
Abstract
Over the last decade, there have been major advances in the understanding of the genetic basis of hyperuricaemia and gout as well as of the pharmacogenetics of urate-lowering therapy. Key findings include the reporting of 28 urate-associated loci, the discovery that ABCG2 plays a central role on extra-renal uric acid excretion, the identification of genes associated with development of gout in the context of hyperuricaemia, recognition that ABCG2 variants influence allopurinol response, and the impact of HLA-B*5801 testing in reducing the prevalence of allopurinol hypersensitivity in high-risk populations. These advances, together with the reducing cost of whole genome sequencing, mean that integrated personalised medicine approaches may soon be possible in clinical practice. Genetic data may inform assessment of disease prognosis in individuals with hyperuricaemia or established gout, personalised lifestyle advice, selection and dosing of urate-lowering therapy, and prevention of serious medication adverse effects. In this article, we summarise the discoveries from genome-wide association studies and discuss the potential for translation of these findings into clinical practice.
Similar content being viewed by others
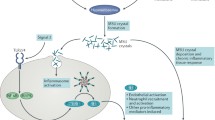


Background
New discoveries regarding the genetic basis of hyperuricaemia and gout, gene-environment interactions, and the pharmacogenetics of urate-lowering therapy (ULT) have increased the possibilities for personalised medicine approaches to be used in clinical practice. In current clinical practice, genetic testing is relatively expensive and performed only where there is a strong clinical need in diagnosis or pharmacogenomics. However, the cost of generating the entire genome sequence for an individual is now less than US$ 1000 and falling. Therefore, future healthcare approaches may include the availability of a curated electronic whole genome sequence so that a genetic result can be available instantly. Herein, we provide an overview of the current knowledge generated by genome-wide association studies (GWAS), and discuss the potential for translation of these findings into integrated personalised medicine approaches for hyperuricaemia and gout.
Genetics of hyperuricaemia and gout: recent discoveries
Serum urate levels and the risk of gout are influenced by a combination of inherited genetic variants and the environment. Heritability is defined as the percent variance in phenotype explained by inherited genetic variants, which can be estimated from studying phenotypic correlations between related individuals, typically twins. Such studies have estimated the heritability of urate to be between 45% and 73% [1,2,3]. In order to characterise the genetic basis of gout GWAS have been employed. A GWAS systematically assesses the genome for common (>1% prevalence) inherited genetic variants involved in disease etiology. These variants typically have a weak effect, with the majority exerting their effect by regulation of gene expression, transcript stability and transcript processing [4].
In gout, the greatest insights have been derived from studies with serum urate as the outcome. The largest GWAS in Europeans involved 110,000 individuals and discovered 28 urate-associated loci [5], 10 of which had previously been reported in smaller GWAS [6,7,8,9,10,11]. These loci are dominated by genes encoding renal and gut uric acid transporters (SLC2A9/GLUT9, ABCG2, SLC22A11/OAT4, SLC22A12/URAT1, SLC17A1/NPT1 and the auxiliary molecule PDZK1; Fig. 1). Statistically convincing sex effects are evident at the loci of strongest effect, with SLC2A9 having a stronger effect in women and ABCG2 in men [5]. The effects of the 28 single nucleotide polymorphisms (SNPs) were similar over multiple ancestral groups (European, African-American, Indian, Japanese) [5]. A GWAS of approximately 33,000 individuals in east Asians identified four loci (SLC2A9, ABCG2, SLC22A12 and MAF) [12], all of which overlap with loci identified in Europeans. Considerably smaller GWAS have been performed in African-American sample sets [13, 14], with SLC2A9 identified in both, with the larger study, that by Tin et al. [8], also detecting SLC22A12 and a novel locus containing the SLC2A12 and SGK1 genes that encode GLUT12 and SGK1, respectively. A GWAS in the Micronesian population of Kosrae detected a genome-wide significant association with serum urate levels only at SLC22A12 [15], and a very small GWAS in Mexican-Americans detected only SLC2A9 [16]. Aside from the Micronesian study, these GWAS emphasise the relative importance of SLC2A9 in the genetic control of urate across ancestral groups.

Influence of uricosuric agents lesinurad (L), benzbromarone (B) and probenecid (P) on the activity of renal uric acid transporters. Bold text, strong effect; normal text, weak to moderate effect; strikethrough, no effect; no text, no data were found. Based on information and data from [71–80]. OAT organic anion transporter; URAT1 urate transporter 1; NPT sodium phosphate transporter. Adapted with permission from Dalbeth et al. [81]
Aside from the uric acid transporter-containing loci the pathways influenced by the other loci remain largely speculative. The locus containing GCKR, encoding the glucokinase regulatory protein, may play a role in urate production via glycolysis (urate is produced as a result of ATP-depletion during hepatic glycolysis). Notably, none of the 18 loci that were newly identified by Köttgen et al. [5] encode known factors involved in uric acid transport. Several of these newly identified loci were within or near genes that encode transcription factors and growth factors, such as genes of the inhibin-activin growth factor network, INHBB and ACVR2A. One of the 18 newly identified loci includes PRPSAP1, which encodes a regulator of purine synthesis. Pathway analysis revealed that many of the 18 loci contained genes connected to glucose metabolism pathways. Emphasising the central role of hyperuricaemia in causing gout, the majority (24/28) of the urate-associated loci have been associated with gout in diverse populations [5, 17, 18]. Those with null associations are more likely a result of test sample sets of insufficient power than to a genuine lack of association, i.e. are false negatives.
SLC2A9, which encodes the GLUT9 protein, explains approximately 3% of variance in urate levels, a very large effect when compared to other complex disease loci. For example, the strongest effect in weight control in Europeans (the obesity associated protein FTO) explains only 0.3% of variance in phenotype [19]. The major SLC2A9 genetic effect associates with isoform expression, whereby the urate-raising causal genetic variant associates with increased expression of an SLC2A9 isoform (SLC2A9-S) that has a 28-residue portion missing from the N-terminus [7, 8]. This isoform is expressed on the apical (urine) side of the renal tubular collecting duct, where it presumably increases reuptake of secreted uric acid, whereas the full-length version (SLC2A9-L) is expressed on the basolateral side, where it is the major basolateral exit route of uric acid into the blood [20]. There has been little progress in identifying functional candidate causal variants to date [21].
In contrast to the great majority of the urate-associated loci, the missense rs2231142 (Q141K) variant in the ABCG2 gene is highly likely to be causal with the 141 K variant reducing the ability of ABCG2 to secrete uric acid by approximately 50% [22]. Expression of most uric acid transporters is relatively high in the kidney or, for SLC22A12/URAT1, restricted to the kidney. However, expression of ABCG2 is also relatively high in the gut [23]. Matsuo et al. [24] created grades of ABCG2 dysfunction based on Q141K and Q126X (a second etiological variant in ABCG2) genotype combinations, with individuals positive for the dysfunctional variants 126X and 141 K having the highest serum urate levels and highest risk for gout. The presence of the 141 K (and 126X) alleles reduces excretion of uric acid through the gut and adds to the circulating urate, overloading the kidney excretion system, and resulting in increased urinary uric acid levels [25].
Rarer variants contributing to the etiology of gout do exist. However, owing to their scarcity they are statistically difficult to detect, unless they have a very strong effect on disease risk. Rare knockout variants in the SLC2A9 and SLC22A12 genes that block the reuptake of urinary filtered uric acid cause hypouricaemia and exercise-induced kidney failure [26,27,28,29]. Examples of uncommon (1–2%), but not rare, genetic variants associated with gout are a coding variant in the ALDH16A1 gene in the Icelandic population (c.1580C > G; odds ratio, 3.7) [30], and the aforementioned knockout variant in the ABCG2 gene in the Japanese population (Q126X; odds ratio, 4.3) [24]. The ABCG2 126X variant disables the uric acid export function of ABCG2; however, the molecular pathogenic basis of the ALDH16A1 variant is unknown. In the coming years the possible contribution of rare penetrant functional variants in ABCG2 to gout will be evaluated [31]. Identification of further uncommon and rare population-specific variants will undoubtedly come from the study of whole genome sequences, although statistical confirmation will require very large sample sets comprising tens of thousands of people with gout.
GWAS in gout have only been performed in relatively small sample sets [5, 30, 32, 33], with the only novel loci reported in Chinese and Japanese GWAS. The Chinese GWAS used hyperuricaemic controls in follow-up testing to demonstrate that the newly discovered loci (BCAS3, RFX3 and KCNQ1) are likely to be involved in pathways leading to presentation with gout in people with hyperuricaemia [32]. The potential role of candidate genes located at these novel loci in gout is not yet clear, although the KCNQ1 association is notable. KCNQ1 is a potassium voltage-gated channel and an established type 2 diabetes susceptibility locus. The Japanese GWAS restricted controls to people with normouricaemia [33], making it more likely that the new loci reported (MYL2-CUX2 and CNIH2) contribute to hyperuricaemia. A follow-up study from the Japanese GWAS identified NIPAL1 (a magnesium transporter) and FAM35A (unknown function) [34]. Both are expressed in the distal tubules of the kidney, suggesting a role in uric acid handling [34].
Replicated candidate gene studies have identified some genes associated with gout. Most prominent are the TLR4 gene and the NLRP3 inflammasome component gene, both involved in the triggering of flares [35,36,37]. Interestingly, non-additive (epistatic) gene-gene interactions between the inflammasome CARD8 C10X variant and a variant in IL-1β associated with IL-1β expression is consistent with an etiology where greater inflammasome activity from reduced CARD8 expression, combined with higher levels of pre-IL-1β expression, leads to increased production of mature IL-1β and an amplified immune response [37]. To our knowledge, the only other replicated candidate gene association in gout, but not serum urate levels, is with the apolipoprotein A1-C3-A4 gene cluster [38, 39].
Translation of genetic discoveries into clinical practice: towards personalised medicine for gout management?
In current clinical practice, there are a few situations for which genetic testing can assist with diagnosis and decisions about management. Monogenic syndromes such as partial hypoxanthine-guanine phosphoribosyltransferase deficiency (Kelley-Seegmiller syndrome), phosphoribosyl pyrophosphate synthetase overactivity, or autosomal dominant tubulointerstitial kidney disease caused by UMOD pathogenic variants are rare, and routine testing for these mutations is not required for the vast majority of individuals with gout. People with glucose-6-phosphate dehydrogenase deficiency are at risk of severe haemolysis and methemoglobinaemia when treated with pegloticase, and screening for this condition (typically by enzyme activity, rather than genotyping) is recommended before commencing pegloticase. In addition, human leukocyte antigen (HLA) variant HLA-B*5801 is an important risk factor for severe allopurinol hypersensitivity syndrome (AHS) [40], and therefore testing for this variant has been recommended in high risk populations (Han Chinese, other Asian populations) prior to commencing allopurinol [41].
These examples are isolated and specific, and current clinical practice does not routinely integrate genetic testing into gout management. A key question is how the new discoveries from GWAS of hyperuricaemia and gout can impact on clinical management of gout. Aside from identification of potential new therapeutic targets, genomic approaches may allow personalised assessment of prognosis, targeted lifestyle interventions, prediction of response to ULT, and prediction of adverse events to commonly used gout medications.
Personalised assessment of prognosis
An important clinical question for individuals with hyperuricaemia is whether gout will develop, and for those presenting with gout, whether there is a risk of future flares and other severe consequences of disease such as tophi and/or joint damage [42]. A number of variables contribute to the risk of progressive disease, with serum urate levels being the most important variable identified to date [43]. However, serum urate levels alone do not reliably predict progression of disease. Risk stratification incorporating genetic testing may allow for more targeted decisions in an individual, for example, whether ULT should be initiated soon after (or even before) the first presentation of gout.
A consistent finding of GWAS has been the observation that, while SLC2A9 variants are most strongly associated with hyperuricaemia, ABCG2 variants are more strongly associated with gout [5]. In combination with evidence that ABCG2 associates with gout using people with asymptomatic hyperuricemia as controls even after adjusting for baseline serum urate levels [44], it can be suggested that, in addition to its effects on serum urate, ABCG2 variants may influence other checkpoints in the pathogenesis of gout, such as crystal formation and/or the inflammatory response to deposited crystals. The possibility that genetic testing has the potential to inform assessment of gout risk in people with hyperuricaemia is further supported by observations from the recent Chinese GWAS reporting three additional SNPs (BCAS3, RFX3, PKCNQ1) associated with gout when compared with asymptomatic hyperuricaemic controls [32].
Some studies have also implicated genetic variants in the development of severe manifestations of disease in those with established gout. A study from Aotearoa New Zealand reported that a non-synonymous SLC2A9 Arg265His variant is associated with tophi in Māori with gout [45]. Two studies, one from Taiwan [42] and one from Aotearoa New Zealand [46, 47], have implicated ABCG2 in the development of tophi in people with gout. In the Aotearoa New Zealand study [47], population-specific effects were observed, with several ABCG2 SNPs (rs2231142 (Q141K) and rs10011796) associated with tophi in Western Polynesian people with gout, independent of highest recorded urate and disease duration.
In order to clarify the importance of these findings to clinical practice, there is a need for large, well-characterised cohorts in different populations that follow individuals through the stages of disease, from hyperuricaemia to crystal deposition, to first presentation most often with flare, to advanced disease with tophus and chronic arthritis. Specifically, these studies will need to address the question of whether testing of genetic variants has additional benefit over standard clinical assessment, including urate levels, imaging assessment of urate deposition, and other known risk factors such as kidney function.
Targeted lifestyle interventions
Lifestyle changes are frequently advocated for prevention and management of gout [48]. Many different dietary changes are advocated, such as reducing intake of beer, sugar-sweetened drinks, and purine-rich foods such as meat, offal and seafood. Increased intake of cherries, omega-3 fatty acids, low fat milk and coffee are also advocated. At present, dietary recommendations are broad, generic and difficult to maintain in the long-term. Moreover, there is little evidence that such dietary changes actually influence serum urate levels in people with gout.
Some recent studies have reported gene-environment interactions in the regulation of serum urate levels or risk of gout. Examples include evidence for a non-additive interaction of sugar-sweetened drinks consumption with a urate-associated variant of SLC2A9 in determining the risk of gout [49], and alcohol intake with LRP2 in determining the risk of hyperuricaemia and gout [49–51]. A genetic risk score including ABCG2, SLC2A9, SLC22A12, SLC22A11 and SLC17A3 has also shown interaction with alcohol intake for gout risk [52].
An interaction between alcohol and the T allele of LRP2 rs2544390 was initially described for serum urate levels in a Japanese cohort with the highest risk of hyperuricaemia in males with TT who consumed five or more drinks per week [51]. In a subsequent study from Aotearoa New Zealand [52], the T allele of rs2544390 was also associated with increased risk of gout in a Polynesian cohort, but was associated with reduced risk of gout in a European cohort. There was a non-additive interaction between any alcohol intake and the risk of gout in the Polynesian cohort; any alcohol intake was associated with a 4.18-fold increased risk in the CC genotype group, compared with a 1.14-fold increased risk in the CT/TT genotype group. These effects were not observed in the European cohort [52].
The population-specific effects for the LRP2-alcohol interaction are instructive as they demonstrate that gene-environment findings in one population may not be translatable to other populations. A further, more fundamental issue is that lifestyle management (such as avoidance of sugar sweetened drinks or reduction of hazardous alcohol intake) may have health benefits beyond the risk of gout in individuals with genetic risk factors for gout. Any approach at personalised lifestyle advice based on genetic data will need to carefully address this issue to ensure that advice can be personalised to both gout and other comorbid conditions.
Prediction of response to ULT
Identification of genetic variations that predict non-response to allopurinol and uricosurics brings the possibility of genetic testing to personalise selection of ULT. ABCG2 is the only gene to date associated with non-response to allopurinol, the most widely used ULT agent. In 2015, a GWAS identified an association between the ABCG2 141 K allele and poor allopurinol response, defined by change in serum urate [53]. This association was replicated in a subsequent study with a stringent definition of poor response, namely serum urate levels ≥ 0.36 mmol/L despite allopurinol > 300 mg per day with adherence confirmed by plasma oxypurinol levels [54]. The 141 K allele frequency varies with ethnicity and ranges from 1% in African to 29% in South East Asian populations. Nevertheless, whether screening for 141 K and choosing alternative ULT in those with 141 K results in more rapid achievement of target urate remains to be determined. Further, the mechanism by which 141 K impairs allopurinol response remains unclear, although alterations in the transport of allopurinol and oxypurinol have been suggested [53].
Genetic variation in aldehyde oxidase (AOX1), encoding the enzyme responsible for the conversion of allopurinol to oxypurinol, and molybdenum cofactor sulfurase, also involved in the conversion, have been investigated. The minor allele of AOX1 rs55754655 (N1135S) leads to a 2- to 4-fold greater AOX1 efficacy than wild-type AOX1, thereby conferring a fast metaboliser phenotype [55]. However, no association has been observed between AOX1 rs55754655 (N1135S) and plasma oxypurinol levels or allopurinol dose required to achieve target serum urate [56]. Other genetic variants within AOX1 and XO have also been reported to be associated with allopurinol dose and change in serum urate [57]. However, larger studies with a more robust definition of allopurinol response and correction for multiple testing are required.
Febuxostat is a ULT drug that also acts via xanthine oxidase inhibition. Febuxostat is metabolised in the liver by conjugation via uridine diphosphate glucuronosyltransferase enzymes and oxidation via cytochrome P450 (CYP) enzymes, including CYP1A2, CYP2C8 and CYP2C9. To date, there are no genetic studies examining febuxostat response. Febuxostat has been identified as an inhibitor of ABCG2 [58]. Whether this has implications for urate lowering and the effects of ABCG2 SNPs on the urate lowering efficacy of febuxostat remains to be determined.
URAT1 (SLC22A12), which mediates the reabsorption of urate on the apical membrane of the proximal tubule in the kidney, is inhibited by the uricosurics probenecid, benzbromarone and lesinurad leading to normalisation of renal urate excretion [59, 60]. In addition, the renal urate transporters OAT1, OAT3, OAT4 (SLC22A11) and GLUT9 (SLC2A9) are variably inhibited by these agents (Fig. 1). There is some evidence that, in people with renal hypouricaemia, a loss of function mutation in URAT1 (W258X; SLC22A12 774G > A) is associated with impaired response to probenecid and benzbromarone [61]. Thus, genetic variants associated with hyperuricaemia and gout may also have pharmacogenetic relevance.
Prediction of adverse effects to commonly used gout medications
The identification of HLA-B*5801 as a major risk factor for AHS has been a major advance in safe prescribing of this medication. This test is now established in clinical practice in high-risk populations, and implementation of this screening has led to a reduction in the prevalence of this life-threatening complication [62].
In European people, HLA-B*5801 is a strong risk factor for AHS but is not sufficient or necessary to explain the disease, in contrast to Han Chinese and other Asian populations [63]. A GWAS of European AHS cases identified a six SNP haplotype (CACGAC) that was significantly associated with AHS (odds ratio, 7.77) [64]. The SNPs in this haplotype are located within the HLA locus on chromosome 6, but are only in partial linkage disequilibrium with HLA-B*5801. In a large Aotearoa New Zealand gout cohort, the CACGAC haplotype occurred at a higher frequency in European patients who experienced mild (non-AHS) allopurinol-related adverse events (13.3% vs. 1.7%; odds ratio, 8.9), but was not associated with allopurinol-related adverse events in Polynesians [65]. These data highlight the potential utility of genetic testing to also predict milder adverse effects in patients on ULT.
Another example of genetic testing with the potential to assess the risk of adverse effects to ULT is testing the cytochrome CYP2C9 poor metaboliser alleles CYP2C9*2 and CYP2C9*3. CYP2C9*3 homozygotes have a markedly longer benzbromarone elimination half-life than other CYP2C9 genotypes, which may increase the risk of benzbromarone-induced hepatotoxicity [66]. The frequency of CYP2C9 poor metaboliser alleles is substantially higher in Europeans compared to Polynesians [67]. Given the role of CYP2C9 in the metabolism of febuxostat, it is possible that those with the poor metaboliser allele are at risk of adverse effects with febuxostat; this is worthy of further investigation.
Genetic testing might also guide the choice of anti-inflammatory medications for flare management. CYP2C9 also metabolises many non-steroidal anti-inflammatory drugs (NSAIDs), including celecoxib, diclofenac, ibuprofen, naproxen and piroxicam. In a study of acute NSAID users, endoscopically documented NSAID-related gastroduodenal bleeding lesions were more common in those with poor metaboliser alleles [68].
Future directions and conclusions
Rapidly progressive technology and disease-specific genetic discoveries have the potential to make personalised medicine a reality in many aspects of gout management, including risk assessment of disease progression, personalised lifestyle advice, selection and dosing of ULT, and prevention of serious medication adverse effects. Although major progress has been made through GWAS, there is a further need for large, well-characterised datasets that include different disease states, detailed pharmacology (including dose information, treatment response, adverse drug reactions) and lifestyle information. A further challenge is population-specific effects, meaning that discoveries in one population may not be translatable to other populations. In order to avoid increasing the disparities that are already evident in gout management [69, 70], study of different populations will be essential, particularly of those with high prevalence of severe disease.
Abbreviations
- AHS:
-
allopurinol hypersensitivity syndrome
- AOX:
-
aldehyde oxidase
- CYP:
-
cytochrome P450
- GWAS:
-
genome-wide association studies
- HLA:
-
human leukocyte antigen
- NSAIDs:
-
non-steroidal anti-inflammatory drugs
- SNP:
-
single nucleotide polymorphism
- ULT:
-
urate-lowering therapy
References
Kalousdian S, Fabsitz R, Havlik R, Christian J, Rosenman R. Heritability of clinical chemistries in an older twin cohort: the NHLBI Twin Study. Genet Epidemiol. 1987;4(1):1–11.
Krishnan E, Lessov-Schlaggar CN, Krasnow RE, Swan GE. Nature versus nurture in gout: a twin study. Am J Med. 2012;125(5):499–504.
Whitfield JB, Martin NG. Inheritance and alcohol as factors influencing plasma uric acid levels. Acta Genet Med Gemellol (Roma). 1983;32(2):117–26.
Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5.
Kottgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, Pistis G, Ruggiero D, O'Seaghdha CM, Haller T, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45(2):145–54.
Li S, Sanna S, Maschio A, Busonero F, Usala G, Mulas A, Lai S, Dei M, Orru M, Albai G, et al. The GLUT9 gene is associated with serum uric acid levels in Sardinia and Chianti cohorts. PLoS Genet. 2007;3(11), e194.
Doring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, Fischer G, Henke K, Klopp N, Kronenberg F, et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008;40(4):430–6.
Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40(4):437–42.
Dehghan A, Kottgen A, Yang Q, Hwang SJ, Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC, et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008;372(9654):1953–61.
Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6), e1000504.
Yang Q, Kottgen A, Dehghan A, Smith AV, Glazer NL, Chen MH, Chasman DI, Aspelund T, Eiriksdottir G, Harris TB, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet. 2010;3(6):523–30.
Okada Y, Sim X, Go MJ, Wu JY, Gu D, Takeuchi F, Takahashi A, Maeda S, Tsunoda T, Chen P, et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat Genet. 2012;44(8):904–9.
Charles BA, Shriner D, Doumatey A, Chen G, Zhou J, Huang H, Herbert A, Gerry NP, Christman MF, Adeyemo A, et al. A genome-wide association study of serum uric acid in African Americans. BMC Med Genomics. 2011;4:17.
Tin A, Woodward OM, Kao WH, Liu CT, Lu X, Nalls MA, Shriner D, Semmo M, Akylbekova EL, Wyatt SB, et al. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum Mol Genet. 2011;20(20):4056–68.
Kenny EE, Kim M, Gusev A, Lowe JK, Salit J, Smith JG, Kovvali S, Kang HM, Newton-Cheh C, Daly MJ, et al. Increased power of mixed models facilitates association mapping of 10 loci for metabolic traits in an isolated population. Hum Mol Genet. 2011;20(4):827–39.
Voruganti VS, Kent Jr JW, Debnath S, Cole SA, Haack K, Goring HH, Carless MA, Curran JE, Johnson MP, Almasy L, et al. Genome-wide association analysis confirms and extends the association of SLC2A9 with serum uric acid levels to Mexican Americans. Front Genet. 2013;4:279.
Phipps-Green AJ, Merriman ME, Topless R, Altaf S, Montgomery GW, Franklin C, Jones GT, van Rij AM, White D, Stamp LK, et al. Twenty-eight loci that influence serum urate levels: analysis of association with gout. Ann Rheum Dis. 2016;75(1):124–30.
Urano W, Taniguchi A, Inoue E, Sekita C, Ichikawa N, Koseki Y, Kamatani N, Yamanaka H. Effect of genetic polymorphisms on development of gout. J Rheumatol. 2013;40(8):1374–8.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–48.
Kimura T, Takahashi M, Yan K, Sakurai H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS One. 2014;9(1), e84996.
Hurba O, Mancikova A, Krylov V, Pavlikova M, Pavelka K, Stiburkova B. Complex analysis of urate transporters SLC2A9, SLC22A12 and functional characterization of non-synonymous allelic variants of GLUT9 in the Czech population: no evidence of effect on hyperuricemia and gout. PLoS One. 2014;9(9), e107902.
Woodward OM, Kottgen A, Coresh J, Boerwinkle E, Guggino WB, Kottgen M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A. 2009;106(25):10338–42.
Huls M, Brown CD, Windass AS, Sayer R, van den Heuvel JJ, Heemskerk S, Russel FG, Masereeuw R. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008;73(2):220–5.
Matsuo H, Takada T, Ichida K, Nakamura T, Nakayama A, Ikebuchi Y, Ito K, Kusanagi Y, Chiba T, Tadokoro S, et al. Common defects of ABCG2, a high-capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med. 2009;1(5):5ra11.
Ichida K, Matsuo H, Takada T, Nakayama A, Murakami K, Shimizu T, Yamanashi Y, Kasuga H, Nakashima H, Nakamura T, et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012;3:764.
Androvitsanea A, Stylianou K, Maragkaki E, Tzanakakis M, Stratakis S, Petrakis I, Giatzakis C, Daphnis E. Vanishing urate, acute kidney injury episodes and a homozygous SLC2A9 mutation. Int Urol Nephrol. 2015;47(6):1035–6.
Stiburkova B, Ichida K, Sebesta I. Novel homozygous insertion in SLC2A9 gene caused renal hypouricemia. Mol Genet Metab. 2011;102(4):430–5.
Windpessl M, Ritelli M, Wallner M, Colombi M. A Novel Homozygous SLC2A9 Mutation Associated with Renal-Induced Hypouricemia. Am J Nephrol. 2016;43(4):245–50.
Gabrikova D, Bernasovska J, Sokolova J, Stiburkova B. High frequency of SLC22A12 variants causing renal hypouricemia 1 in the Czech and Slovak Roma population; simple and rapid detection method by allele-specific polymerase chain reaction. Urolithiasis. 2015;43(5):441–5.
Sulem P, Gudbjartsson DF, Walters GB, Helgadottir HT, Helgason A, Gudjonsson SA, Zanon C, Besenbacher S, Bjornsdottir G, Magnusson OT, et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat Genet. 2011;43(11):1127–30.
Stiburkova B, Miyata H, Zavada J, Tomcik M, Pavelka K, Storkanova G, Toyoda Y, Takada T, Suzuki H. Novel dysfunctional variant in ABCG2 as a cause of severe tophaceous gout: biochemical, molecular genetics and functional analysis. Rheumatology (Oxford). 2016;55(1):191–4.
Li C, Li Z, Liu S, Wang C, Han L, Cui L, Zhou J, Zou H, Liu Z, Chen J, et al. Genome-wide association analysis identifies three new risk loci for gout arthritis in Han Chinese. Nat Commun. 2015;6:7041.
Matsuo H, Yamamoto K, Nakaoka H, Nakayama A, Sakiyama M, Chiba T, Takahashi A, Nakamura T, Nakashima H, Takada Y, et al. Genome-wide association study of clinically defined gout identifies multiple risk loci and its association with clinical subtypes. Ann Rheum Dis. 2016;75(4):652–9.
Nakayama A, Nakaoka H, Yamamoto K, Sakiyama M, Shaukat A, Toyoda Y, Okada Y, Kamatani Y, Nakamura T, Takada T, et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann Rheum Dis. 2017;76(5):869–77.
Qing YF, Zhou JG, Zhang QB, Wang DS, Li M, Yang QB, Huang CP, Yin L, Pan SY, Xie WG, et al. Association of TLR4 Gene rs2149356 polymorphism with primary gouty arthritis in a case-control study. PLoS One. 2013;8(5), e64845.
Rasheed H, McKinney C, Stamp LK, Dalbeth N, Topless RK, Day R, Kannangara D, Williams K, Smith M, Janssen M, et al. The Toll-Like Receptor 4 (TLR4) Variant rs2149356 and Risk of Gout in European and Polynesian Sample Sets. PLoS One. 2016;11(1), e0147939.
McKinney C, Stamp LK, Dalbeth N, Topless RK, Day RO, Kannangara DR, Williams KM, Janssen M, Jansen TL, Joosten LA, et al. Multiplicative interaction of functional inflammasome genetic variants in determining the risk of gout. Arthritis Res Ther. 2015;17:288.
Cardona F, Tinahones FJ, Collantes E, Escudero A, Garcia-Fuentes E, Soriguer FJ. Contribution of polymorphisms in the apolipoprotein AI-CIII-AIV cluster to hyperlipidaemia in patients with gout. Ann Rheum Dis. 2005;64(1):85–8.
Rasheed H, Phipps-Green AJ, Topless R, Smith MD, Hill C, Lester S, Rischmueller M, Janssen M, Jansen TL, Joosten LA, et al. Replication of association of the apolipoprotein A1-C3-A4 gene cluster with the risk of gout. Rheumatology (Oxford). 2016;55(8):1421–30.
Hung SI, Chung WH, Liou LB, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS, et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A. 2005;102(11):4134–9.
Khanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, Pillinger MH, Merill J, Lee S, Prakash S, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431–46.
Dalbeth N, Stamp L. Hyperuricaemia and gout: time for a new staging system? Ann Rheum Dis. 2014;73(9):1598–600.
Shiozawa A, Szabo SM, Bolzani A, Cheung A, Choi HK. Serum Uric Acid and the Risk of Incident and Recurrent Gout: A Systematic Review. J Rheumatol. 2017;44(3):388–96.
Merriman TR, Phipps-Green A, Boocock J, Riches P, Tausche A-K, Radstake T, Janssen M, Joosten LAB, Jansen TL, So A et al. Pleiotropic effect of ABCG2 in gout. In: American College of Rheumatology Annual Scientific Meeting. Washington DC, USA: Arthritis Rheumatol; 2016;68(suppl 10).
Hollis-Moffatt JE, Gow PJ, Harrison AA, Highton J, Jones PB, Stamp LK, Dalbeth N, Merriman TR. The SLC2A9 nonsynonymous Arg265His variant and gout: evidence for a population-specific effect on severity. Arthritis Res Ther. 2011;13(3):R85.
Tu HP, Ko AM, Chiang SL, Lee SS, Lai HM, Chung CM, Huang CM, Lee CH, Kuo TM, Hsieh MJ, et al. Joint effects of alcohol consumption and ABCG2 Q141K on chronic tophaceous gout risk. J Rheumatol. 2014;41(4):749–58.
He W, Phipps-Green A, Stamp LK, Merriman TR, Dalbeth N. Population-specific association between ABCG2 variants and tophaceous disease in people with gout. Arthritis Res Ther. 2017:in press.
Khanna D, Khanna PP, Fitzgerald JD, Singh MK, Bae S, Neogi T, Pillinger MH, Merill J, Lee S, Prakash S, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1447–61.
Batt C, Phipps-Green AJ, Black MA, Cadzow M, Merriman ME, Topless R, Gow P, Harrison A, Highton J, Jones P, et al. Sugar-sweetened beverage consumption: a risk factor for prevalent gout with SLC2A9 genotype-specific effects on serum urate and risk of gout. Ann Rheum Dis. 2014;73(12):2101–6.
Hamajima N, Naito M, Okada R, Kawai S, Yin G, Morita E, Higashibata T, Tamura T, Nakagawa H, Matsuo H. Significant interaction between LRP2 rs2544390 in intron 1 and alcohol drinking for serum uric acid levels among a Japanese population. Gene. 2012;503(1):131–6.
Rasheed H, Phipps-Green A, Topless R, Hollis-Moffatt JE, Hindmarsh JH, Franklin C, Dalbeth N, Jones PB, White DH, Stamp LK. Association of the lipoprotein receptor-related protein 2 gene with gout and non-additive interaction with alcohol consumption. Arthritis Res Ther. 2013;15(6):R177.
Tu HP, Chung CM, Min-Shan Ko A, Lee SS, Lai HM, Lee CH, Huang CM, Liu CS, Ko YC. Additive composite ABCG2, SLC2A9 and SLC22A12 scores of high-risk alleles with alcohol use modulate gout risk. J Hum Genet. 2016;61(9):803–10.
Wen C, Yee S, Liang X, Hoffmann T, Kvall M, Banda Y, Jorgenson E, Schaefer C, Risch N, Giacomini K. Genome-wide association study identifies ABCG2 (BRCP) as an allopurinol transporter and a determinant of drug response. Clin Pharm Ther. 2015;97(5):518–25.
Roberts RL, Wallace MC, Phipps-Green AJ, Topless R, Drake JM, Tan P, Dalbeth N, Merriman TR, Stamp LK: ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. 2016, doi: 10.1038/tpj.2015.101.
Hartmann T, Terao M, Garattini E, Teutloff C, Alfaro J, Jones J, Leimkuhler S. The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug Metab Dispos. 2012;40(5):856–64.
Roberts R, Zhang M, Marinaki A, Stamp L. Does genetic variability in aldehyde oxidoreductase and molybdenum cofactor sulfurase predict non-response to allopurinol? Aliment Pharmacol Ther. 2010;32:310–1.
Carroll M, Smith D, Shaak T. Genomic sequencing of uric acid metabolizing and clearing genes in relationship to xanthine oxidase inhibitor dose. Rheumatol Int. 2016;37(3):445–53. doi:10.1007/s00296-016-3592-2.
Miyata H, Takeda T, Toyoda Y, Matsuo H, Ichida K, Suzuki H. Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. Front Pharmacol. 2016, doi: 10.3389/pharf.2016.00518.
Shin H, Takeda M, Enomoto A, Fujimura M, Miyazaki H, Anzai N, Endou H. Interactions of urate transporter URAT1 in human kidney with uricosuric drugs. Nephrology (Carlton). 2011;16(2):156–62.
Miner J, Tan P, Hyndman D, Liu S, Iverson C, Nanavati P, Hagerty D, Manhard K, Shen Z, Girardet J-L, et al. Lerinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibiton of urate transporters in the kidney. Arthritis Res Ther. 2016;18(1):214.. doi:10.1186/s13075-016-1107-x.
Ichida K, Hosoyamada M, Hisatome I, Enomoto A, Hikita M, Endou H, Hosoya T. Clinical and Molecular Analysis of Patients with Renal Hypouricemia in Japan-Influence of URAT1 Gene on Urinary Urate Excretion. J Am Soc Nephrol. 2004;15:164–73.
Ko TM, Tsai CY, Chen SY, Chen KS, Yu KH, Chu CS, Huang CM, Wang CR, Weng CT, Yu CL, et al. Use of HLA-B*58:01 genotyping to prevent allopurinol induced severe cutaneous adverse reactions in Taiwan: national prospective cohort study. BMJ. 2015;351:h4848.
Lonjou C, Borot N, Sekula P, Ledger N, Thomas L, Halevy S, Naldi L, Bouwes-Bavinck JN, Sidoroff A, de Toma C, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008;18(2):99–107.
Genin E, Schumacher M, Roujeau JC, Naldi L, Liss Y, Kazma R, Sekula P, Hovnanian A, Mockenhaupt M. Genome-wide association study of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis in Europe. Orphanet J Rare Dis. 2011;6:52.
Roberts RL, Wallace MC, Harrison A, Dalbeth N, Merriman TR, Stamp LK. A human leukocyte antigen locus haplotype confers risk for allopurinol-related adverse effects in Caucasian patients with gout. Pharmacogenet Genomics. 2015;25(8):412–5.
Uchida S, Shimada K, Misaka S, Imai H, Katoh Y, Inui N, Takeuchi K, Ishizaki T, Yamada S, Ohashi K, et al. Benzbromarone pharmacokinetics and pharmacodynamics in different cytochrome P450 2C9 genotypes. Drug Metab Pharmacokinet. 2010;25(6):605–10.
Roberts RL, Wallace MC, Wright DF, Cadzow M, Dalbeth N, Jones PB, Stamp LK, Harrison AA, Black MA, Merriman TR. Frequency of CYP2C9 polymorphisms in Polynesian people and potential relevance to management of gout with benzbromarone. Joint Bone Spine. 2014;81(2):160–3.
Pilotto A, Seripa D, Franceschi M, Scarcelli C, Colaizzo D, Grandone E, Niro V, Andriulli A, Leandro G, Di Mario F, et al. Genetic susceptibility to nonsteroidal anti-inflammatory drug-related gastroduodenal bleeding: role of cytochrome P450 2C9 polymorphisms. Gastroenterology. 2007;133(2):465–71.
Jackson G, Dalbeth N, Te Karu L, Winnard D, Gow P, Gerard C, Minko N. Variation in gout care in Aotearoa New Zealand: a national analysis of quality markers. N Z Med J. 2014;127(1404):37–47.
Singh JA, Bharat A, Khanna D, Aquino-Beaton C, Persselin JE, Duffy E, Elashoff D, Khanna PP. Racial differences in health-related quality of life and functional ability in patients with gout. Rheumatology (Oxford). 2017;56(1):103–12.
Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Annu Rev Physiol. 2015;77:323–45.
Miner J, Tan PK, Hyndman D, Liu S, Iverson C, Nanavati P, Hagerty DT, Manhard K, Shen Z, Girardet JL, et al. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Res Ther. 2016;18(1):214.
Hagos Y, Stein D, Ugele B, Burckhardt G, Bahn A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J Am Soc Nephrol. 2007;18(2):430–9.
Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P, et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5(10), e197.
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417(6887):447–52.
van Aubel RA, Smeets PH, Peters JG, Bindels RJ, Russel FG. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol. 2002;13(3):595–603.
Sweet DH, Chan LM, Walden R, Yang XP, Miller DS, Pritchard JB. Organic anion transporter 3 (Slc22a8) is a dicarboxylate exchanger indirectly coupled to the Na + gradient. Am J Physiol Renal Physiol. 2003;284(4):F763–769.
Enomoto A, Takeda M, Shimoda M, Narikawa S, Kobayashi Y, Kobayashi Y, Yamamoto T, Sekine T, Cha SH, Niwa T, et al. Interaction of human organic anion transporters 2 and 4 with organic anion transport inhibitors. J Pharmacol Exp Ther. 2002;301(3):797–802.
Aslamkhan A, Han YH, Walden R, Sweet DH, Pritchard JB. Stoichiometry of organic anion/dicarboxylate exchange in membrane vesicles from rat renal cortex and hOAT1-expressing cells. Am J Physiol Renal Physiol. 2003;285(4):F775–783.
Jutabha P, Kanai Y, Hosoyamada M, Chairoungdua A, Kim DK, Iribe Y, Babu E, Kim JY, Anzai N, Chatsudthipong V, et al. Identification of a novel voltage-driven organic anion transporter present at apical membrane of renal proximal tubule. J Biol Chem. 2003;278(30):27930–8.
Dalbeth N, Merriman T. Crystal ball gazing: new therapeutic targets for hyperuricaemia and gout. Rheumatology (Oxford). 2009;48(3):222–6.
Acknowledgements
Not applicable.
Funding
ND, LS and TM are supported by the Health Research Council of New Zealand (grant number 14-527).
Availability of data and materials
Not applicable.
Authors’ contributions
All authors drafted the manuscript, and approved the final version of the manuscript.
Competing interests
ND has received consulting fees, speaker fees or grants from Takeda, Teijin, Menarini, Pfizer, Ardea Biosciences, AstraZeneca, Fonterra, Crealta and Cymabay. LKS has received consulting fees from AstraZeneca. TRM has received consulting fees or grants from Ardea Biosciences and AstraZeneca.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dalbeth, N., Stamp, L.K. & Merriman, T.R. The genetics of gout: towards personalised medicine?. BMC Med 15, 108 (2017). https://doi.org/10.1186/s12916-017-0878-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-017-0878-5