
Abstract
Background
Pyruvate decarboxylase (PDC) is a well-known pathway for ethanol production, but has not been demonstrated for high titer ethanol production at temperatures above 50 °C.
Result
Here we examined the thermostability of eight PDCs. The purified bacterial enzymes retained 20% of activity after incubation for 30 min at 55 °C. Expression of these PDC genes, except the one from Zymomonas mobilis, improved ethanol production by Clostridium thermocellum. Ethanol production was further improved by expression of the heterologous alcohol dehydrogenase gene adhA from Thermoanaerobacterium saccharolyticum.
Conclusion
The best PDC enzyme was from Acetobactor pasteurianus. A strain of C. thermocellum expressing the pdc gene from A. pasteurianus and the adhA gene from T. saccharolyticum was able to produce 21.3 g/L ethanol from 60 g/L cellulose, which is 70% of the theoretical maximum yield.
Similar content being viewed by others
Background
Plant lignocellulosic biomass represents the most abundant renewable resource on the earth which is produced at an approximate rate of 150–170 × 109 tons annually [1]. It is therefore one of the most attractive substrates for sustainable production of second-generation biofuels and organic chemicals. Among all the processes to overcome the recalcitrance of cellulosic biomass, consolidated bioprocessing (CBP) is the most promising configuration because it accomplishes biomass solubilization and fermentation in one step without adding enzymes [2,3,4]. Clostridium thermocellum is a good candidate organism for CBP due to its ability to rapidly ferment cellulose and produce ethanol. Several approaches have been pursued aimed at engineering C. thermocellum to produce ethanol at higher yield [5] including by-product elimination [6,7,8,9], adaptive evolution [10] and gene overexpression [11,12,13,14]; however, CBP for ethanol production from cellulose using C. thermocellum is still not an economical process according to the target performance metrics for cost-effective production of ethanol from lignocellulose of 90% of theoretical yield and 40 g/L titer [15].
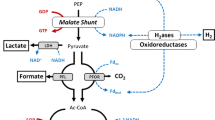
One of the limiting steps for the ethanol production in C. thermocellum is the pyruvate ferredoxin oxidoreductase (PFOR, Fig. 1). The PFOR enzyme transfers electrons from pyruvate to ferredoxin, which must be subsequently transferred to a nicotinamide cofactor (NAD+ or NADP+) in order to be used for ethanol production. Electron transfer from ferredoxin to NAD(P) + is an exergonic reaction which is frequently coupled to another endergonic reaction for energy conservation [16]. This energy conservation, however, comes at the price of thermodynamic driving force [17]. Another potential problem with the PFOR-based pathway is its reliance on the ALDH reaction. The ΔrG′m value of this reaction is − 0.1 kJ/mol (Fig. 1) and would thus require either a high ratio of acetyl-CoA to acetaldehyde or NAD(P)H to NAD(P) (or both) in order to proceed towards ethanol production.

Two ethanol production pathways from pyruvate. PFOR pyruvate ferredoxin oxidoreductase, ALDH acetaldehyde dehydrogenase, ADH alcohol dehydrogenase, PDC pyruvate decarboxylase, FNOR ferredoxin-NAD(P)+ oxidoreductase, Fdo oxidized ferredoxin, FdR reduced ferredoxin. All the ΔrG′m values were calculated by eQuilibrator [51] and are in units of kJ/mol. ‘m’ to mark the 1 mM concentration that is used for all reactants. For purposes of calculation, CO2 is assumed to be in the gas phase
An alternative pathway for ethanol production involves direct conversion of pyruvate to acetaldehyde by the enzyme pyruvate decarboxylase (PDC). This pathway is simpler than the PFOR pathway because it requires fewer enzymatic reactions (two vs. four for the PFOR pathway Fig. 1). Furthermore, both steps of the PDC pathway are thermodynamically favorable at standard conditions. Similar to the native pathway, the PDC pathway is also redox balanced. This pathway has been widely studied in Saccharomyces cerevisiae [18] and Zymomonas mobilis [19], but is found in a range of other organisms including Acetobacter pasteurianus (note that the PDC pathway in A. pasteurianus can also be used to produce acetate) [20], Kluyveromyces marxianus [21], Zymobacter palmae [22], Ogataea polymorpha [23, 24] and Gluconobacter oxydans [25].
The pyruvate decarboxylase gene from Z. mobilis has been used to enhance ethanol production in a variety of mesophilic organism including Escherichia coli [26], Klebsiella oxytoca [27], Bacillus subtilis [28], Lactobacillus casei [29] and cyanobacteria Synechococcus sp. [30]. When the gene was overexpressed in Clostridium cellulolyticum, a mesophilic cellulolytic clostridia, cellulose consumption was improved but the ethanol yield did not increase [31].
There are a number of thermophilic species of yeast that produce ethanol using the PDC pathway, but the upper temperature limit for ethanol production in these organisms appears to be around 45–48 °C [23, 24]. Although the Pdc proteins from a variety of organisms are tolerant to incubation at temperatures up to 65 °C [22], attempts to express the pdc gene in thermophilic organisms have met with limited success. Expression of the Gluconobacter oxydans pdc gene in Geobacillus thermoglucosidasius resulted in an increase in ethanol yield from 49% of theoretical (0.25 g ethanol/g glucose) to 69% of theoretical (0.35 g ethanol/g glucose), but this effect was only observed during growth at 45 °C and disappeared when the temperature was raised to 52 °C [32]. Kannuchamy et al. recently reported expression of the Z. mobilis pdc gene in C. thermocellum which increased ethanol titer from 1.5 g/L (26% of theoretical yield) to 3.0 g/L (53% of theoretical yield), however these yield values are significantly lower than the highest reported ethanol yields for C. thermocellum of 75% [10].
Here, we tested the thermostability of eight pyruvate decarboxylase candidates in vitro and then overexpressed them in C. thermocellum, with the goal of identifying the best candidate for ethanol production.
Results and discussion
Biochemical characterization of the PDC candidates
Eight thermostable pyruvate decarboxylase candidates were selected based on previous reports of thermostability (Table 1).
For our measurements of the thermostability of PDC enzymes, we used the method of Raj et al. [22] which is one of the more widely-used methods for this purpose. Candidate genes were cloned and expressed in E. coli. The resulting purified proteins were incubated at different temperatures from 30 to 60 °C for 30 min and then chilled on ice for 10 min before the assay. PDC activity was determined at 30 °C (Additional file 1). Relative activity was determined using the activity at 30 °C as the basis (Fig. 2). Among all eight of the candidates, only four (ZpPdc, ZmPdc, GoPdc and ApPdc) still had more than 30% of activity after 30 min incubation at 55 °C, the growth temperature for C. thermocellum. Organism name abbreviations are described in Table 1.

Thermostability of eight pyruvate decarboxylase candidates. Enzyme activity at 30 °C is set to 100%. Thermostability was tested at the temperatures of 45, 50, 55 and 60 °C. The data represents the average of three individual rounds of protein purification and assay. Error bars are smaller than the size of the data point markers and are therefore not shown
Heterologous expression of pdc genes in C. thermocellum
Plasmid pDGO143 was used to express all of the candidate pdc genes under control of the native promotor P2638 from C. thermocellum and their activities from cell extracts were measured (Additional file 2) [13]. After transformation into C. thermocellum, four colonies were selected for each candidate pdc gene, and analyzed by batch fermentation in MTC-5 medium with 10 g/L cellobiose. Three candidates, ZpPdc, GoPdc and ApPdc showed an increase in ethanol titer of 20, 16 and 14% respectively. For the others, the effect was less than 5% (Fig. 3). This result was consistent with the in vitro thermostability result (Fig. 2). Generally, proteins that showed high thermostability also showed increased ethanol production. The one exception was ZmPdc, which exhibited high levels of thermostability in vitro, but did not increase ethanol production in C. thermocellum.

The ethanol titer of C. thermocellum with different pyruvate decarboxylase candidates. Four colonies were selected for each candidate pdc gene and evaluated by batch fermentation in MTC-5 medium with 10 g/L cellobiose. Data for each colony is represented by a single point. 10 μg/mL thiamphenicol was added to maintain the plasmid. Strain LL1004 is wild type C. thermocellum. The box includes the 25th to 75th percentile of data; whiskers represent 1.5 times the interquartile range
Our results conflict with the results of Kannuchamy et al. [37], who reported that Pdc from Z. mobilis improves ethanol production in C. thermocellum. We do not have a good explanation for this discrepancy. We tried to obtain strain and/or plasmid stocks from the authors of that work, but were told that they no longer exist and were thus not able to perform a more detailed side-by-side comparison.
In C. thermocellum, the bifunctional enzyme AdhE is primarily responsible for both the ADH and ALDH reactions (Fig. 1) in C. thermocellum [38, 39]. It has been theorized that this bifunctional enzyme can channel acetaldehyde between the ALDH and ADH domains, which limits the leakage of acetaldehyde, which is volatile and toxic [40, 41]. This channeling may reduce the ability of the ADH domain of AdhE to accept free acetaldehyde as a substrate, and could explain the high KM values that have been measured for the ADH reaction in AdhE [39]. Since the product of the pyruvate decarboxylase reaction is acetaldehyde, we decided to express a dedicated ADH enzyme. Furthermore, it has been shown that AdhA from Thermoanaerobacterium saccharolyticum is able to increase ethanol yield in C. thermocellum [42].
The adhA and pdc genes were cloned into a plasmid. Protein expression was optimized by targeted modification of the RBS region using a web-based tool [43, 44]. A plasmid containing only the adhA gene was also constructed as a control. The new plasmids were used to transform the wild type strain of C. thermocellum (LL1004). Two colonies of each transformation were selected and then cultured in MTC-5 medium with 30 g/L cellobiose. The ethanol yields (percentage of the theoretical maximum yield of four moles of ethanol per mole of cellobiose consumed) of the colonies with ZpPdc, GoPdc and ApPdc are shown in Fig. 4a.

The ethanol production of C. thermocellum with different pyruvate decarboxylase genes and T. saccharolyticum alcohol dehydrogenase. a Two colonies were selected for each candidate pdc gene and evaluated by batch fermentation in MTC-5 medium with 30 g/L cellobiose. Three biological replicates were measured. b Ten colonies with AdhA and ApPdc were cultured in MTC-5 medium with 30 g/L cellobiose. Data for each colony is represented by a single point. All the P values were calculated based on the control strain LL1004 with only AdhA. 10 μg/mL thiamphenicol was added to maintain the plasmid. The box includes the 25th to 75th percentile of data; whiskers represent 1.5 times the interquartile range
Among these pdc genes, the strains with ApPdc produced the highest titer. Although strains expressing the other two Pdc proteins also show similar yields, their titers were much lower compared to the strain expressing ApPDC. To determine the colony-to-colony variation, eight more colonies with ApPdc were selected and cultured in MTC-5 medium with 30 g/L cellobiose (Fig. 4b; Additional file 3). Most of the colonies produced more than 200 mM ethanol, 54% more than the control strain (AdhA without Pdc). The strain expressing AdhA and ApPdc exerted an inhibitory effect on growth which reduced the growth rate by ~ 12% (Additional file 4). Colony LL1398 (expressing ApPDC and AdhA) was the best ethanol producing strain, based on the 30 g/L cellobiose fermentation experiment, and was selected for further study.
Confirmation of PDC activity in C. thermocellum
Transcription of the pdc gene was confirmed by qPCR (Additional file 5: Figure S1). Both the adhA and pdc genes showed high levels of expression (relative to the recA reference gene). However, we were unable to accurately measure PDC activity from cell extract of C. thermocellum. The background activity was quite high because C. thermocellum has a number of enzymes which could interfere with the PDC activity assay, including lactate dehydrogenase, pyruvate ferredoxin oxidoreductase and pyruvate formate lyase. To avoid this interference, we purified the ApPdc from C. thermocellum using a 6× His-tag. The purified enzyme had a specific activity of 0.28 ± 0.04 U/mg, which is about 100 times lower than the activity of the ApPdc cloned and purified from E. coli (31.07 ± 1.50 U/mg). One possible explanation for this difference is that in C. thermocellum, the protein was kept at 55 °C for a longer period of time (i.e. the time when the organism was growing, before the protein was harvested), compared to E. coli, which was grown at 37 °C. Another possible explanation is that Pdc acquires post-translational modifications in E. coli that it does not receive in C. thermocellum. Yet another possibility is that a large portion of the enzyme is degraded by native protease enzymes in C. thermocellum, but not in E. coli.
Batch culture of the strain with ApPdc and AdhA
In a bioreactor batch fermentation of strain LL1398 (expressing ApPDC and AdhA) with 60 g/L cellulose (Avicel PH105), 95% of the substrate was consumed (Fig. 5; Additional file 6). The final ethanol titer was 21.3 ± 0.2 g/L and the yield was 0.35 gEtOH/gGlu eq, which is 70% of the maximum theoretical yield. The main by-products were lactate (1.1 ± 0.1 g/L), acetate (2.0 ± 0.2 g/L) and formate (1.2 ± 0.1 g/L).

Cellulose consumption and product formation by the strain with AdhA and ApPDC at 55 °C and pH 6.5 in a bioreactor. The strain was grown in pH-controlled bioreactors, with 60 g/L crystalline cellulose as the sole carbon source. Error bars on each data point represent one standard deviation (n = 3 fermentations)
The carbon recovery based on the T = 100 h sample was calculated and the total carbon recovery was 100.7% (Table 2). Besides biomass (8.0%), the main fermentation by-products were extracellular amino acids which accounted for 11.8% of the carbon. Glutamate (1.7 ± 0.3 g/L), valine (0.6 ± 0.2 g/L), arginine (0.5 ± 0.1 g/L) and alanine (0.4 ± 0.02 g/L) were the most abundant amino acids. In comparison to the amino acid production of wild type and some engineered C. thermocellum strains, the concentrations of amino acids of the glutamate family (glutamate and arginine) were much higher. In contrast, the concentrations of amino acids of the pyruvate family (alanine and valine) were much lower [10, 45]. The changes in amino acid concentration may be the result of pdc and adhA overexpression.
Whole-genome sequencing
To identify secondary mutations, two colonies from strains expressing ZpPdc, GoPdc and ApPdc were sent for whole genome sequencing (Additional file 7). Fermentation profiles of these strains are shown in Additional file 8. No mutations were found in the pdc or adhA genes. Most of mutant genes have no clear function. The functions of mutant genes Clo1313_0217 (UDP-N-acetylglucosamine pyrophosphorylase), Clo1313_1397 (copper amine oxidase-like domain-containing protein) and Clo1313_2174 (RNA polymerase sigma-I factor) do not seem to be directly related to ethanol production. However, two other mutations have annotations related to protease activity. One is Clo1313_0988, which is annotated as a proteinase inhibitor. The mutation in the RBS region of Clo1313_0988 is unique to strain LL1398 (the highest ethanol producer). Based on the calculated RBS activity [43, 44], the mutation should result in increased translation of the Clo1313_0988 gene. If this proteinase inhibitor is targeting proteinases that target Pdc or AdhA, its overexpression might improve the life-time of these proteins. To test this hypothesis, we cloned the Clo1313_0988 gene into one of the plasmid expressing the ApPdc and adhA genes and transformed it into C. thermocellum. However, we did not observe a significant change in ethanol production.
The other is protease-related mutation is Clo1313_1489, which is annotated as a peptidase. The mutation in Clo1313_1489 is a transposon insertion that presumably leads to a loss of function, which could improve PDC activity if Clo1313_1489 is responsible for PDC degradation, however this hypothesis awaits experimental confirmation.
Plasmid stability
In this work, pdc and adhA genes were expressed on a plasmid, however we observed that in some cases up to 45% of the cells had lost the plasmid at the end of the fermentation (CFU assay result, Additional file 9). One possible solution to overcome the problem of plasmid instability would be to integrate the pdc and adhA genes on the genome.
Conclusion
In this work, the pyruvate decarboxylase from A. pasteurianus was heterologously expressed in C. thermocellum. With the help of an alcohol dehydrogenase from T. saccharolyticum, both the ethanol yield and titer were significantly increased. The final engineered strain, LL1398, produced ethanol from cellulose at 70% of the maximum theoretical yield and titer of 21.3 g/L. This technique could readily be applied to other thermophilic organisms.
Methods
Strain construction and molecular techniques
All the chemicals ordered were from Sigma-Aldrich (St. Louis, MO) or Fischer Scientific (Pittsburgh, PA) and were of molecular grade unless otherwise stated. Primers were ordered from Integrated DNA Technologies (Coralville, IA). Strains, plasmids, and primers can be found in Additional files 10, 11, 12. Transformation of plasmid DNA into to C. thermocellum was performed as previously described [46, 47]. Plasmid sequences are available from Genbank (see Accession Numbers in Additional file 11). Genetic modification was confirmed by PCR and whole genome sequencing.
Media and growth
Strains were grown anaerobically at 55 °C for all experiments. For biochemical characterization and transformation, strains were grown in CTFUD medium with 5 g/L cellobiose in a COY (Ann Arbor, MI) anaerobic chamber (85% N2, 10% CO2, and 5% H2) [46]. For quantification of fermentation products, strains were grown in sealed serum bottles on 30 g/L cellobiose in 50 mL of defined MTC-5 medium with a N2 headspace. The medium recipe and bottle preparation has been described previously [10]. Bioreactor fermentations were carried out in 1.5-L (1-L working volume) Sartorius Biostat A-plus Sartorius Stedim (Sartorius Stedim, Bohemia, NY) bioreactors in modified MTC-5 medium without MOPS buffer, with the temperature maintained at 55 °C and stirred at 150 rpm. The pH was controlled at 6.5 with a Mettler-Toledo pH probe (Columbus, OH) by the addition of 8 N KOH. The vitamin supplementation solution contained pyridoxamine dihydrochloride 0.04 g/L, PABA 0.008 g/L, d-biotin 0.004 g/L, vitamin B-12 0.004 g/L. The vitamin supplementation solution was filter sterilized and added after autoclaving the bioreactor. The bioreactor was inoculated with 5% v/v transfer of a fresh seed culture grown on 5 g/L Avicel PH105 in MTC-5 (0.5% v/v). The headspace of the bioreactor was flushed with an anaerobic gas mixture (80% N2 and 20% CO2) prior to inoculation. Thiamphenicol (dissolved in dimethyl sulfoxide) was added to the medium to a final concentration of 15 µg/mL as a selective agent to maintain the plasmid.
For growth rate measurements, 200 µL cultures were grown in 96-well pre-sterilized polystyrene plates in an anaerobic chamber (85% N2, 10% CO2, and 5% H2). Absorbance measurements (OD600) were taken every 10 min for 36 h using a Biotek plate reader (BioTek Instruments Inc., Winooski, VT). Growth rates were determined based on the slope of log-transformed absorbance data. Growth rates were determined from the mean of five independent biological replicates.
Heterologous protein expression in E. coli
Target genes were amplified by PCR with Q5 DNA polymerase (New England Biolabs, Ipswich, MA USA). Genomic DNA from each target organism was used as the template. The primers used for each gene are listed in Additional file 12. The target genes were inserted into plasmid pD861-CH (DNA2.0 Inc., Menlo Park, CA, USA) and tagged with a C-terminal His6 cassette. The vector was transformed into E. coli BL21(DE3).
Cells were grown aerobically in TB medium at 37 °C with a stirring speed of 225 rpm. When the OD600 reached 0.6, 4 mM rhamnose was added to induce the expression of the target gene. The cells were then grown aerobically for 4 h before harvesting by centrifugation. The cell pellets were washed with buffer (50 mM Tris–HCl, 0.5 mM DTT pH 7.5) and stored at − 80 °C.
Preparation of cell extracts
Escherichia coli cells were prepared as described in “Confirmation of PDC activity in C. thermocellum ”. C. thermocellum cells were harvested by centrifugation when the absorbance at 600 nm reached a value of 0.6.
The cell pellet was resuspended in lysis buffer (1 × BugBuster reagent (EMD Millipore, Darmstadt, Germany) with 0.2 mM dithiothreitol). The cells were lysed with Ready-Lyse lysozyme (Epicentre, Madison, WI, USA), and DNase I (New England Biolabs, Ipswich, MA, USA) was added to reduce the viscosity. After incubation for 30 min at room temperature, the resulting solution was centrifuged at 10,000×g for 5 min. The supernatant was used as cell extract for enzyme assays or purification.
Protein purification
All purification steps were performed at room temperature as described previously [17]. His-tag affinity spin columns (His SpinTrap; GE Healthcare BioSciences, Pittsburgh, PA, USA) were used to purify the protein. The column was first equilibrated with binding buffer (50 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH 7.5). Cell extracts (in 50 mM sodium phosphate, 500 mM NaCl, 20 mM imidazole, pH 7.5) were applied to the column, and then the column was washed twice with wash buffer (50 mM sodium phosphate, 500 mM NaCl, 50 mM imidazole, 20% ethanol, pH 7.5). The His-tagged protein was eluted with elution buffer (50 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.5).
For large volume purification, Ni Sepharose high performance affinity media (GE Healthcare BioSciences, Pittsburgh, PA, USA) and Glass Econo-Column® Columns 2.5 × 10 cm (BioRad, Hercules, CA, USA) were used.
Pyruvate decarboxylase assay
The pyruvate decarboxylase activity was measured using a coupled assay with yeast alcohol dehydrogenase [26]. The reaction mix contained: 50 mM pH 7.0 Tris–HCl buffer (The pH was adjusted at 25 °C), 2 mM MgCl2, 0.2 mM thiamine pyrophosphate, 0.3 mM NADH, 10 mM pyruvate and 1 U/mL of yeast alcohol dehydrogenase from Sigma (A3263). Pyruvate was used to start the reaction and the consumption of NADH was followed spectrophotometrically at 340 nm (molar extinction coefficient ε of NADH = 6.22 mM−1 cm−1). The protein concentration was determined using the Bradford protein reagent with bovine serum albumin as the standard (BioRad, Hercules, CA).
For the thermostability assay, the Pdc protein was mixed with the reaction mix without NADH and Adh and then incubated at various temperatures for 30 min. The pH of reaction mix was set to 7.0 at 25 °C. Then the incubated mix was chilled on ice for 15 min before the assay. Both NADH and Adh protein could be used to start the reaction. In cases where Adh protein was used to start the reaction, the rate of the reaction was determined after the reaction had reached steady state (i.e. the brief, transient initial rate was ignored).
Analytical methods
Acetate, formate, ethanol, glucose, and cellobiose were determined by high pressure liquid chromatography (HPLC, Waters, Milford, MA) with refractive index detection using an Aminex HPX-87H column (Bio-Rad, Hercules, CA) with a 2.5 mM sulfuric acid solution as the mobile phase. Pellet nitrogen was determined using a Shimadzu TOC-VCPH total organic carbon analyzer with added total nitrogen unit (Shimadzu Scientific Instruments, Columbia, MD), calibrated using an acidified glycine standard [48]. 1 mL samples were centrifuged at 15,000×g for 10 min, the supernatant was discarded, and the pellet was rinsed twice using equal volumes of deionized purified water (MilliQ). Residual Avicel PH105 concentration was determined in units of glucose equivalents by quantitative saccharification as previously described [45]. Supernatant protein was determined with the Bradford assay (Thermo Scientific, Rockford, IL) with bovine serum albumin (BSA) (Thermo Scientific, Rockford, IL) as a standard. The concentrations of secreted amino acid were determined using an Aracus amino acid analyzer (membraPure, Berlin, Germany) as previously described [8]. Briefly, after separation using a T111 Li-cation exchange column, amino acids were derivatized with ninhydrin. Derivatized amino acids were detected at a wavelength of 570 nm, and concentrations were calculated by comparing peak areas to those of a standard amino acid mixture.
Carbon balance calculations
Carbon balances were calculated as described previously [49]. The molar concentration of Avicel PH105 was calculated based on glucose monomers with a formula weight of 163 g/mole and 2 C3 equivalents per glucose. For glucose, extracellular sugar (non-glucose), and isobutanol, each mole was assumed to be equivalent to 2 C3 units. Ethanol, acetate, malate and lactate were assumed to be equivalent to 1 C3 unit. For amino acids, the number of C3 units was based on calculations from Stephanopoulos et al. 1998 [50]. Alanine (1 C3 equivalent) and valine (2 C3 equivalents) account for the majority of the amino acid carbon. For biomass, 1 mol of pellet carbon was assumed to be equivalent to 1/3 mol of C3 units and based upon measured pellet nitrogen values [48]. Extracellular protein was converted to C3 equivalents by assuming that extracellular protein is 45% carbon by mass.
Quantitative PCR
Quantitative PCR was performed as previously described [13]. Cultures were harvested to mid-exponential phase (OD600 between 0.6 and 1.0) and then treated with RNA protect bacteria reagent (Qiagen Catalog Number 76506) per kit instructions. The primers used for qPCR are listed in Additional file 12: Table S3. Gene expression in all strains were normalized against C. thermocellum recA expression [52].
Abbreviations
- ADH:
-
alcohol dehydrogenase
- ALDH:
-
aldehyde dehydrogenase
- BSA:
-
bovine serum albumin
- CBP:
-
consolidated bioprocessing
- CFU:
-
colony forming unite
- MOPS:
-
3-morpholino-propane-1-sulfonic acid
- PDC:
-
pyruvate decarboxylase
- PFOR:
-
pyruvate ferredoxin oxidoreductase
- FNOR:
-
ferredoxin-NAD(P)+ oxidoreductase
- Fdo:
-
oxidized ferredoxin
- FdR:
-
reduced ferredoxin
- SNV:
-
single nucleotide variation
References
Pauly M, Keegstra K. Cell-wall carbohydrates and their modification as a resource for biofuels. Plant J. 2008;54:559–68.
Lynd LR, Weimer PJ, Van Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev. 2002;66:506–77.
Lynd L, van Zyl W, McBride J, Laser M. Consolidated bioprocessing of cellulosic biomass: an update. Curr Opin Biotechnol. 2005;16:577–83.
Olson DG, McBride JE, Shaw AJ, Lynd LR. Recent progress in consolidated bioprocessing. Curr Opin Biotechnol. 2012;23:396–405. doi:10.1016/j.copbio.2011.11.026.
Olson DG, Sparling R, Lynd LR. Ethanol production by engineered thermophiles. Curr Opin Biotechnol. 2015;33:130–41.
Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, et al. High ethanol titers from cellulose by using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microbiol. 2011;77:8288–94.
Biswas R, Zheng T, Olson DG, Lynd LR, Guss AM. Elimination of hydrogenase active site assembly blocks H2 production and increases ethanol yield in Clostridium thermocellum. Biotechnol Biofuels. 2015;8:20.
Rydzak T, Lynd LR, Guss AM. Elimination of formate production in Clostridium thermocellum. J Ind Microbiol Biotechnol. 2015;42:1263–72.
Papanek B, Biswas R, Rydzak T, Guss AM. Characterization of Clostridium thermocellum strains with disrupted fermentation end-product pathways. Metab Eng. 2015;32:49–54.
Tian L, Papanek B, Olson DG, Rydzak T, Holwerda EK, Zheng T, et al. Simultaneous achievement of high ethanol yield and titer in Clostridium thermocellum. Biotechnol Biofuels. 2016;9:116.
Lo J, Olson DG, Murphy SJ-L, Tian L, Hon S, Lanahan A, et al. Engineering electron metabolism to increase ethanol production in Clostridium thermocellum. Metab Eng. 2017;39:71–9.
Olson DG, Hörl M, Fuhrer T, Cui J, Zhou J, Maloney MI, et al. Glycolysis without pyruvate kinase in Clostridium thermocellum. Metab Eng. 2016;39:169–80. doi:10.1016/j.ymben.2016.11.011.
Hon S, Lanahan AA, Tian L, Giannone RJ, Hettich RL, Olson DG, et al. Development of a plasmid-based expression system in Clostridium thermocellum and its use to screen heterologous expression of bifunctional alcohol dehydrogenases (adhEs). Metab Eng Commun. 2016;3:120–9. doi:10.1016/j.meteno.2016.04.001.
Deng Y, Olson DG, Zhou J, Herring CD, Shaw AJ, Lynd LR. Redirecting carbon flux through exogenous pyruvate kinase to achieve high ethanol yields in Clostridium thermocellum. Metab Eng. 2013;15:151–8.
Dien BS, Cotta MA, Jeffries TW. Bacteria engineered for fuel ethanol production: current status. Appl Microbiol Biotechnol. 2003;63:258–66.
Wang S, Huang H, Moll J, Thauer RK. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 2010;192:5115–23.
Tian L, Lo J, Shao X, Zheng T, Olson DG, Lynd R. Ferredoxin: NAD+ oxidoreductase of Thermoanaerobacterium saccharolyticum and its role in ethanol formation. Appl Environ Microbiol. 2016;82:7134–41.
Pronk J, Steensma H, Van Dijken J. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast. 1996;12:1607–33.
Hoppner TC, Doelle HW. Purification and kinetic characteristics of pyruvate decarboxylase and ethanol dehydrogenase from Zymomonas mobilis in relation to ethanol production. Eur J Appl Microbiol Biotechnol. 1983;17:152–7.
Chandra Raj K, Ingram LO, Maupin-furlow JA. Pyruvate decarboxylase: a key enzyme for the oxidative metabolism of lactic acid by Acetobacter pasteurianus. Arch Microbiol. 2001;176:443–51.
Gunawan C, Satianegara G, Chen AK, Breuer M, Hauer B, Rogers PL, et al. Yeast pyruvate decarboxylases: variation in biocatalytic characteristics for (R)-phenylacetylcarbinol production. FEMS Yeast Res. 2007;7:33–9.
Raj KC, Talarico LA, Ingram LO, Maupin-furlow JA. Cloning and characterization of the Zymobacter palmae pyruvate decarboxylase gene (pdc) and comparison to bacterial homologues. Microbiology. 2002;68:2869–76.
Kata I, Semkiv MV, Ruchala J, Dmytruk KV, Sibirny AA. Overexpression of the genes PDC1 and ADH1 activates glycerol conversion to ethanol in the thermotolerant yeast Ogataea (Hansenula) polymorpha. Yeast. 2016;33:471–8.
Ishchuk OP, Voronovsky AY, Stasyk OV, Gayda GZ, Gonchar MV, Abbas CA, et al. Overexpression of pyruvate decarboxylase in the yeast Hansenula polymorpha results in increased ethanol yield in high-temperature fermentation of xylose. FEMS Yeast Res. 2008;8:1164–74.
van Zyl LJ, Schubert W, Tuffin MI, Cowan DA, Van Zyl LJ, Schubert W, et al. Structure and functional characterization of pyruvate decarboxylase from Gluconacetobacter diazotrophicus. BMC Struct Biol. 2014;14:21.
Ingram LO, Conway T, Clark DP, Sewell GW, Preston JF. Genetic engineering of ethanol production in Escherichia coli. Appl Environ Microbiol. 1987;53:2420–5.
Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram L. M5A1 for ethanol production from xylose and metabolic engineering of Klebsiella oxytoca M5A1 for ethanol production from xylose and glucose. Appl Environ Microbiol. 1991;57:2810–5.
Barbosa MDF, Ingram LO. Expression of the Zymomonas mobilis alcohol dehydrogenase II (adhB) and pyruvate decarboxylase (pdc) genes in Bacillus. Curr Microbiol. 1994;28:279–82.
Gold RS, Meagher MM, Tong S, Hutkins RW, Conway T. Cloning and expression of the Zymomonas mobilis “production of ethanol” genes in Lactobacillus casei. Curr Microbiol. 1996;33:256–60.
De Deng M, Coleman JR. Ethanol synthesis by genetic engineering in cyanobacteria. Appl Environ Microbiol. 1999;65:523–8.
Guedon Emmanuel, Desvaux M, Petitdemange H. Improvement of cellulolytic properties of Clostridium cellulolyticum by metabolic engineering. Society. 2002;68:53–8.
Van Zyl LJ, Taylor MP, Eley K, Tuffin M, Cowan DA, Van Zyl LJ, et al. Engineering pyruvate decarboxylase-mediated ethanol production in the thermophilic host Geobacillus thermoglucosidasius. Appl Microbiol Biotechnol. 2014;98:1247–59.
Stevenson BJ, Liu JW, Ollis DL. Directed evolution of yeast pyruvate decarboxylase 1 for attenuated regulation and increased stability. Biochemistry. 2008;47:3013–25.
Limtong S, Sringiew C, Yongmanitchai W. Production of fuel ethanol at high temperature from sugar cane juice by a newly isolated Kluyveromyces marxianus. Bioresour Technol. 2007;98:3367–74.
Gocke D, Graf T, Brosi H, Frindi-Wosch I, Walter L, Müller M, et al. Comparative characterisation of thiamin diphosphate-dependent decarboxylases. J Mol Catal B Enzym. 2009;61:30–5.
Pohl M, Mesch K, Rodenbrock A, Kula M. Stability investigations on on the pyruvate decarboxylases from Zymomonas mobilis. Biotechnol Appl Biochem. 1995;22:95–105.
Kannuchamy S, Mukund N, Saleena LM. Genetic engineering of Clostridium thermocellum DSM1313 for enhanced ethanol production. BMC Biotechnol. 2016;16:34. doi:10.1186/s12896-016-0260-2.
Lo J, Zheng T, Hon S, Olson DG, Lynd LR. The bifunctional alcohol and aldehyde dehydrogenase gene, adhE, is necessary for ethanol production in Clostridium thermocellum and Thermoanaerobacterium saccharolyticum. J Bacteriol. 2015;197:JB.02450-14. doi:10.1128/JB.02450-14.
Zheng T, Olson DG, Tian L, Bomble YJ, Himmel ME, Lo J, et al. Cofactor specificity of the bifunctional alcohol and aldehyde dehydrogenase (AdhE) in wild-type and mutants of Clostridium thermocellum and Thermoanaerobacterium saccharolyticum. J Bacteriol. 2015;197:JB.00232-15. doi:10.1128/JB.00232-15.
Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu Rev Biochem. 2001;70(149):80.
James CL, Viola RE. Production and characterization of bifunctional enzymes. Substrate channeling in the aspartate pathway. Biochemistry. 2002;41:3726–31.
Hon S, Olson DG, Holwerda EK, Lanahan AA, Murphy SJL, Maloney MI, et al. The ethanol pathway from Thermoanaerobacterium saccharolyticum improves ethanol production in Clostridium thermocellum. Metab. Eng. 2017;42:175–84.
Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–50.
Espah Borujeni A, Channarasappa AS, Salis HM. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014;42:2646–59.
Holwerda EK, Thorne PG, Olson DG, Amador-Noguez D, Engle NL, Tschaplinski TJ, et al. The exometabolome of Clostridium thermocellum reveals overflow metabolism at high cellulose loading. Biotechnol Biofuels. 2014;7:155.
Olson DG, Lynd LR. Transformation of Clostridium thermocellum by electroporation. Methods Enzymol. 2012;510:317–30.
Guss AM, Olson DG, Caiazza NC, Lynd LR. Dcm methylation is detrimental to plasmid transformation in Clostridium thermocellum. Biotechnol Biofuels. 2012;5:30.
Holwerda EK, Ellis LD, Lynd LR. Development and evaluation of methods to infer biosynthesis and substrate consumption in cultures of cellulolytic microorganisms. Biotechnol Bioeng. 2013;110:2380–8.
Ellis LD, Holwerda EK, Hogsett D, Rogers S, Shao X, Tschaplinski T, et al. Closing the carbon balance for fermentation by Clostridium thermocellum (ATCC 27405). Bioresour Technol. 2012;103:293–9.
Stephanopoulos G, Aristidou AA, Nielsen J. Metabolic engineering: principles and methodologies. Cambridge: Academic press; 1998.
Flamholz A, Noor E, Bar-Even A, Milo R. EQuilibrator—the biochemical thermodynamics calculator. Nucleic Acids Res. 2012;40:770–5.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC T method. Methods. 2001;25:402–8. doi:10.1006/meth.2001.1262.
Authors’ contributions
LT and DGO designed experiments, LT and SJK performed experiments; LT, AMG, DGO and LRL analyzed data and wrote the manuscript. JTB measured the secreted amino acids. XL performed quantitative saccharification of the fermentation samples. SH constructed the adhA plasmid and help in plasmids construction. All authors read and approved the final manuscript.
Acknowledgements
We thank Johannes P. van Dijken for useful discussions regarding selection of PDC candidates and enzyme assay conditions.
Competing interests
Lee R. Lynd is a founder of the Enchi Corporation, which has a financial interest in C. thermocellum. The authors declare that they have no competing interests.
Funding
The bioenergy science center is a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE office of science.
The genomic resequencing work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE office of science user facility, is supported by the office of science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Notice: This manuscript has been authored by Dartmouth College under Contract No. DE-AC05-00OR22725 with U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable world-wide license to publish or reproduce the published form of this manuscript or allow others to do so, for United States Government purposes (end of notice).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
12934_2017_783_MOESM3_ESM.xlsx
Additional file 3. Fermentation product and sugar residules of 10 Clostridium thermocellum colonies overexpressing AdhA and ApPDC.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tian, L., Perot, S.J., Hon, S. et al. Enhanced ethanol formation by Clostridium thermocellum via pyruvate decarboxylase. Microb Cell Fact 16, 171 (2017). https://doi.org/10.1186/s12934-017-0783-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0783-9