and clinical outcomes of mAbs, including sample size, genetic differences between the study populations, and drug dosage. For instance, it was mentioned that KRAS mutations act as a predictor of resistance to therapy with panitumumab [82] or microsatellite instability acts as a predictive marker in treatment with immune checkpoint inhibitors [90, 107]. Furthermore, determining fixed dosing of anticancer therapies offers considerable advantages, including lower costs, reduced dose preparation errors, and ease of dose preparation [108]. Considering the pharmacokinetic data, such as half-life, allows administration scheduling of each mAb and optimal anti-tumor effects [77]. The toxicity of mAbs and their combination with toxicity agents is another factor determining the success of mAb. Although the toxicity in using anti-angiogenesis agents in combination with chemotherapy is manageable, an increase in toxicity profile is associated with the usage of chemotherapy agents. However, there are some toxicities related to anti-angiogenesis agents. For example, hypertension and proteinuria are the most common side effects of bevacizumab [109, 110].
There are some challenges in the designing, manufacturing, formulation, and stabilization of mAbs. For example, posttranslational modifications of amino acids such as methionine oxidation, aspartic acid isomerization, and asparagine deamidation can lead to a lack of potency and/or heterogeneity if they are involved in the recognition and interaction with the target. Moreover, tryptophan can induce molecular aggregation, resulting in a lack of solubility and immunogenicity [111]. Thus, considering these biophysical properties will help to produce favorable therapeutic mAbs.
Protein A chromatography is known as the golden standard for mAb purification. The main challenge in using this technology is related to resins high costs and their low lifetime compared to others. Furthermore, protein A ligands cannot bind all types of IgG and other therapies such as antibody fragments without the Fc region. Different ligands such as a combination of protein A and new affinity ligands and protein L could develop at the commercial level in mAb purification to address this challenge [112]. Also, applying harsh conditions such as pH and salt to eliminate viral contaminations can lead to antibody instability and loss [113, 114].
Another consideration in mAbs is their formulation and clinical use. Non-invasive administration, including nasal, pulmonary or oral, exposes therapeutic mAbs to enzymatic and chemical degradation in the gastrointestinal (GI) tract. Due to relatively large molecular weight and polar surface charge, the bioavailability of therapeutic mAbs through non-invasive routes is poor. Several strategies, including nanoparticles, liposomes, and microencapsulation, have been developed to control mAb release and stability, prolonging their half-life. Moreover, these nanocarriers can increase tumor penetration of mAbs [115].
CAR-T cell therapy
Overview of CAR-T cell
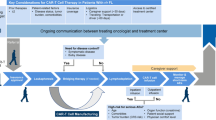
Adoptive T-cell therapy (ACT) has long been used for cancer therapy in which adaptive T-cells are transferred into the patients. The first ACT was developed for melanoma by isolation and expansion of tumor-infiltrating lymphocytes (TILs) from patients in the 1990s [116]. In TIL therapy, tumor biopsy is fragmented and plated in the presence of interleukine-2 (IL-2) for 2–4 weeks. The cultured TILs are selected and obtained according to the T-cell phenotype and reactivity to tumor cells for infusion into the patient [117]. Despite promising beneficial effects, many hurdles limit immunotherapy based on TILs. Due to TILs limited numbers in many tumors, their isolation is a time-consuming procedure [118]. Although the extracted TILs are tumor-specific, a remarkable portion of extracted cells has suppressive functions rather than anti-tumor activity [119]. Furthermore, immunotherapy with TILs is MHC-restricted and based on the recognition of TAAs by MHC, while the majority of tumor cells, mostly solid tumors, downregulate the expression of MHC [120]. Thus, genetically engineered T-cells have been emerged to overcome these hurdles. Figure 2 shows the generation process of CAR-T cells.

Overview of CAR-T cell therapy. The blood of the patient is collected and T cells are isolated. T cells are then genetically altered to express specific receptors for binding to certain targets on the cancer cells. The generated CAR-T cells are cultivated and expanded in vitro. Finally, CAR-T cells are infused into the patient’s bloodstream to kill the tumor cells
Chimeric antigen receptors (CARs) are bioengineered and hybrid of T-cell and antibody receptors composed of 4 distinct regions, including an extracellular domain, a hinge, a transmembrane (TM) domain, and an intracellular signaling domain (endodomain). The extracellular domain which recognizes antigen is a single-chain fragment variant (scFv) contains the variable light and heavy chain regions of an antibody separated via a flexible linker and is connected to the transmembrane domain by a hinge (spacer) [121]. The TM domain, a hydrophobic alpha-helix structure, guarantees the stability and expression of the receptor [121, 122]. The endodomain undergoes conformational changes following antigen recognition, triggering downstream signaling pathways to induce immune responses [123]. According to the composition and structure of the endodomain, five generations of CAR-T cells have been developed (Fig. 3). The first-generation CARs contain only CD3ζ intracellular domain. Owing to a lack of co-stimulatory and interleukin signals, the anti-tumor activity of them is limited [124]. To enhance their functions, the scientists added a co-stimulatory domain, including 4-1BB (CD137) or CD28, and developed the second-generation CAR-T cells [125]. A third-generation of CARs contains two co-stimulatory domains, such as both 4-1BB and CD28 [126]. The fourth-generation CAR-T cells, T-cells redirected for universal cytokine-mediated killing (TRUCKs), are designed based on the second-generation CARs which contain additional domains for cytokine secretion, such as IL-2. The fifth-generation or next-generation CARs are also based on second-generation CARs which include intracellular domains of cytokine receptors, such as IL-2Rβ [127].

Five generations of CAR-T cells. The first-generation CAR-T cells only contain one intracellular signal domain CD3ζ. The second-generation CARs consist of a co-stimulatory domain, including 4-1BB (CD137) or CD28, whereas the third-generation ones have two co-stimulatory domains. The fourth-generation CAR-T cells, based on the second-generation CARs, can induce cytokine production. The fifth-generation CAR-T cells are also based on the second-generation CARs, containing intracellular domains of cytokine receptors, such as IL-2Rβ chain fragment
CAR-T cell therapy for CRC
CAR-T cell therapy is becoming an interesting candidate in the treatment of cancer. Although most CAR-T cell therapy studies focus on the treatment of hematologic malignancies, such as lymphoma, with targeting CD19 antigen, demand for their application in the treatment of solid tumors in clinical trials is high. Different TAAs were translated into clinical trials for the treatment of CRC, including carcinoembryonic antigen (CEA), EGFR, mesothelin (MSLN), mucin 1 (MUC1), Natural killer group 2 member D (NKG2D) and its ligand, human epidermal growth factor receptor 2 (HER2), c-met, and CD133 (Table 2).
CEA
CEA, a glycoprotein belonging to the carcinoembryonic antigen cell adhesion molecules (CEACAM) family, is produced in the GI tract of humans in the early stages of embryonic development, whereas its expression decreases before birth and remains at a low level in the adult [128]. However, CEA expression is elevated in CRC and tissue CEA overexpression is associated with poorer prognosis and can act as a tumor marker in CRC [129]. Thus, CEA is an attractive target of CRC immunotherapy.
Chi et al. investigated the anti-tumor efficacy of CEA-specific CAR-T cell, a second-generation CAR in which 4‐1BB acted as a costimulatory domain, in combination with recombinant human IL‐12. They indicated that IL-12 markedly increases proliferation, activation, and cytotoxicity of CEA‐CAR‐T cells following treating CEA-positive HT‐29 cells. Combination with IL-12 enhances the expression levels of IFN‐γ, IL-2, CD25, and CD69, as markers for activation of CAR-T cells. Intravenous administration of 1 × 107 CEA‐CAR‐T cells in combination with 1500 U/mouse IL-12 showed stronger anti-tumor function compared with CEA‐CAR‐T cell administration in mice model. This combination regimen increased the persistence of CEA‐CAR‐T cells and enhanced secretion of IFN‐γ, TNF‐α, and IL-2 [130]. Blat et al. designed a CEA-targeting CAR regulatory T cells (Tregs) to suppress colitis-associated CRC. They showed that CEA-targeting CAR Tregs could reduce the progress of induced colitis toward CRC in a mouse model and decrease tumor burden [131]. In a phase I clinical trial, Zhang et al. investigated the safety and efficacy of CAR-T cell therapy in the treatment of CEA-positive CRC patients. They designed two CARs: a second-generation CAR with CD28 signaling domain and a third-generation CAR with CD28 and CD137 domains. Of the ten patients, two patients indicated tumor shrinkage and seven patients had stable disease in which two patients showed stable condition for more than 30 weeks after CAR-T cell therapy. The serum analysis revealed that CEA levels declined in most of the patients. In high-dose receiving, the persistence of CARs was observed. Furthermore, the cytotoxicity and cytokine secretion of the third-generation CARs was not better than the second-generation ones [132].
GUCY2C
Guanylyl Cyclase C (GUCY2C), a membrane-bound receptor, is regularly expressed by apical surfaces of the intestinal epithelium and in the brain [133]. Binding of cognate ligands to the GUCY2C produces cGMP as a second messenger, leading to regulation of colon homeostasis, obesity, and tumorigenesis [134]. It has been shown that GUCY2C is universally overexpressed in the primary and metastatic CRC [135, 136], making it a tempting immunotherapy target.
Magee et al. designed a third-generation GUCY2C-specific CAR-T cell composed of scFv, the CD8α hinge region, the TM and endodomain of CD28, and the endodomain of CD137 and CD3ζ and tested its efficacy in a metastatic CRC mice model. They found that GUCY2C CAR-T cells reduced the number of metastatic tumors in mice's lungs and the treated-mice showed reduced morbidity and improved survival without inducing autoimmunity. Moreover, treatment with GUCY2C CAR-T cells did not show toxicity and accumulation in the mice intestine [137]. This group also demonstrated that the protection of GUCY2C CAR-T cells against lung metastasis is long-term [138].
NKG2D
NKG2D is regularly expressed on the natural killer (NK) cells, CD8 + T-cells, some CD4 + T-cells, and γẟ T-cells which provides activating and costimulatory signals [139]. In humans, NKG2D interacts with 8 NKG2D ligands (NKG2DLs), including MHC class I-related chain A and B (MICA and MICB) and six unique long 16 (UL16)-binding proteins (ULBP1–6). Although NKG2DLs are undetectable or at low levels on healthy tissues, it is upregulated upon infection, DNA damage, and cell transformation [140, 141]. Binding of NKG2DLs to NKG2D activates immune cells, triggering cells' proliferation, producing proinflammatory cytokines, and eliminating target cells.
Deng et al. constructed a third-generation CAR composed of the CD8α signal sequence, the extracellular region of human NKG2D, the CD8α hinge, the TM and endodomain of CD 28, and the endodomains of CD3ζ and CD137. NKG2D CAR-T cells indicated cytotoxicity against human CRC in vitro in a dose-dependent manner and secreted high levels of IFN-γ and IL-2. Administration of 1 × 107 NKG2D CAR-T cells markedly inhibited tumor growth, reduced tumor size, and prolonged survival in a xenograft CRC mice model without any severe side effects in vital organs [142]. In another study, Xiao et al. used an RNA electroporation approach to provide NKG2D RNA for NK cells and assessed its anti-tumor functions. NKG2D RNA CAR therapy remarkably enhanced the cytolytic function of NK cells against tumor cells in vitro and significantly reduced tumor progression and prolonged survival time in a CRC mice model. Treatment of patients with metastatic CRC using NKG2D RNA CAR reduced ascites generation and tumor cell number in ascites samples [143]. Zhang et al. constructed NKG2D CAR T cells to target Rae1, an NKG2DL, for inhibiting tumor vasculature. They showed that NKG2D CAR T cells reduced tumor angiogenesis in a colon cancer mice model [144].
Other targets
Wei et al. a third-generation CAR-T cell for targeting epithelial cell adhesion molecule (EpCAM) which is overexpressed in different cancers. EpCAM CAR-T cells were able to show cytotoxicity against EpCAM positive CRC cells and secrete cytokines, such as IFN‐γ and TNF‐α. This adaptive therapy also significantly suppressed tumor growth and formation in a xenograft model without systemic toxicity [145].
Tumor-associated glycoprotein (TAG)-72 is another TAA that has been targeted for the treatment of CRC using CAR-T cells. TAG-72 is oncofetal mucin highly expressed in most of the human epithelial adenocarcinomas and its expression is mainly restricted to tumor cells [146]. Hege et al. constructed a first-generation TAG-72 CAR-T cell and tested its safety and efficacy in patients with metastatic CRC. They demonstrated that TAG-72 CAR-T cell therapy is relatively safe, whereas their persistence is limited owing to the lack of costimulatory domains [147].
Challenges of CAR-T cells
CAR-T cell therapy is faced with various challenges in the treatment of solid tumors. For example, physical barriers, including the surrounding stroma and cells, hinder the sufficient infiltration of CARs to the tumors. To overcome these barriers, Wang et al. designed FAP-CAR T cells to target fibroblasts in the tumor microenvironment (TME), leading to reduce the number of tumor fibroblasts and inhibition of tumor growth [148]. Targeting heparan sulfate proteoglycans, as a member of extracellular matrix (ECM) which limits cells homing to the tumors, with heparanase (HPSE)-expressing CARs also overcomes the physical barrier and improves CARs infiltration to the tumor [149] (Fig. 1).
The other concern in using CAR-T cells is their on-target/off-tumor toxicity. Since most tumor antigens are found both on cancerous cells and normal tissues, constructed CARs cannot distinguish the normal ones. Thus, identifying specific tumor antigens always is the CAR-T cell therapy challenge. For instance, a case-study of a patient with metastatic cancer expressing HER-2 revealed that antigen-specific CARs could result in organ dysfunctions, rapid respiratory distress, and death. Accumulation of anti-ERBB2 CARs in the lung and recognizing ERBB2-expressing normal cells and subsequent release of IFN-γ and TNF-α led to pulmonary toxicity and edema [150]. This toxicity is associated with the release of cytokines, also known as cytokine release syndrome. In another study, it was reported that targeting CEA in patients with colon cancer by CAR-T cells led to severe transient colitis because of CEA recognition in normal intestinal tissue [151] (Fig. 2).
Immunosuppressive TME also reduces CARs efficacy. TME consists of immune cells, including myeloid-derived suppressor cells (MDSCs), Tregs, and tumor-associated macrophages (TAMs), and molecular factors, including checkpoint inhibitory proteins, IL-10, and TGF-β, that inhibit anti-tumor functions of CAR-T cells [152]. There is evidence that blockade of the inhibitory pathways can augment CAR-T cell therapy. For example, the combination of an anti-PD-1 antibody with HER2 CAR T cells significantly enhanced tumor eradication in a mouse model [153] (Fig. 3).
Conclusions
Standard treatments for CRC, including surgery, radiotherapy, and chemotherapy, have many side effects owing to their non-specificity toward normal cell, leading to toxicity. Thus, alternative and safety treatments are crucial for CRC patients. In this regard, vaccines, cytokines, adjuvant, and neoadjuvant therapy to stimulate immune responses against tumor antigens have been developed as immunotherapy agents against CRC. Taken together, immunotherapy using both mAbs and CAR-T cells are effective with minimal side effects and toxicity. Preclinical and clinical studies demonstrated that CRC cells are vulnerable to specific mAbs. Furthermore, clinical trials using CARs could have promising therapeutic outcomes in patients with CRC. Although some challenges and concerns are using both mAbs and CAR-T cells, combinational therapies will improve the anti-tumor effects and optimize their functions.
Availability of data and materials
Not applicable.
Abbreviations
- CRC:
-
Colorectal cancer; mAbs: Monoclonal antibodies
- CAR-T cells:
-
Chimeric antigen receptor T cells
- TAAs:
-
Tumor-associated antigens
- TME:
-
Tumor microenvironment
- VEGF:
-
Vascular endothelial growth factor
- EGFR:
-
Epidermal growth factor receptor
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- HER2/neu:
-
Human epidermal growth factor receptor-2
- MUC5AC:
-
Mucin 5AC
- DR5:
-
Death receptor
- CTLA-4:
-
Cytotoxic T-lymphocyte associated protein 4
- PD-L1:
-
Programmed death-ligand 1
- PlGF:
-
Placental growth factors
- TCR:
-
T cell receptors
- MHC:
-
Major histocompatibility complex
- APC:
-
Antigen-presenting cell
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- GI:
-
Gastrointestinal
- ACT:
-
Adoptive T-cell therapy
- TILs:
-
Tumor-infiltrating cells
- TM:
-
Transmembrane
- scFv:
-
Single-chain fragment variant
- CEA:
-
Carcinoembryonic antigen
- MSLN:
-
Mesothelin
- MUC1:
-
Mucin 1
- NKG2D:
-
Natural killer group 2 member D
- Tregs:
-
Regulatory T cells
- GUCY2C:
-
Guanylyl cyclase C
- TAG-72:
-
Tumor-associated glycoprotein-72
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Veisi Malekshahi Z, Hashemi Goradel N, Shakouri Khomartash M, Maleksabet A, Kadkhodazadeh M, Kardar GA, et al. CEA plasmid as therapeutic DNA vaccination against colorectal cancer. Iran J Immunol. 2019;16:235–45.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Thanikachalam K, Khan G. Colorectal cancer and nutrition. Nutrients. 2019;11:164.
Martin FL, Martinez EZ, Stopper H, Garcia SB, Uyemura SA, Kannen V. Increased exposure to pesticides and colon cancer: Early evidence in Brazil. Chemosphere. 2018;209:623–31.
Hashemi Goradel N, Heidarzadeh S, Jahangiri S, Farhood B, Mortezaee K, Khanlarkhani N, et al. Fusobacterium nucleatum and colorectal cancer: a mechanistic overview. J Cell Physiol. 2019;234:2337–44.
Khiavi MA, Safary A, Somi MH. Recent advances in targeted therapy of colorectal cancer: impacts of monoclonal antibodies nanoconjugates. BioImpacts BI. 2019;9:123.
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422.
Goulart A, Ferreira C, Rodrigues A, Coimbra B, Sousa N, Leão P. The correlation between serum vascular endothelial growth factor (VEGF) and tumor VEGF receptor 3 in colorectal cancer. Ann Surg Treat Res. 2019;97:15–20.
Mokhtari M, Ardestani MM, Movahedipour M. An immunohistochemical study of EGFR expression in colorectal cancer and its correlation with lymph nodes status and tumor grade. J Res Med Sci Off J Isfahan Univ Med Sci. 2012;17:741.
Shiratsuchi I, Akagi Y, Kawahara A, Kinugasa T, Romeo K, Yoshida T, et al. Expression of IGF-1 and IGF-1R and their relation to clinicopathological factors in colorectal cancer. Anticancer Res. 2011;31:2541–5.
Wang X-Y, Zheng Z-X, Sun Y, Bai Y-H, Shi Y-F, Zhou L-X, et al. Significance of HER2 protein expression and HER2 gene amplification in colorectal adenocarcinomas. World J Gastrointest Oncol. 2019;11:335.
Vonlaufen A, Wiedle G, Borisch B, Birrer S, Luder P, Imhof BA. Integrin α v β 3 expression in colon carcinoma correlates with survival. Mod Pathol. 2001;14:1126–32.
Pothuraju R, Rachagani S, Krishn SR, Chaudhary S, Nimmakayala RK, Siddiqui JA, et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol Cancer. 2020;19:1–14.
Devetzi M, Kosmidou V, Vlassi M, Perysinakis I, Aggeli C, Choreftaki T, et al. Death receptor 5 (DR5) and a 5-gene apoptotic biomarker panel with significant differential diagnostic potential in colorectal cancer. Sci Rep. 2016;6:36532.
Omura Y, Toiyama Y, Okugawa Y, Yin C, Shigemori T, Kusunoki K, et al. Prognostic impacts of tumoral expression and serum levels of PD-L1 and CTLA-4 in colorectal cancer patients. Cancer Immunol Immunother. 2020;69:2533–46.
Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointest Oncol. 2015;6:208.
Ge P, Wang W, Li L, Zhang G, Gao Z, Tang Z, et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed Pharmacother. 2019;118:109228.
Roelands J, Kuppen PJK, Vermeulen L, Maccalli C, Decock J, Wang E, et al. Immunogenomic classification of colorectal cancer and therapeutic implications. Int J Mol Sci. 2017;18:2229.
Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. 2018;9:398.
Hu G, Wu P, Cheng P, Zhang Z, Wang Z, Yu X, et al. Tumor-infiltrating CD39+ γ δ Tregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology. 2017;6:e1277305.
Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. M2 macrophages confer resistance to 5-fluorouracil in colorectal cancer through the activation of CCL22/PI3K/AKT signaling. Onco Targets Ther. 2019;12:3051.
Lan J, Sun L, Xu F, Liu L, Hu F, Song D, et al. M2 macrophage-derived exosomes promote cell migration and invasion in colon cancer. Cancer Res. 2019;79:146–58.
Marech I, Ammendola M, Sacco R, Sammarco G, Zuccalà V, Zizzo N, et al. Tumour-associated macrophages correlate with microvascular bed extension in colorectal cancer patients. J Cell Mol Med. 2016;20:1373–80.
Nosho K, Sukawa Y, Adachi Y, Ito M, Mitsuhashi K, Kurihara H, et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J Gastroenterol. 2016;22:557.
Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7.
Buss NAPS, Henderson SJ, McFarlane M, Shenton JM, De Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 2012;12:615–22.
Presta LG. Engineering of therapeutic antibodies to minimize immunogenicity and optimize function. Adv Drug Deliv Rev. 2006;58:640–56.
Harding FA, Stickler MM, Razo J, DuBridge R. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–65.
Mallbris L, Davies J, Glasebrook A, Tang Y, Glaesner W, Nickoloff BJ. Molecular insights into fully human and humanized monoclonal antibodies: What are the differences and should dermatologists care? J Clin Aesthet Dermatol. 2016;9:13.
Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7(1):9–14.
Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27:1–30.
Tsumoto K, Isozaki Y, Yagami H, Tomita M. Future perspectives of therapeutic monoclonal antibodies. Immunotherapy. 2019;11:119–27.
Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen Y-J, Ciombor KK, et al. NCCN guidelines insights: colon cancer, version 2.2018. J Natl Compr Cancer Netw. 2018;16:359–69.
Hashemi Goradel N, Ghiyami-Hour F, Jahangiri S, Negahdari B, Sahebkar A, Masoudifar A, et al. Nanoparticles as new tools for inhibition of cancer angiogenesis. J Cell Physiol. 2018;233:2902–10.
Goradel NH, Asghari MH, Moloudizargari M, Negahdari B, Haghi-Aminjan H, Abdollahi M. Melatonin as an angiogenesis inhibitor to combat cancer: Mechanistic evidence. Toxicol Appl Pharmacol. 2017;335:56–63.
Goradel NH, Mohammadi N, Haghi-Aminjan H, Farhood B, Negahdari B, Sahebkar A. Regulation of tumor angiogenesis by microRNAs: State of the art. J Cell Physiol. 2019;234:1099–110.
Costache MI, Ioana M, Iordache S, Ene D, Costache CA, Săftoiu A. VEGF expression in pancreatic cancer and other malignancies: a review of the literature. Rom J Intern Med. 2015;53:199–208.
Bendardaf R, Buhmeida A, Hilska M, Laato M, Syrjänen S, Syrjänen K, et al. VEGF-1 expression in colorectal cancer is associated with disease localization, stage, and long-term disease-specific survival. Anticancer Res. 2008;28:3865–70.
Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017.
Ellis LM, Kirkpatrick P. Bevacizumab. Nat Rev Drug Discov. 2005;4:S8–9.
Hey SP, Gyawali B, D’Andrea E, Kanagaraj M, Franklin JM, Kesselheim AS. A systematic review and meta-analysis of bevacizumab in first-line metastatic breast cancer: lessons for research and regulatory Enterprises. JNCI J Natl Cancer Inst. 2020;112:335–42.
Marchetti C, Muzii L, Romito A, Panici PB. First-line treatment of women with advanced ovarian cancer: focus on bevacizumab. Onco Targets Ther. 2019;12:1095.
Seto T, Azuma K, Yamanaka T, Sugawara S, Yoshioka H, Wakuda K, et al. Randomized phase III study of continuation maintenance bevacizumab with or without pemetrexed in advanced nonsquamous non–small-cell lung cancer: COMPASS (WJOG5610L). J Clin Oncol. 2020;38:793–803.
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60–5.
Saltz LB, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26:2013–9.
Bennouna J, Sastre J, Arnold D, Österlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14:29–37.
Baraniskin A, Buchberger B, Pox C, Graeven U, Holch JW, Schmiegel W, et al. Efficacy of bevacizumab in first-line treatment of metastatic colorectal cancer: a systematic review and meta-analysis. Eur J Cancer. 2019;106:37–44.
Verdaguer H, Tabernero J, Macarulla T. Ramucirumab in metastatic colorectal cancer: evidence to date and place in therapy. Ther Adv Med Oncol. 2016;8:230–42.
Chiorean EG, Hurwitz HI, Cohen RB, Schwartz JD, Dalal RP, Fox FE, et al. Phase I study of every 2-or 3-week dosing of ramucirumab, a human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2 in patients with advanced solid tumors. Ann Oncol. 2015;26:1230–7.
Spratlin JL, Cohen RB, Eadens M, Gore L, Camidge DR, Diab S, et al. Phase I pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010;28:780.
Garcia-Carbonero R, Rivera F, Maurel J, Ayoub J-PM, Moore MJ, Cervantes A, et al. An open-label phase II study evaluating the safety and efficacy of ramucirumab combined with mFOLFOX-6 as first-line therapy for metastatic colorectal cancer. Oncologist. 2014;19:350.
Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blin. Lancet Oncol. 2015;16:499–508.
Trichonas G, Kaiser PK. Aflibercept for the treatment of age-related macular degeneration. Ophthalmol Ther. 2013;2:89–98.
Kim ES, Serur A, Huang J, Manley CA, McCrudden KW, Frischer JS, et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc Natl Acad Sci. 2002;99:11399–404.
Khayat D, Tejpar S, Spano J-P, Verslype C, Bloch J, Vandecaveye V, et al. Intravenous aflibercept administered in combination with irinotecan, 5-fluorouracil and leucovorin in patients with advanced solid tumours: results from the expansion cohort of a phase I study. Eur J Cancer. 2013;49:790–7.
Lockhart AC, Rothenberg ML, Dupont J, Cooper W, Chevalier P, Sternas L, et al. Phase I study of intravenous vascular endothelial growth factor trap, aflibercept, in patients with advanced solid tumors. J Clin Oncol. 2010;28:207.
Townsley CA, Siu LL, San Pedro-Salcedo M, Liu L, Wakelee HA. A phase I study of aflibercept, pemetrexed (P), and cisplatin (C) in patients with advanced solid tumors. J Clin Oncol. 2010;28:2536.
Denda T, Sakai D, Hamaguchi T, Sugimoto N, Ura T, Yamazaki K, et al. Phase II trial of aflibercept with FOLFIRI as a second-line treatment for Japanese patients with metastatic colorectal cancer. Cancer Sci. 2019;110:1032–43.
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499–506.
Lee SH. Tanibirumab (TTAC-0001): a fully human monoclonal antibody targets vascular endothelial growth factor receptor 2 (VEGFR-2). Arch Pharm Res. 2011;34:1223.
Lee SJ, Lee SY, Lee WS, San Yoo J, Sun J-M, Lee J, et al. Phase I trial and pharmacokinetic study of tanibirumab, a fully human monoclonal antibody to vascular endothelial growth factor receptor 2, in patients with refractory solid tumors. Invest New Drugs. 2017;35:782–90.
Kienast Y, Klein C, Scheuer W, Raemsch R, Lorenzon E, Bernicke D, et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin Cancer Res. 2013;19:6730–40.
Schaefer W, Regula JT, Bähner M, Schanzer J, Croasdale R, Dürr H, et al. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc Natl Acad Sci. 2011;108:11187–92.
Hidalgo M, Martinez-Garcia M, Le Tourneau C, Massard C, Garralda E, Boni V, et al. First-in-human phase I study of single-agent vanucizumab, a first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin Cancer Res. 2018;24:1536–45.
Bendell JC, Sauri T, Gracián AC, Alvarez R, López-López C, García-Alfonso P, et al. The McCAVE trial: VANUCIZUMAB plus mFOLFOX-6 Versus Bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal carcinoma (mCRC). Oncologist. 2020;25:e451.
Costa R, Shah AN, Santa-Maria CA, Cruz MR, Mahalingam D, Carneiro BA, et al. Targeting epidermal growth factor receptor in triple negative breast cancer: new discoveries and practical insights for drug development. Cancer Treat Rev. 2017;53:111–9.
Sforza V, Martinelli E, Ciardiello F, Gambardella V, Napolitano S, Martini G, et al. Mechanisms of resistance to anti-epidermal growth factor receptor inhibitors in metastatic colorectal cancer. World J Gastroenterol. 2016;22:6345.
Krasinskas AM. EGFR signaling in colorectal carcinoma. Patholog Res Int 2011;2011.
Li N, Bu X, Wu P, Wu P, Huang P. The, “HER2–PI3K/Akt–FASN Axis” regulated malignant phenotype of colorectal cancer cells. Lipids. 2012;47:403–11.
Huang C-W, Tsai H-L, Chen Y-T, Huang C-M, Ma C-J, Lu C-Y, et al. The prognostic values of EGFR expression and KRAS mutation in patients with synchronous or metachronous metastatic colorectal cancer. BMC Cancer. 2013;13:599.
Gomez D, De Rosa A, Addison A, Brooks A, Malik HZ, Cameron IC. Cetuximab therapy in the treatment of metastatic colorectal cancer: the future frontier? Int J Surg. 2013;11:507–13.
Gridelli C, Maione P, Ferrara ML, Rossi A. Cetuximab and other anti-epidermal growth factor receptor monoclonal antibodies in the treatment of non-small cell lung cancer. Oncologist. 2009;14:601–11.
Bokemeyer C, Bondarenko I, Hartmann JT, De Braud F, Schuch G, Zubel A, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–46.
Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8.
Matsuda A, Yamada T, Jamjittrong S, Shinji S, Ohta R, Sonoda H, et al. Comparison between biweekly and weekly cetuximab in patients with metastatic colorectal cancer: a meta-analysis. Anticancer Res. 2020;40:3469–76.
Battaglin F, Puccini A, Djaballah SA, Lenz H-J. The impact of panitumumab treatment on survival and quality of life in patients with RAS wild-type metastatic colorectal cancer. Cancer Manag Res. 2019;11:5911.
Wainberg Z, Hecht JR. Panitumumab in colon cancer: a review and summary of ongoing trials. Expert Opin Biol Ther. 2006;6:1229–35.
Veronese ML, O’Dwyer PJ. Monoclonal antibodies in the treatment of colorectal cancer. Eur J Cancer. 2004;40:1292–301.
Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol. 2014;25:1346–55.
Lievre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5.
Prewett M, Tonra JR, Rajiv B, Hooper AT, Makhoul G, Finnerty B, et al. Antitumor activity of a novel, human anti-epidermal growth factor receptor (EGFR) monoclonal antibody (IMC-11F8) in human tumor xenograft models. AACR. 2004;64:1235.
Elez E, Hendlisz A, Delaunoit T, Sastre J, Cervantes A, Varea R, et al. Phase II study of necitumumab plus modified FOLFOX6 as first-line treatment in patients with locally advanced or metastatic colorectal cancer. Br J Cancer. 2016;114:372–80.
Lewis AL, Chaft J, Girotra M, Fischer GW. Immune checkpoint inhibitors: a narrative review of considerations for the anaesthesiologist. Br J Anaesth. 2020;124:251–60.
Huyghe N, Baldin P, Van den Eynde M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: what is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep. 2020;8:11–24.
Jácome AA, Eng C. Role of immune checkpoint inhibitors in the treatment of colorectal cancer: focus on nivolumab. Expert Opin Biol Ther. 2019;19:1247–63.
Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015;67:4–17.
O’Neil BH, Wallmark JM, Lorente D, Elez E, Raimbourg J, Gomez-Roca C, et al. Safety and antitumor activity of the anti–PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLoS ONE. 2017;12:e0189848.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Le DT, Kavan P, Kim TW, Burge ME, Van Cutsem E, Hara H, et al. KEYNOTE-164: pembrolizumab for patients with advanced microsatellite instability high (MSI-H) colorectal cancer. Am Soc Clin Oncol. 2018;36:3514.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5:43–51.
Chaudhari PB. Nivolumab-Pearls of evidence. Indian J Med Paediatr Oncol Off J Indian Soc Med Paediatr Oncol. 2017;38:520.
Yamamoto N, Nokihara H, Yamada Y, Shibata T, Tamura Y, Seki Y, et al. Phase I study of Nivolumab, an anti-PD-1 antibody, in patients with malignant solid tumors. Invest New Drugs. 2017;35:207–16.
Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36:773–9.
Inman BA, Longo TA, Ramalingam S, Harrison MR. Atezolizumab: a PD-L1–blocking antibody for bladder cancer. Clin Cancer Res. 2017;23:1886–90.
Antoniotti C, Borelli B, Rossini D, Pietrantonio F, Morano F, Salvatore L, et al. AtezoTRIBE: a randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer. 2020;20:1–9.
Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20:849–61.
Zhao Y, Yang W, Huang Y, Cui R, Li X, Li B. Evolving roles for targeting CTLA-4 in cancer immunotherapy. Cell Physiol Biochem. 2018;47:721–34.
Kooshkaki O, Derakhshani A, Hosseinkhani N, Torabi M, Safaei S, Brunetti O, et al. Combination of ipilimumab and nivolumab in cancers: from clinical practice to ongoing clinical trials. Int J Mol Sci. 2020;21:4427.
Morse MA, Overman MJ, Hartman L, Khoukaz T, Brutcher E, Lenz H, et al. Safety of nivolumab plus low-dose ipilimumab in previously treated microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer. Oncologist. 2019;24:1453.
Ribas A, Hanson DC, Noe DA, Millham R, Guyot DJ, Bernstein SH, et al. Tremelimumab (CP-675,206), a cytotoxic T lymphocyte–associated antigen 4 blocking monoclonal antibody in clinical development for patients with cancer. Oncologist. 2007;12:873–83.
Chung KY, Gore I, Fong L, Venook A, Beck SB, Dorazio P, et al. Phase II study of the anti-cytotoxic T-lymphocyte–associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J Clin Oncol. 2010;28:3485–90.
Rocha Lima CM, Bayraktar S, Flores AM, MacIntyre J, Montero A, Baranda JC, et al. Phase Ib study of drozitumab combined with first-line mFOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Cancer Invest. 2012;30:727–31.
Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F, et al. Phase II study of the anti–insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non–small-cell lung cancer. J Clin Oncol. 2009;27:2516–22.
Françoso A, Simioni PU. Immunotherapy for the treatment of colorectal tumors: focus on approved and in-clinical-trial monoclonal antibodies. Drug Des Devel Ther. 2017;11:177.
Le DT, Uram JN, Wang H, Bartlett B, Kemberling H, Eyring A, et al. Programmed death-1 blockade in mismatch repair deficient colorectal cancer. J Clin Oncol. 2016;34:103.
Mathijssen RHJ, De Jong FA, Loos WJ, Van Der Bol JM, Verweij J, Sparreboom A. Flat-fixed dosing versus body surface area-based dosing of anticancer drugs in adults: does it make a difference? Oncologist. 2007;12:913.
Kreisl TN, Zhang W, Odia Y, Shih JH, Butman JA, Hammoud D, et al. A phase II trial of single-agent bevacizumab in patients with recurrent anaplastic glioma. Neuro Oncol. 2011;13:1143–50.
Lafayette RA, McCall B, Li N, Chu L, Werner P, Das A, et al. Incidence and relevance of proteinuria in bevacizumab-treated patients: pooled analysis from randomized controlled trials. Am J Nephrol. 2014;40:75–83.
Almagro JC, Daniels-Wells TR, Perez-Tapia SM, Penichet ML. Progress and challenges in the design and clinical development of antibodies for cancer therapy. Front Immunol. 2018;8:1751.
Ramos-de-la-Peña AM, González-Valdez J, Aguilar O. Protein A chromatography: challenges and progress in the purification of monoclonal antibodies. J Sep Sci. 2019;42:1816–27.
Ulmer N, Vogg S, Müller-Späth T, Morbidelli M. Purification of human monoclonal antibodies and their fragments. In: Steinitz M, editor. Human monoclonal antibodies. Methods in Molecular Biology; vol. 1904, 2019. pp. 63–88.
Arosio P, Barolo G, Müller-Späth T, Wu H, Morbidelli M. Aggregation stability of a monoclonal antibody during downstream processing. Pharm Res. 2011;28:1884–94.
Sifniotis V, Cruz E, Eroglu B, Kayser V. Current advancements in addressing key challenges of therapeutic antibody design, manufacture, and formulation. Antibodies. 2019;8:36.
Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science (80-). 1986;233:1318–21.
Lee S, Margolin K. Tumor-infiltrating lymphocytes in melanoma. Curr Oncol Rep. 2012;14:468–74.
Hayes C. Cellular immunotherapies for cancer. Irish J Med Sci 2020;1–17.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9.
Comoli P, Chabannon C, Koehl U, Lanza F, Urbano-Ispizua A, Hudecek M, et al. Development of adaptive immune effector therapies in solid tumors. Ann Oncol. 2019;30:1740–50.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering car-t cells. Biomark Res. 2017;5:1–6.
Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11:855–73.
Su X, Vale R. Mechanisms of chimeric antigen receptor (CAR) signaling during T cell activation. Biophys J. 2018;114:107a–8a.
Smith AJ, Oertle J, Warren D, Prato D. Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: summary and perspective. J Cell Immunother. 2016;2:59–68.
Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric antigen receptor T cells: a race to revolutionize cancer therapy. Transfus Med Hemotherapy. 2019;46:15–24.
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13:1–19.
Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. 2019;120:26–37.
Rodrigues JG, Balmaña M, Macedo JA, Poças J, Fernandes Â, De-Freitas-Junior JCM, et al. Glycosylation in cancer: Selected roles in tumour progression, immune modulation and metastasis. Cell Immunol. 2018;333:46–57.
Tong G, Xu W, Zhang G, Liu J, Zheng Z, Chen Y, et al. The role of tissue and serum carcinoembryonic antigen in stages I to III of colorectal cancer—a retrospective cohort study. Cancer Med. 2018;7:5327–38.
Chi X, Yang P, Zhang E, Gu J, Xu H, Li M, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019;8:4753–65.
Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. 2014;22:1018–28.
Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther. 2017;25:1248–58.
Valentino MA, Lin JE, Snook AE, Li P, Kim GW, Marszalowicz G, et al. A uroguanylin-GUCY2C endocrine axis regulates feeding in mice. J Clin Invest. 2011;121:3578–88.
Aka AA, Rappaport JA, Pattison AM, Sato T, Snook AE, Waldman SA. Guanylate cyclase C as a target for prevention, detection, and therapy in colorectal cancer. Expert Rev Clin Pharmacol. 2017;10:549–57.
Frick GS, Pitari GM, Weinberg DS, Hyslop T, Schulz S, Waldman SA. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn. 2005;5:701–13.
Carrithers SL, Barber MT, Biswas S, Parkinson SJ, Park PK, Goldstein SD, et al. Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc Natl Acad Sci. 1996;93:14827–32.
Magee MS, Kraft CL, Abraham TS, Baybutt TR, Marszalowicz GP, Li P, et al. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology. 2016;5:e1227897.
Magee MS, Abraham TS, Baybutt TR, Flickinger JC, Ridge NA, Marszalowicz GP, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res. 2018;6:509–16.
Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. 2018;51:55–61.
Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–41.
Antonangeli F, Soriani A, Cerboni C, Sciumè G, Santoni A. How mucosal epithelia deal with stress: role of NKG2D/NKG2D ligands during inflammation. Front Immunol. 2017;8:1583.
Deng X, Gao F, Li N, Li Q, Zhou Y, Yang T, et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res. 2019;9:945.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27:1114–25.
Zhang T, Sentman CL. Mouse tumor vasculature expresses NKG2D ligands and can be targeted by chimeric NKG2D-modified T cells. J Immunol. 2013;190:2455–63.
Zhang B-L, Li D, Gong Y-L, Huang Y, Qin D-Y, Jiang L, et al. Preclinical evaluation of chimeric antigen receptor-modified T cells specific to epithelial cell adhesion molecule for treating colorectal cancer. Hum Gene Ther. 2019;30:402–12.
Thor A, Ohuchi N, Szpak CA, Johnston WW, Schlom J. Distribution of oncofetal antigen tumor-associated glycoprotein-72 defined by monoclonal antibody B72.3. Cancer Res. 1986;46:3118–24.
Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22.
Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–66.
Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–9.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51.
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–6.
Xia A-L, Wang X-C, Lu Y-J, Lu X-J, Sun B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: challenges and opportunities. Oncotarget. 2017;8:90521.
John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin cancer Res. 2013;19:5636–46.
Acknowledgements
Not applicable.
Funding
This work was supported by Zhejiang Provincial Science and Technology Projects (No. LGD19H160001), National Natural Science Foundation of China (Nos. 81772537, 81374014, and 81903597), and Zhejiang Provincial Natural Science Foundation (No. LQ16H310003).
Author information
Authors and Affiliations
Contributions
Study conception and design: K-TJ. Article screening: BC. Drafting of the manuscript: K-TJ, BC, Y-YL, H-RL. Critical revision: J-PY. Approve final version: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, KT., Chen, B., Liu, YY. et al. Monoclonal antibodies and chimeric antigen receptor (CAR) T cells in the treatment of colorectal cancer. Cancer Cell Int 21, 83 (2021). https://doi.org/10.1186/s12935-021-01763-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-01763-9