
Abstract
Background
Uric acid is the primary byproduct of purine metabolism. Hyperuricemia is associated with body mass index (BMI), sex, and multiple complex diseases including gout, hypertension (HTN), renal disease, and type 2 diabetes (T2D). Multiple genome-wide association studies (GWAS) in individuals of European ancestry (EA) have reported associations between serum uric acid levels (SUAL) and specific genomic loci. The purposes of this study were: 1) to replicate major signals reported in EA populations; and 2) to use the weak LD pattern in African ancestry population to better localize (fine-map) reported loci and 3) to explore the identification of novel findings cognizant of the moderate sample size.
Methods
African American (AA) participants (n = 1,017) from the Howard University Family Study were included in this study. Genotyping was performed using the Affymetrix® Genome-wide Human SNP Array 6.0. Imputation was performed using MACH and the HapMap reference panels for CEU and YRI. A total of 2,400,542 single nucleotide polymorphisms (SNPs) were assessed for association with serum uric acid under the additive genetic model with adjustment for age, sex, BMI, glomerular filtration rate, HTN, T2D, and the top two principal components identified in the assessment of admixture and population stratification.
Results
Four variants in the gene SLC2A9 achieved genome-wide significance for association with SUAL (p-values ranging from 8.88 × 10-9 to 1.38 × 10-9). Fine-mapping of the SLC2A9 signals identified a 263 kb interval of linkage disequilibrium in the HapMap CEU sample. This interval was reduced to 37 kb in our AA and the HapMap YRI samples.
Conclusions
The most strongly associated locus for SUAL in EA populations was also the most strongly associated locus in this AA sample. This finding provides evidence for the role of SLC2A9 in uric acid metabolism across human populations. Additionally, our findings demonstrate the utility of following-up EA populations GWAS signals in African-ancestry populations with weaker linkage disequilibrium.
Similar content being viewed by others


Background
In humans, uric acid is the primary byproduct of purine metabolism and has long been associated with the development of gouty arthritis [1, 2]. Since the late 1800 s, it has been postulated that hyperuricemia plays a role in gout, kidney dysfunction, and vascular tone [3]. Over the past several decades, evidence linking uric acid to body mass index (BMI), insulin resistance, the metabolic syndrome, [4, 5], dietary intake of food substances high in purine [1], dietary fructose intake [2, 6, 7], renal disease and hypertension [8–12] has been expanding.
Clustering of uric acid, gout, renal disease, and hypertension has been known to have familial links since the late 1800 s [11, 12], suggesting a hereditary component to these traits. Furthermore, varying levels of uric acid in human populations, in addition to being attributable to dietary habits, are likely the result of evolutionary mutations that took place greater than 8 million years ago [13, 14]. These mutations have lead to the genetic variation we see in modern human populations [1, 15].
Mounting evidence generated from genome-wide association studies (GWAS) have linked uric acid to specific genomic loci [4, 16–18]. However, the GWAS reporting association of uric acid with specific genetic loci (PDZK1, GCKR, SLC16A9, SLC22A11, SLC22A12, LRRC16A, WDR1, RAF1P1, ZNF5188, and ABCG2), have been conducted in individuals of European [4, 17, 18], and Asian [19] descent. Given the paucity of GWAS in populations of African-ancestry, we chose to focus this manuscript on three main objectives in the following order of priority 1) to replicate major signals for uric acid reported in EA populations; 2) to use the weak LD patterns in African-ancestry populations to better localize (fine-map) reported loci and 3) to explore the identification of novel findings cognizant of the moderate sample size as well as the higher rates of obesity, renal disease, T2D, HTN, and decreased glomerular filtration rate in African Americans [20].
Methods
Ethics Statement
Declaration of assurance of ethical conduct of research was granted by the Howard University Institutional Review Board. All participants provided written informed consent for specimen collection and analysis. This study adhered to the tenets of the Declaration of Helsinki.
Study Sample
The study population has been described previously [21]. Briefly, participants included in this study were derived from the Howard University Family Study (HUFS), a population-based study of related and unrelated African Americans from the Washington, D.C. metropolitan area. The primary aims of HUFS included: 1) enrollment and examination of a randomly ascertained sample of 350 African American families with members in multiple generations from the Washington, D.C. metropolitan area; 2) characterization of participants for anthropomorphic measures (including height, weight, body composition measures and measures of obesity, blood pressure and related physiological intermediates, and diabetes-related and lipid-related variables); and 3) storing high-quality DNA to conduct studies to identify genes or genomic regions linked and/or associated with common, complex traits. Recruitment was conducted via door-to-door canvassing, community events, and advertisement in regional papers. A population-based approach was used to establish an unascertained sample with which to study multiple, common diseases. In a second phase of recruitment, additional unrelated individuals from the same geographic area were enrolled to facilitate nested case-control studies. Enrollment procedures (forms, measurements, and laboratory assays) for unrelated individuals were identical to those for families. The total number of recruited individuals was 2,028, of which 1,976 remained after data cleaning. From this sample, we created a subset of 1,055 unrelated adults (≥ 20 years of age).
Phenotyping
A baseline physical examination and an interview-based demographic questionnaire were conducted. Blood was drawn for biochemical assays for creatinine, glucose, uric acid, and several other molecular phenotypes. Weight was measured on an electronic scale to the nearest 0.1 kg with the participant wearing light clothes. Height was measured with a stadiometer to the nearest 0.1 cm with participants in bare feet. Body mass index was calculated as (weight in kg)/(height in m)2. Blood pressure was measured while participants were seated using an oscillometric device (Omron Healthcare, Inc., Bannockburn, Illinois). The readings were taken with a ten minute interval between readings. Reported systolic and diastolic blood pressure readings were the averages of the second and third readings. Participants with systolic blood pressure ≥ 140 mm Hg, diastolic blood pressure ≥ 90 mm Hg, or on prescribed antihypertensive drug therapy were defined as hypertensive. Serum creatinine levels were estimated on fasting sample using the modified Jaffé method. Estimated glomerular filtration (eGFR) was calculated using the simplified Modification of Diet in Renal Disease Study equation: eGFR = 186 × (serum creatinine)-1.154 × age-0.203(× 0.742 if female)(× 1.210 if Black) [22]. EGFR was measured in ml/min/1.73 m2 and creatinine was measured in mg/dl. Participants with fasting plasma glucose ≥ 7.0 mmol (126 mg/dl) were defined as having type 2 diabetes. Individuals with prediabetes (fasting plasma glucose between 5.6 mmol and 7.0 mmol) were given unknown case/control status. Serum uric acid levels were determined using the COBAS Integra Uric Acid assay, version 2 (Roche Diagnostics, Indianapolis, Indiana).
Genotyping
The Affymetrix® Genome-Wide Human SNP array 6.0 was used to conduct genome-wide genotyping [23]. Genetic material was processed and hybridized according to the manufacturer's instructions. Following processing, the chips were scanned and genotype calls were determined using the Birdseed 2 algorithm [23, 24]. The individual sample call rate had to be ≥ 95% for inclusion (no samples excluded). SNPs with call rates < 95% (n = 41,885) across all individuals, minor allele frequency ≤ 0.01 (n = 19,154), or a Hardy-Weinberg equilibrium (HWE) test p-value < 1 × 10-3 (n = 6,317) were excluded. This analysis included the 808,465 autosomal SNPs that passed these filters. The average call rate for SNPs in this group of individuals was 99.5% and the agreement of blind duplicates was 99.74%.
Imputation
Imputation of missing SNPs was performed using MACH, version 1.0.16 http://www.sph.umich.edu/csg/abecasis/MACH/[25] using a two stage approach. We downloaded the HapMap combined phase II+III raw genotype files for NCBI build 36, release 27 from http://hapmap.ncbi.nlm.nih.gov/downloads/genotypes/2009-02_phaseII+III/forward/non-redundant/[26]. For both the CEU and YRI samples, we retained only those individuals marked as founders. SNP inclusion criteria for imputation were that they had a MAF ≥ 0.01, a missingness rate ≤ 5%, and an individual missingness rate of ≤ 5%. These criteria resulted in 2,327,370 CEU and 2,598,198 YRI reference SNPs. Imputation was performed separately for these two reference panels. In the first stage, haplotype phases for the reference data were inferred using the settings -rounds 50 -states 200. In the second stage, imputation was conditioned upon the maximum-likelihood estimates of the crossover map and the error rate map. Imputation error was calibrated by ascertaining the threshold of posterior probability associated with a 10% error rate for the CEU reference panel and a 5% error rate for the YRI reference panel averaged over 6,800 SNPs for which we masked the experimentally determined genotypes. Imputed genotypes had to pass quality control filters requiring MAF ≥ 0.01, SNP missingness rate ≤ 10%, and HWE test p-value ≥ 0.001. For imputed genotypes that differed when using the CEU and YRI reference panels, we accepted the imputed genotype using the YRI reference panel. We successfully imputed 1,558,391 SNPs, yielding 2,366,856 experimentally determined and imputed autosomal SNPs. Quality control and data management were performed using PLINK, which is freely available and can be downloaded from http://pngu.mgh.harvard.edu/purcell/plink/[27]. More detailed descriptions of these procedures have been described previously [28, 29].
Assessment of Population Stratification
Assessment of population stratification was done via nonparametric clustering of genotypes using AWClust [30]. From the sample of 1,055 unrelated individuals, 37 individuals were identified as outliers and were excluded from analysis. Possible inflation of the type I error rate due to population stratification was investigated using genomic control [31]. EIGENSOFT was used to assess population structure [32]. A previously published scree plot [21] illustrates the two principal components (PCs) used as covariates in the analysis.
Association Analysis
Of the 1,018 included participants, one was missing the serum uric acid measurement, leaving a final analyzed sample of 1,017 individuals. Descriptive and multivariate analyses were conducted using R version 2.10.0 [33]. Serum uric acid values were not normally distributed. Therefore, serum uric acid values were transformed using a Box-Cox transformation (Box-Cox parameter lambda = 0.54). In the multivariate analysis, serum uric acid was analyzed for association with potential covariates age, sex, BMI, T2D, HTN, and eGFR. In genetic analysis, serum uric acid was analyzed as a continuous variable using linear regression, adjusting for age, sex, BMI, HTN, T2D, eGFR, and the first two PCs of the genotypes as covariates. PLINK, version 1.07 [27], was used to conduct the association analyses.
Results
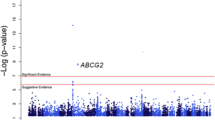
Clinical characteristics of the participants of our study are displayed in Table 1. In the univariate analysis, age (p = 4.68 × 10-9), sex (p = 4.83 × 10-35), BMI (p = 6.98 × 10-7), eGFR (p = 1.33 × 10-26), HTN (p = 6.93 × 10-16), and T2D (p = 0.021) were identified as significant covariates. In the multivariate linear regression analysis, sex, BMI, HTN, and eGFR (but not age and T2D) were significantly associated with uric acid levels (Table 2). The distribution of p-values for genetic association with serum uric acid is illustrated in Figure 1. The genomic control inflation factor was 0.9964, indicating no inflation of the type I error rate due to population stratification (Additional File 1). The top 25 ranked SNPs unadjusted for covariates are provided in Additional File 2 the 25 top ranked SNPs adjusted for age and sex are provided in Additional File 3 while the top 25 ranked SNPs adjusting for all covariates are provided in Additional File 4.

Distribution of p -values from the multiple linear regression of SNPs, associated with uric acid. The red line indicates the genome-wide significance level 1 × 10-8 and the blue line indicates the suggestive significance level 1 × 10-6.
The four genome-wide significant associations were for SNPs rs3775948, rs7663032, rs6856396, and rs6449213 (Additional File 4). These 4 SNPs were among the eleven most frequently reported SNPs in SLC2A9 associated with serum uric acid in European-ancestry populations (Additional File 5) [16, 34–40]. These four SNPs are all located in a linkage disequilibrium (LD) block on chromosome 4 (Figure 2) in the gene SLC2A9 (GeneID 56606). The next 8 highest ranking SNPs were also located in SLC2A9 (Additional File 4). The association at rs3775948 explained 2.6% of the phenotypic variance in our sample.

Local genetic architecture. Association p-values from multiple linear regression are shown based on physical position (NCBI build 36, dbSNP build 126). The light blue curve depicts the recombination rate from the combined Hap Map Phase II data. Linkage disequilibrium based on the HUFS sample is color-coded red for r 2 to the top SNP ≥ 0.8, orange for r 2 < 0.8 and ≥ 0.5, blue for r 2 < 0.5 and ≥ 0.2, and gray for r 2 < 0.20. Green arrows indicate the direction of transcription.
Serum uric acid levels were significantly different between males and females (Wilcoxon rank sum test, p = 4.55 × 10-35), with males having a mean serum uric acid level of 6.25 mg/dl compared to 5.02 mg/dl for females. The effect size estimates between females and males at rs6449213 trended toward being significantly different (Welch's t-test, p = 0.068). The top 25 ranked SNPs for serum uric acid in males and females, unadjusted for covariates, are provided in Additional Files 6 and 7, respectively. The top 25 ranked SNPs for serum uric acid, adjusted for age, BMI, hypertension, eGFR, T2D, and the top two PCs, in males and females are provided in Additional Files 8 and 9, respectively.
Fine Mapping
Using an r 2 cutoff of ≥ 0.3, LD with rs6449123 extends across an interval of approximately 263 kb in the CEU population and 231 kb in the YRI population. Restricting the r 2 cutoff to ≥0.5, LD with rs6449213 in the CEU population remains unchanged while, in sharp contrast, the LD interval in YRI is reduced to approximately 37 kb. All of the 10 top ranking SNPs fall within the 263 kb range; on the other hand, and quite remarkably 9 of the 10 top ranked SNPs lie within the 37 kb interval and 3 out of the 4 SNPs that achieved genome-wide significance lie in an approximately 1.3 kb interval (Figure 3).

Comparison of linkage disequilibrium in the HapMap Phase II CEU and YRI data and our HUFS sample.
Discussion
Our findings replicate those of other investigators who found association between variants in SLC2A9 and SUAL [4, 17, 18, 38]. To our knowledge, our study is the first to report association between SLC2A9 and uric acid in a large sample of admixed African Americans. The high expression of SLC2A9 in the epithelial cells of the proximal tubule and the atypical membranes of the kidneys [41], along with evidence that SLC2A9 is responsible for transport/re-absorption of uric acid and to a lesser extent glucose and fructose [42], provides biological plausibility for the high p-values for the SNPs in this gene in association with uric acid levels.
Hyperuricemia has been implicated in multiple physiologic outcomes including hypertension and renal dysfunction. Hyperuricemia is suspected to influence the development of hypertension via its role in vascular endothelial cell dysfunction and activation of the renin-angiotensin system [10]. Furthermore, experimental models demonstrating the causative effects of hyperuricemia in the development of hypertension were produced in rats with oxonic acid-induced hyperuricemia. These rats developed salt-resistant hypertension after induction of hyperuricemia, which resolved following reduction of uric acid to normal levels [43].
Hyperuricemia has also been demonstrated to increase the odds of developing acute renal dysfunction after cardiovascular surgery and increased the odds of developing chronic renal disease 4-fold and 3-fold, respectively, [44, 45]. This information, coupled with evidence that hyperuricemia causes epithelial dysfunction in renal vessels [46], also supports the association we found between higher uric acid levels and reduced eGFR. The SNP rs6449213 has not only been associated with uric acid levels but this association was demonstrated to be influenced by sex and BMI [36], which may help explain the associations we found between uric acid, sex, and BMI in this study.
The findings of our fine-mapping analysis demonstrate the advantage of using African-ancestry populations in follow-up analyses of GWAS signals originally discovered in European-ancestry populations. Replication analysis, using follow-up samples of increasing numbers of individuals with European ancestry (more specifically, populations with similar linkage disequilibrium patterns), allows for assessment of replication and refinement of effect size estimates. In contrast, using follow-up samples of individuals with ancestry differing from the discovery sample (specifically, populations with weaker linkage disequilibrium patterns) potentially allows for resolution of the location of the GWAS signals through the weaker linkage disequilibrium patterns in the follow-up population. Capitalizing on the weaker linkage disequilibrium in African Americans compared to EA populations, we were able to fine-map the SLC2A9 signal from 263 kb to 37 kb.
Serum uric acid's association with sex is confirmed in our study. The association between decreased serum uric acid and the effect allele of rs6449213 in SLC2A9 was sex-specific, replicating the findings of other investigators [36, 37]. Specifically, rs6449213 reached genome-wide significance in females but not in males. Given that rs6449213 has a smaller effect on serum uric acid in males than in females and that sex-stratified analysis reduces sample size, it is possible that the lack of association of rs6449213 with serum uric acid in males and the marginal p-value for the test of effect size estimates in females vs. males in our study both reflect false negatives finding.
A major limitation of our study is the moderate sample size compared to other GWAS studies. This suggests that loci with small effects may have been missed. The paucity of GWAS data on large numbers of African-Americans limits our ability to replicate our findings in a population with a similar substructure at this time. Despite these limitations, it is noteworthy that we replicated several of the reported association variants in the gene, SLC2A9. The effect size of this association (2.6% of the phenotypic variance averaged across sexes) is large compared to those from GWAS in general [40] but comparable to estimates from several studies of individuals of European ancestry [16, 35, 38, 39].
Conclusions
We found that SLC2A9 was significantly associated with serum uric acid in this population-based sample of African Americans, with a stronger effect in females than in males. Additionally, we observed significant association between uric acid levels and BMI, sex, eGFR, and hypertension. These observations deserve more in-depth evaluation in other human populations.
Abbreviations
- (ABCG2):
-
Adenosine triphosphate-Binding Cassette, Subfamily G, Member 2
- (AA):
-
African American
- (BMI):
-
Body Mass index
- (CEU):
-
CEPH Utah residents with ancestry from northern and western Europe
- (GWAS):
-
Genome-wide Association Study
- (GFR):
-
Glomerular Filtration Rate
- (GCKR):
-
Glucokinase Regulatory Protein
- (Hap Map):
-
International Hap Map Consortium
- (HWE):
-
Hardy-Weinberg equilibrium
- (HUFS):
-
Howard University Family Study
- (HTN):
-
Hypertension
- (LRRC16A):
-
Leucine-Rich Repeat-Containing Protein 16A
- (PDZK1):
-
PDZ Domain-Containing Protein 1
- (PC):
-
Principal component
- (SLC2A9):
-
Solute Carrier Family 2, Member 9
- (SLC16A9):
-
Solute Carrier Family 16, Member 9,
- (SLC22A11):
-
Solute Carrier Family 22, Member 11
- (SLC2A12):
-
Solute Carrier Family 22, Member 12
- (T2D):
-
Type 2 Diabetes
- (WDR1):
-
WD repeat domain 1
- (YRI):
-
Yoruba in Ibadan, Nigeria
References
Johnson RJ, Rideout BA: Uric acid and diet--insights into the epidemic of cardiovascular disease. N Engl J Med. 2004, 350 (11): 1071-1073. 10.1056/NEJMp048015.
Mene P, Punzo G: Uric acid: bystander or culprit in hypertension and progressive renal disease. Journal of Hypertension. 2008, 26: 2085-2092. 10.1097/HJH.0b013e32830e4945.
Feig DI, Kang D-H, Johnson RJ: Uric Acid and cardiovascular disease. The New England Journal of Medicine. 2009, 359 (17): 1811-1821. 10.1056/NEJMra0800885.
van der Harst P, Bakker SJL, de Boer R, Wolffenbuttel BHR, Johnson T, Caulfield MJ, Navis g: Replication of the five novel loci for uric acid concentrates and potential mediating mechanisms. Human Molecular Genetics. 2010, 19 (2): 387-395. 10.1093/hmg/ddp489.
Cirillo P, Sato W, Reungjui S, Heinig M, Gersch M, Sautin Y, Nakagawa T, Johnson RJ: Uric acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 2006, 17 (12 Suppl 3): S165-168. 10.1681/ASN.2006080909.
Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, Ouyang X, Feig DI, Block ER, Herrera-Acosta J, et al: A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006, 290 (3): F625-631. 10.1152/ajprenal.00140.2005.
Heinig M, Johnson RJ: Role of uric acid in hypertension, renal disease, and metabolic syndrome. Cleve Clin J Med. 2006, 73 (12): 1059-1064. 10.3949/ccjm.73.12.1059.
Avram Z, Krishnan E: Hyperuricemia--where nephrology meets rheumatology. Rheumatology. 2008, 47: 960-964. 10.1093/rheumatology/ken070.
Johnson RJ, Segal MS, Srinivas T, Ejaz A, Mu W, Roncal C, Sanchez-Lozada LG, Gersch M, Rodriguez-Iturbe B, Kang DH, et al: Essential hypertension, progressive renal disease, and uric acid: a pathogenetic link?. J Am Soc Nephrol. 2005, 16 (7): 1909-1919. 10.1681/ASN.2005010063.
Johnson RJ, Feig DI, Herrera-Acosta J, Kang DH: Resurrection of uric acid as a causal risk factor in essential hypertension. Hypertension. 2005, 45 (1): 18-20.
Mahomed FA: On chronic Bright's disease, and its essential symptoms. Lancet. 1879, I: 399-401. 10.1016/S0140-6736(02)45936-3.
Haig A: On uric acid and arterial tension. BMJ. 1889, I: 288-291. 10.1136/bmj.1.1467.288.
Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ: Uric acid, evolution and primitive cultures. Semin Nephrol. 2005, 25 (1): 3-8. 10.1016/j.semnephrol.2004.09.002.
Oda M, Satta Y, Takenaka O: Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Bol Evol. 2002, 19: 640-653.
Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, Mazzali M, Johnson RJ: Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension. 2002, 40 (3): 355-360. 10.1161/01.HYP.0000028589.66335.AA.
Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, et al: Meta-Analysis of 28,141 Individuals Identifies Common Variants within Five New Loci That Influence Uric Acid Concentrations. PLoS Genet. 2009, 5 (6): e1000504-10.1371/journal.pgen.1000504.
Wallace C, Newhouse SJ, Braund P, Zhang F, Tobin M, Falchi M, Ahamadi K, Dobson RJ, Marcano ACB, Hajat C, et al: Genome-wide association study identifies genes for biomarkers of cardiovascular disease: Serum urate and dyslipidemia. The American Journal of Human genetics. 2008, 82: 139-149. 10.1016/j.ajhg.2007.11.001.
Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P, et al: SLC2A9 Is a High-Capacity Urate Transporter in Humans. PLoS Med. 2008, 5 (10): e197-10.1371/journal.pmed.0050197.
Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N: Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 42 (3): 210-215. 10.1038/ng.531.
Hsu C-y, Iribarren C, McCulloch CE, Darbinian J, Go AS: Risk factors for end stage renal disease: 25-Year follow-up. Archives of Internal Medicine. 2009, 169 (4): 342-350. 10.1001/archinternmed.2008.605.
Adeyemo A, Gerry N, Chen G, Herbert A, Doumatey A, Huang H, Zhou J, Lashley K, Chen Y, Christman M, et al: A genome-wide association study of hypertension and blood pressure in African Americans. PLoS Genet. 2009, 5 (7): e1000564-10.1371/journal.pgen.1000564.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D, Group ftMoDiRdS: A more accurate method to estimate the glomerular filtration rate from serum creatinine: A new prediction equation. Annals of Internal Medicine. 1999, 130 (6): 461-470.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PIW, Maller JB, Kirby A, et al: Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008, 40 (10): 1166-1174. 10.1038/ng.238.
Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al: Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008, 40 (10): 1253-1260. 10.1038/ng.237.
Li Y, Abecasis G: MACH. Edited by: Michigan Uo. Ann Arbor: The Regents of the University of Michigan; MACH 1.0
HapMap Consortium: Index of/downloads/genotypes/2009-02_phaseII+III/forward/non-redundant. 2009, NCBI, NLM, 2009
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al: PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. The American Journal of Human genetics. 2007, 81 (3): 559-575. 10.1086/519795.
Shriner D, Adeyemo A, Chen G, Rotimi CN: Practical considerations for imputation of untyped markers in admixed populations. Genetic epidemiology. 2010, 34 (3): 258-265.
Shriner D, Adeyemo A, Gerry NP, Herbert A, Chen G, Doumatey A, Huang H, Zhou J, Christman MF, Rotimi CN: Transferability and fine-mapping of genome-wide associated loci for adult height across human populations. PLoS One. 2009, 4 (12): e8398-10.1371/journal.pone.0008398.
Gao X, Starmer J: AWclust: point-and-click software for non-parametric population structure analysis. BMC Bioinformatics. 2008, 9 (1): 77-10.1186/1471-2105-9-77.
Devlin B, Roeder K: Genomic control for association studies. Biometrics. 1999, 55: 997-1004. 10.1111/j.0006-341X.1999.00997.x.
Price A, Patterson N, Plenge R, Weinblatt M, Shadick N, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006, 38 (8): 904-909. 10.1038/ng1847.
R Development Core Team: R: A language and environment for statistical computing. Edited by: Computing RFfS. 2009, Vienna: R Foundation for Statistical Computing
Dehghan A, Kottgen A, Yang Q, Hwang S-J, Kao WL, Rivadeneria F, Boerwinkle E, Levy D, Hofman A, Astor BC, et al: Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet. 2008, 372 (9654): 1953-1961. 10.1016/S0140-6736(08)61343-4.
Zemunik T, Boban M, Lauc G, Janković S, Rotim K, Vatavuk Z, Benćić G, Đogaš Z, Boraska V, Torlak V, et al: Genome-wide association study of biochemical traits in Korcula Island, Croatia. Croatian Medical Journal. 2009, 50: 23-33. 10.3325/cmj.2009.50.23.
Brandstätter A, Kiechl S, Kollerits B, Hunt S, Heid IM, Coassin S, Willeit J, Adams TD, Illig T, Hopkins pN, et al: Sex Specific association of the putative fructose transporter SLCA29 variants with uric acid levels is modified by BMI. Diabetes Care. 2008, 31 (8): 1662-1667.
Doring A, Gieger C, Mehta D, Gohlke H, Prokisch H, Coassin S, Fischer G, Henke K, Klopp N, Kronenberg F, et al: SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat Genet. 2008, 40: 430-436. 10.1038/ng.107.
McArdle PF, Parsa A, Chang Y-P, Weir M, O'Connell JR, Mitchell BD, Shuldiner AR: A common non-synonymous variant in GLUT9 is a determinant of serum uric acid levels in old order Amish. Arthritis Rheum. 2008, 58 (9): 2874-2881. 10.1002/art.23752.
Wallace C, Newhouse S, Braund P, Zhang F, Tobin M, Falchi M, Ahmadi K, Dobson R, Marcano A, Hajat C, et al: Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia. Am J Hum Genet. 2008, 82: 139-149. 10.1016/j.ajhg.2007.11.001.
National Human Genome Research Institute: A Catalog of Genome-Wide Association Studies. 2010, National Institutes of Health, June 09, 2010 Last Update edn
Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH: Identification and Characterization of Human Glucose Transporter-like Protein-9 (GLUT9). Journal of Biological Chemistry. 2004, 279 (16): 16229-16236. 10.1074/jbc.M312226200.
Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CNA, Knott SA, Kolcic I, Polasek O, Graessler J, et al: SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008, 40 (4): 437-442. 10.1038/ng.106.
Mazzali M, Hughes J, Kim YG, Jefferson JA, Kang DH, Gordon KL, Lan HY, Kivlighn S, Johnson RJ: Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001, 38 (5): 1101-1106. 10.1161/hy1101.092839.
Ejaz AA, Beaver TM, Shimada M, Sood P, Lingegowda V, Schold JD, Kim T, Johnson RJ: Uric acid: a novel risk factor for acute kidney injury in high-risk cardiac surgery patients?. Am J Nephrol. 2009, 30 (5): 425-429. 10.1159/000238824.
Obermayr RP, Temml C, Gutjahr G, Knechtelsdorfer M, Oberbauer R, Klauser-Braun R: Elevated Uric Acid Increases the Risk for Kidney Disease. J Am Soc Nephrol. 2008, 19 (12): 2407-2413. 10.1681/ASN.2008010080.
Sanchez-Lozada LG, Tapia E, Santamaria J, Avila-Casado C, Soto V, Nepomuceno T, Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J: Mild hyperuricemia induces vasoconstriction and maintains glomerular hypertension in normal and remnant kidney rats. Kidney Int. 2005, 67 (1): 237-247. 10.1111/j.1523-1755.2005.00074.x.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/4/17/prepub
Acknowledgements
The study was supported by grants S06GM008016-320107 to CR and S06GM008016-380111 to AA, both from the NIGMS/MBRS/SCORE Program. Participant enrollment was carried out at the Howard University General Clinical Research Center (GCRC), which is supported by grant number 2M01RR010284 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Additional support was provided by the Coriell Institute for Biomedical Sciences. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, in the Center for Research in Genomics and Global Health (Z01HG200362). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Conception: AA, AH, MC, CR. Study design: BC, DS, AD, GC, JZ, HH, AH, NP, MC, AA, CR Sample processing and data management: HH, AD, NG, ZH. Genotyping: AD, AH, NG, HH, MC. Imputation: DS, AA, GC, AH. Analyzed the Data: BC, DS, AH, JZ, GC, NG. Interpretation: BC, DS, AD, AA, GC, CR, Drafted the manuscript: BC, DS. Revised manuscript: AA, CR. All authors read and approved the final manuscript.
Electronic supplementary material
12920_2010_212_MOESM1_ESM.PNG
{kind=link}
Additional file 1: Supplementary Figure S1. Quantile-quantile plot for genomic control. The red line indicates the expected distribution. The inflation factor (λGC) is shown. (PNG 70 KB)
12920_2010_212_MOESM4_ESM.DOC
Additional file 4: Table 3. Top 25 SNPs for serum uric acid, adjusted for age, sex, BMI, HTN, eGFR, T2D, and the top two PCs. (DOC 62 KB)
12920_2010_212_MOESM5_ESM.DOC
Additional file 5: Table 4. Previously reported GWAS associations between specific SNPs and serum uric acid levels. (DOC 97 KB)
12920_2010_212_MOESM6_ESM.DOC
Additional file 6: Supplementary Table S3. Top 25 SNPs for serum uric acid in males, unadjusted for covariates. (DOC 70 KB)
12920_2010_212_MOESM7_ESM.DOC
Additional file 7: Supplementary Table S4. Top 25 SNPs for serum uric acid in females, unadjusted for covariates. (DOC 70 KB)
12920_2010_212_MOESM8_ESM.DOC
Additional file 8: Supplementary Table S5. Top 25 SNPs for serum uric acid in males, adjusted for age, BMI, HTN, eGFR, T2D, and the top two PCs. (DOC 72 KB)
12920_2010_212_MOESM9_ESM.DOC
Additional file 9: Supplementary Table S6. Top 25 SNPs for serum uric acid in females, adjusted for age, BMI, HTN, eGFR, T2D, and the top two PCs. (DOC 68 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Charles, B.A., Shriner, D., Doumatey, A. et al. A genome-wide association study of serum uric acid in African Americans. BMC Med Genomics 4, 17 (2011). https://doi.org/10.1186/1755-8794-4-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-4-17