
Abstract
Background
Practical approaches for multigene transformation and gene stacking are extremely important for engineering complex traits and adding new traits in transgenic crops. Trait deployment by gene stacking would greatly simplify downstream plant breeding and trait introgression into cultivars. Gene stacking into pre-determined genomic sites depends on mechanisms of targeted DNA integration and recycling of selectable marker genes. Targeted integrations into chromosomal breaks, created by nucleases, require large transformation efforts. Recombinases such as Cre-lox, on the other hand, efficiently drive site-specific integrations in plants. However, the reversibility of Cre-lox recombination, due to the incorporation of two cis-positioned lox sites, presents a major bottleneck in its application in gene stacking. Here, we describe a strategy of resolving this bottleneck through excision of one of the cis-positioned lox, embedded in the marker gene, by nuclease activity.
Methods
All transgenic lines were developed by particle bombardment of rice callus with plasmid constructs. Standard molecular approach was used for building the constructs. Transgene loci were analyzed by PCR, Southern hybridization, and DNA sequencing.
Results
We developed a highly efficient gene stacking method by utilizing powerful recombinases such as Cre-lox and FLP-FRT, for site-specific gene integrations, and nucleases for marker gene excisions. We generated Cre-mediated site-specific integration locus in rice and showed excision of marker gene by I-SceI at ~20 % efficiency, seamlessly connecting genes in the locus. Next, we showed ZFN could be used for marker excision, and the locus can be targeted again by recombinases. Hence, we extended the power of recombinases to gene stacking application in plants. Finally, we show that heat-inducible I-SceI is also suitable for marker excision, and therefore could serve as an important tool in streamlining this gene stacking platform.
Conclusions
A practical approach for gene stacking in plant cell was developed that allows targeted gene insertions through rounds of transformation, a method needed for introducing new traits into transgenic lines for their rapid deployment in the field. By using Cre-lox, a powerful site-specific recombination system, this method greatly improves gene stacking efficiency, and through the application of nucleases develops marker-free, seamless stack of genes at pre-determined chromosomal sites.
Similar content being viewed by others

Background
Practical approaches for gene stacking are a critical need of crop biotechnology as the future requires multigene engineering for expressing complex traits, and periodic introduction of new traits into the previously engineered crops. The conventional methods of plant transformation integrate genes into undetermined chromosomal sites. As a result, breeding of transgenes into a single line becomes exponentially complex with the increase of transgenic donor lines or the number of independently segregating transgenes. To avoid segregation of transgenes, methods for targeted gene integration into the pre-determined genomic sites or gene stacking are needed. Gene stacking can resolve the complexity of multi-trait breeding, and ensure timely deployment of new traits into diverse crop varieties [1]. Targeted gene integration can be practiced by two distinct approaches, homologous (and non-homologous) recombination into the chromosomal double-stranded breaks (DSB) induced by synthetic or rare nucleases [2–4], and site-specific recombination (SSR) into the pre-integrated recombination sites [5–7]. Since the repair of DSBs could occur through one of the multiple pathways [8], precise targeted gene integration is practically a rare outcome, requiring development and screening of a high number of transgenic clones [3, 4]. SSR, on the other hand, is a simple, predictable reaction leading to a high number of transgenic clones that contain precise site-specific integration [9–13].
A number of SSR systems are known, Cre-lox among them stands out for its high efficiency in complex genome engineering in eukaryotic cells including plants [14–17]. So far, four SSR systems have been successfully used for directing site-specific integration (SSI) in plants, that include Cre-lox, FLP-FRT, R-RS, and Bxb1 [9, 12, 18, 19]. The efficiency of Cre-lox and FLP-FRT combined with the strategy to select SSI clones greatly improves the transformation pipeline through high SSI recovery rates and enhanced production of stable lines [13, 20–22]. However, Cre-lox and FLP-FRT recombination are reversible as they generate two recombination sites (loxP or FRT) in SSI (cis-positioned) that can recombine to reverse the structure. While, mechanisms of controlling the reversibility of recombination are highly effective [9, 10, 13, 23], they are not suitable for the iterative applications. Previously, we developed a selection marker gene (SMG)-free site-specific integration approach based on the use of FLP-FRT for gene integration followed by the use of Cre-lox for SMG-deletion [24]. This approach generated marker-free lines in a single generation that transmitted the marker-free SSI to the progeny at >95 % efficiency. While, this approach is suitable for integrating DNA constructs containing multiple genes, it cannot easily be used for the repeated rounds of transformation that would be needed for adding new traits to the ‘stacked site’.
Here, we modified the ‘marker-free site-specific integration’ approach by introducing nucleases for SMG deletion. Concomitant cuts on a chromosome could delete the intervening fragment, and the broken ends of the chromosome could join through DNA repair process. The repaired site incorporates sequence changes that are no longer targeted by the same nuclease [8]. Therefore, a given nuclease can be used again for SMG deletion. If one of the two cis-positioned lox or FRT is present within this fragment, it will also be deleted, leaving a single site for gene stacking. This modified method for marker-free site-specific integration is suitable for iterative gene stacking. Each of the steps in this method, individually, have been demonstrated by earlier studies. Cre or FLP-mediated site-specific gene integration, and nuclease mediated DNA deletion have all been demonstrated in plants [9–13, 25–28]. However, these diverse molecular mechanisms have not been integrated in a single platform. Where Cre-lox is well-known for generating highly precise recombination products [16], nuclease-induced DSB repair could create insertion-deletions (indels) at the site [8]. Therefore, for this gene stacking approach to be successful, it is important to establish that nuclease-induced indels would not alter the adjacent sequences, too often, or interfere with Cre-lox components in the iterative site-specific integrations. The focus of the present work is to develop the proof-of-concept for the proposed marker-free gene stacking strategy by characterizing the deletion sites induced by I-SceI and CCR5-ZFN at the Cre-mediated SSI locus in rice, and demonstrate the second round of site-specific integration in the same site. We first generated SSI lines using a founder line, T5, and subjected them to SMG deletion by I-SceI, and carried out a second round of site-specific integration by FLP-FRT and characterized CCR5-ZFN mediated DSB/repair sites. We also tested the utility of heat-inducible I-SceI and ZFN in DNA deletions. Overall, this work shows that Cre-lox and FLP-FRT can be used for gene stacking in plants, and their power extended to iterative genome engineering applications.
Results
Molecular strategy
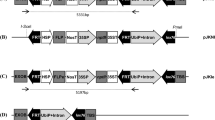
The strategy of iterative gene stacking is based on the use of an efficient site-specific recombination (SSR) system and a pair of nucleases to direct transgene integration and selection marker gene (SMG) deletion, respectively (Fig. 1). We propose using Cre-lox or FLP-FRT for transgene integration for their robust activity and high specificity in plants [14, 15]. Cre or FLP-mediated gene integration, however, results in the incorporation of two cis-positioned lox or FRT sites (Fig. 1a-c), which can excise out the integrated DNA in the subsequent rounds of transformation. Therefore, SMG deletion is carried out by nuclease, which removes one of the two cis-positioned recombination sites along with the SMG fragment (Fig. 1d). The single recombination site in the SMG-free SSI site serves as the target for the next round of site-specific integration. It should be noted that SMG removal is an integral step in gene stacking, therefore, no extra effort is added in this strategy. We used efficient nucleases, I-SceI and CCR5-ZFN, in this study; however, other nuclease technologies, if found to be efficient, can also be used. As shown in Fig. 1, the strategy utilizes promoter/gene trap to isolate site-specific integration events (Fig. 1a-c), which are subjected to nuclease-mediated marker deletion to generate marker-free SSI (Fig. 1d). This process could be repeated to achieve gene stacking into the selected chromosomal sites.

Gene stacking approach based on the use of Cre-lox for gene integration and a pair of nucleases for marker deletion. a Gene stacking site consists of a loxP fused to promoter-less marker gene (NPT*) followed by the first nuclease target site, e.g., I-SceI site. The GS construct could also include the first set of gene(s)-of-interest (GOI-1); b The first donor vector, pDonor1, contains loxP flanked construct consisting of the second promoter-less marker gene (HPT*), the second nuclease target site, e.g., ZFN site, GOI-2, the first nuclease target site, and the promoter for activating NPT gene at the GS site; c Co-delivery of pDonor1 and cre gene generates site-specific integration (SSI) locus, which is selectable due to the activation of NPT gene, and contains two I-SceI target sites for marker excision; d Delivery of I-SceI gene into SSI cells leads to excision of NPT gene, seamlessly connecting GOI-1 and GOI-2; e The second donor vector, pDonor2, is similar to pDonor1 except the first marker gene, NPT, is included and nuclease target sites are exchanged. Targeting of pDonor2 into marker-free SSI-1 line will generate SSI-2 that is selectable due to HPT activation, and converted to marker-free SSI-2 by ZFN activity
Development of site-specific integration lines
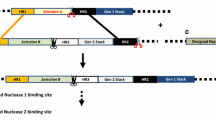
To develop SSI locus containing I-SceI and ZFN target sites, pNS27 was developed for DNA integration into T5 site, which contains a lox76 site for Cre-mediated gene integration, and expresses Cre activity (Fig. 2a). pNS27 contains promoter-less Bar gene flanked by I-SceI target sites, GFP gene, CCR5 site (ZFN target), and promoter-less NPT II gene fused to FRT for FLP-mediated site-specific integration (Fig. 2b). Delivery of pNS27 into T5 calli generated 5 bialaphos-resistant SSI lines, S1 – S5, from ten bombarded calli plates. All except one (S2) were confirmed to contain the expected SSI structure by PCR, sequencing, and Southern hybridization (Fig. 2c-e; sequencing data not shown). Two of these (S1 and S4) were used for the subsequent work.

Site-specific integration into T5 locus. The development of T5 line is described earlier [28]. a T5 site contains lox76 (right arm mutant) between maize ubiquitin-1 (Ubi) promoter and cre coding sequence, and a hygromycin selection marker (HPT); b Donor plasmid pNS27 (pUC backbone), designed for site-specific integration into T5 site, contains between loxP (wild-type) and lox75 (left arm mutant), a promoter-less Bar gene (*), GFP gene (35S:GFP), promoter-less NPT gene, and FRT site. Bar gene is flanked by I-SceI target sites, and a single CCR5 site is present downstream of NPT gene. Each gene contains nos 3′ transcriptional termination (nosT) sequence (not shown); c Introduction of pNS27 into T5 cells, that already express Cre activity, results in the integration of the donor construct into T5 site via lox75 x lox76 recombination, generating precise site-specific integration (SSI) that contains loxP downstream of Ubi, and double-mutant lox78 upstream of cre (black triangles); d PCR analysis of T5-SSI lines (S1 – S5) using primers shown above; d Southern hybridization of EcoR1 (e) digested genomic DNA of SSI lines with GFP and nosT fragments. M, 1-kb DNA ladder. The expected PCR and EcoRI fragments from precise SSI site are shown in (c)
I-SceI-mediated marker deletion from SSI locus
S1 and S4 were retransformed by co-bombardment with pUb:ISceI and pSS1 (the selection plasmid containing 35S:NPT gene), and the calli were selected on geneticin. The resistant lines were analyzed for the deletion of Bar gene from the SSI locus. A total of 72 geneticin resistant callus lines (UI) were isolated, each of which was subjected to PCR using primers around the Bar gene (Fig. 3a-b). The concomitant DSBs created by I-SceI are expected to delete the Bar gene, and the broken ends would be repaired through the DNA repair process. Since homologous DNA sequences are not present at the broken ends, repair through non-homologous end joining mechanism would occur that incorporates insertions – deletions (indels) at the repaired site [8]. The uncut SSI locus is expected to generate 1.9 and 2.8 kb amplicons with primers a1-b and a2-b; however, precise deletion of Bar would reduce the amplicon size to 0.9 and 1.8, respectively (Fig. 3a-b). Fourteen UI lines (19.4 %) showed 0.9 and 1.8 kb bands without the presence of the parental bands in PCR with a1 - b and a2 - b, indicating near-perfect repair of the cut site (Fig. 3c; Table 1). Sequencing of 0.9 kb fragments showed that the excision-repair in these lines occurred without the loss of sequences immediately adjacent to the I-SceI sites, confirming near-perfect repair of the site (Fig. 3d-e). These 14 lines contained 4 types (1 – 4) of the repaired sequences, with type 1 and 2 found frequently, and type 3 and 4 found in one line each (Fig. 3e). Specifically, 12 lines contained either type 1 (TAGGGATAATCCCTA) or type 2 (TAGGGATTATCCCTA) sequence that differed in the 5′-overhang (5′-ATAA) generated by the I-SceI cut. Since the overhangs from the two sites (upstream and downstream of Bar; Fig. 3d) are complementary in 2 of 4 bases, they could initiate microhomology based recombination [29, 30]. Type 3 and 4, on the other hand, contain very short deletions (≤10 bases) at the repair site. The rest of the lines either contained no change or imperfect deletion (Fig. 3c; Table 1). Seven lines (9.7 %) contained short indels at the excision site, indicated by amplification of bands larger or smaller than 0.9 or 1.8 kb. Twenty five lines (48.6 %) did not amplify any product, indicating the presence of large indels at the excision site, and 26 lines (36 %) amplified the parental 1.9 and/or 2.8 kb band expected from the intact SSI site (no change). While, large indels could not be analyzed due to the lack of PCR amplification, analysis of short indels showed that DSB sites frequently incorporated vector DNA sequences, while one line contained rice mitochondrial DNA (data not shown).

I-SceI-mediated marker excision from site-specific integration (SSI) locus. a The structure of T5-SSI containing I-SceI targets (magenta bars) around Bar gene; b Introduction of I-SceI activity by retransformation of the SSI line, S1, excises Bar gene to generate marker-free SSI; c PCR on UI lines (S1 line retransformed with Ubi:ISceI construct) amplifies ‘excision’ footprints (0.9 or 1.8 kb), indicative of precise excision (#) or indels (*); d Sequence of SSI locus around Bar gene containing loxP upstream and GFP downstream. Red fonts represent I-SceI targets (nick site indicated by ^); e Sequences of the 0.9 kb excision ‘footprints’ obtained from UI lines. Concomitant DSB on the two I-SceI sites followed by joining generates near-perfect repair without the loss of sequences immediately adjacent to the I-SceI targets. The numbers indicate the four repair sequences (1–4) observed in perfect excision lines; f Southern blot analysis of UI lines on KpnI (K)-digested genomic DNA using GFP and Bar probe. Arrow points out the expected excision band (3.5 kb) from near-perfect excision locus. S1, parental SSI line; g PCR analysis of UI lines to detect the presence of Ubi:ISceI gene. Note the absence of the expected 0.8 kb band in UI lines. P, positive control (pUbi:ISceI)
Southern blot analysis was done on the genomic DNA isolated from the regenerated plants of the UI lines. Probing of the KpnI-digested genomic DNA with GFP fragment generated the expected ~2 kb band from S1 parent, which upon deletion of Bar fragment that contains a KpnI site is expected to increase to 3.5 kb (Fig. 3a-b). Southern data corroborated with the PCR data as the UI lines that were found to contain the near-perfect deletion in PCR showed 3.5 kb band upon hybridization with GFP, and those having imperfect deletions (short/large indels) showed variable sizes, while the lines found to contain the parental locus showed the intact 2 kb band (Fig. 3f). Re-probing this blot with the Bar fragment confirmed complete deletion of Bar gene from the ‘near-perfect’ UI lines (Fig. 3f). In the PCR to amplify I-SceI gene, surprisingly, none of the UI lines were found to contain full I-SceI construct as indicated by the absence the expected 0.8 kb amplicon or the presence of smaller amplicons indicating truncated/rearranged I-SceI fragments (Fig. 3g). The exclusion of the functional I-SceI gene in UI lines points to the potential toxicity of the strong constitutive I-SceI activity in rice. Therefore, deletion of Bar gene in rice cells essentially occurred by the transient I-SceI expression. This data also explains the failure of Bar deletion in a large proportion of UI lines (36 %). A BLAST search for I-SceI target site identity in rice genome found partial matches with 14 loci, in which 10–15 bases out of 18 base I-SceI sequence (≤70 %) matched. Two of these loci that showed 15 and 13 base match on chromosome 10 were analyzed. PCR/sequencing of these sites (NC_008403.2|:22808202–22809528 and NC_008403.2|:20914800–20915400) in 6 different UI lines, three of which contained perfect Bar deletion, found no sequence change (Data not shown). In summary, I-SceI mediated marker deletion was found to occur in 46 of 72 lines (63 %), 14 of which (19.4 %) showed perfect deletion without the loss of the adjacent sequences at the SSI locus. Marker-deletion in the SSI lines essentially occurred by the transient I-SceI activity as strong constitutive expression appeared to be toxic to rice genome.
Subsequent rounds of site-specific integration by FLP-FRT system
The gene stacking strategy shown in Fig. 1 can be practiced by a single SSR system. However, T5 locus used in the present work is not designed for iterative gene stacking, therefore, we included an FRT site in pNS27 construct for FLP-mediated site-specific integrations into marker-free SSI lines. The T5-SSI locus contains a promoter-less NPT gene fused to FRT site. Therefore, site-specific integration into FRT site will be selectable through NPT activation. This strategy of promoter - gene fusion based selection in which an FRT site is located between the promoter and NPT gene (Fig. 4a-c) was demonstrated in our previous work [13]. Using the same strategy, we showed that the FRT located in the SSI site, e.g., S1 locus, can be targeted for gene stacking by subsequent transformation. A new donor vector, pNS35 that contains FRT-flanked Ubi promoter, was delivered by co-bombardment with pUbiFLPe into S1 line, and the geneticin-resistant lines were analyzed by PCR and Southern hybridization for the presence of SSI structure arising from FRT x FRT recombination (Fig. 4b-c). The use of FLPe is critical in obtaining site-specific integration as this variant of FLP is several fold more efficient in both DNA excisions and integrations [13, 31, 32]. From the bombardment of 10 plates, 5 SSI lines (A – E) were recovered that expressed GFP and GUS (data not shown), and contained the predicted 1.4 kb Ubi:NPT fusion in PCR with primers c and a2 (Fig. 4c-d). Southern hybridization of the HindIII (H) digested DNA of these lines with Ubi probe showed the presence of the expected ~3.2 and 2 kb bands in each line, while the parent S1 line contained only 2 kb band. Two of the SSI lines (A and D) contained additional bands, while the remaining three contained only the expected bands, indicating the absence of extra-SSI fragments (Fig. 4e). Overall, this analysis confirmed the reactivity of the FRT site in the SSI locus and validated its suitability as the target for the next round of gene stacking.

Site-specific integration into T5-SSI by FLP-FRT recombination. a Structure of the T5-SSI obtained through Cre-mediated site-specific integration (see Fig. 2), note the presence of promoter-less (*) NPT gene and FRT site for the next round of site-specific integration; b Donor vector, pNS35, contains FRT flanked maize ubiquitin promoter (Ubi) oriented to trap NPT gene in the marker-free SSI site, and the gus gene as gene-of-interest; c Delivery of pNS35 and FLPe gene generates SSI locus through FRT x FRT recombination. This SSI is selectable due to the activation of NPT gene; d PCR analysis of geneticin resistant SSI lines showing the presence of the expected 1.4 kb SSI footprint; e Southern hybridization of SSI lines (A – E) on HindIII-digested genomic DNA probed with Ubi fragment. HindIII (H) sites, fragment sizes, primer sites are shown in the SSI structure (c). DNA sizes are shown in kb
Testing ZFN activity in rice genome
As shown in Fig. 1, two nucleases are required for the alternating use in the gene stacking process. The second nuclease tested in this study is ZFN that targets human CCR5 sequence [33]. A single CCR5 site consisting of 33 nucleotides is located in the SSI locus (Fig. 5a). To test the activity of ZFN at this site, S1 line was retransformed with pUbi:ZFN, and the transformed callus lines (UZ lines) were analyzed by PCR using primers e and f (Fig. 4a). This PCR is expected to generate 1.2 kb amplicon from the SSI locus, and upon DSB/repair at CCR5 site, could either generate the same size amplicon (very short indels or no change), variable size amplicon (short indels) or no amplicon (large indels). A total of 68 UZ lines were analyzed, 38 of which (55.8 %) did not generate any amplicon, indicating the presence of large indels. The sequencing of the amplicons obtained (sizes ranging from 0.8 to 1.1 kb) from the remaining 30 lines, showed that 21 lines contained no sequence change, and nine contained very short or short indels at the CCR5 site (30 %) (Table 1). Four of these lines contained a very short insertion or deletion, with three lines containing only 3–14 bp deletion and 1 line containing 3 bp insertion (Fig. 5a). Overall, this analysis indicated that CCR5-ZFN efficiently creates DSB at the CCR5 site placed in the rice genome, as 47 out of 68 lines (69 %) contained indels at the CCR5 site. However, most of these lines contained large indels and possibly incorporated sequences of the introduced plasmid DNA. To distinguish between large deletions and insertions, PCR on 24 of these lines was conducted using primers g and f that is expected to amplify 2.1 kb fragment from the SSI locus (Fig. 4a). Thirteen of these lines amplified fragments of variable sizes, indicating large (>1 kb) deletions at the CCR5 site, since the analysis was done on callus, which could consist of more than one cell line, most of these samples also generated the parental 2.1 kb band (Fig. 5b). Although insertion was observed frequently in this study, most likely it occurred due to the presence of high quantities of exogenous DNA in the form of the introduced plasmids. Finally, the integration of ZFN gene in these lines was tested by PCR, which showed that the majority of the lines (46 out of the 68) contained ZFN gene, however, a fair number of lines (22 lines) did not contain ZFN gene, in spite of showing indels at the CCR5 site. Therefore, transient ZFN activity could also induce DSB at CCR5 site in the rice genome. It should be noted that a relatively small quantity of Ubi:ZFN plasmid was bombarded (100 ng per shot), which could explain a high proportion of lines lacking stable ZFN integration.

ZFN and heat-inducible I-SceI activity. a Sequences of CCR5 site amplified by PCR using e – f primers (see Fig. 4a). Sequences obtained from the parental S1 line and four UZ lines (transformed with pUbi:ZFN). Deletion is shown by dots and insertion by small case letters. Red fonts indicate CCR5 sequence; b PCR analysis of UZ lines with g – f primers to detect large deletions at CCR5 site. Note the presence of <2.1 kb bands in different lanes; c PCR analysis of HI lines (1–3, S1 line retransformed with heat-inducible I-SceI gene). HI samples maintained at room temperature (RT) or exposed to heat-shock (HS) were analyzed to detect the excision of Bar gene using primers a2 - b (see Fig. 3a). Note the presence of 1.8 kb excision footprint in HS samples; d Sequences of 1.8 kb excision footprints from HS samples. Red fonts indicate I-SceI target sequences
Testing heat-inducible nuclease activity for marker deletion
The nuclease activity can be introduced into SSI lines by retransformation, genetic crosses or inducing gene activity. The use of inducible nucleases is particularly attractive for reducing effort in the process. Previously, we showed that heat-inducible Cre or FLPe are effective in excising SMG from rice by exposing seedlings to 42 °C for 3 h [24, 34]. Here, we assessed the efficacy of heat-inducible I-SceI and ZFN in deleting SMG from the T5-SSI locus (Fig. 2c). HS:ISceI or HS:ZFN constructs were introduced into S1 line by retransformation, and the resulting clones (HI or HZ) were confirmed by PCR to contain the respective nuclease gene (Data not shown). Calli of 7 HI and 23 HZ lines were incubated at 42 °C for 3 h, transferred to room temperature for 72 h, and ground for DNA extraction. Due to the presence of two I-SceI targets, I-SceI activity could be detected by PCR using a2-b primers (Fig. 3a-b), whereas, ZFN activity can only be detected by sequencing the PCR product as only one ZFN target (CCR5) is present (Fig. 4a). Three out of 7 HI lines generated 1.8 kb deletion fragment with a2 - b primers in the heat-treated samples (Fig. 5c). Sequencing of these fragments showed near-perfect deletion of Bar gene (Fig. 5d), confirming the efficacy of heat-inducible I-SceI. The remaining HI lines showed the parental 2.8 kb band in the heat-treated samples, indicating poor I-SceI induction or gene silencing. PCR analysis of 23 heat-treated HZ lines with e – f primers (Fig. 4a) found 1.2 kb amplicon in 10 lines, <1.0 kb amplicon in 2 lines, and no amplification in 11 lines (data not shown), indicating large indels in the latter 13 lines. The sequencing of 1.2 kb amplicons of the former ten lines, however, did not show indels at the CCR5 site in any of the lines. A number of these lines failed to amplify the expected 1.2 kb band even without heat-treatment, indicating promiscuous ZFN activity. Therefore, more work is needed to determine the efficiency of HS:ZFN for inducible marker excision in plants.
Discussion
The current methods of multigene transformations include transfer of large T-DNA or co-transformation of multiple vectors [35, 36], which can deliver multiple genes in a single attempt but are not suitable for the periodic introduction of new genes into the engineered sites. Targeted gene integrations in plants by synthetic nucleases has been reported in recent years [37–42], the mechanism of which is the repair of chromosomal breaks through homologous recombination. However, since the repair could also occur by joining of the broken chromosomal ends through non-homologous recombination, targeted gene integrations are practically infrequent [4, 39]. Targeted integrations at 3–5 % efficiency has been reported in crop plants [3]; however, it is generally accepted that isolation of targeted events require large transformation efforts even when a selection strategy is employed to isolate targeted insertions [4, 42].
Efficient gene stacking approaches could become widely practiced technologies. The use of Cre-lox system, in combination with the selection strategies, is extremely successful in producing high quality SSI clones without significantly lowering plant transformation efficiencies [9, 11]. Similarly, FLP-FRT is effective in producing SSI clones at high efficiency in plants [12, 13]. These SSR systems are reliable as they have been widely used and found to be effective in a number of plant species [14]. However, the reversibility of recombination, especially the recombination leading to SSI, remained a major bottleneck in developing iterative site-specific gene integration approaches. The use of mutant lox sites for controlling reversibility would allow only two rounds of integrations as the recombination between a set of mutant lox sites generates a loxP [9], which can be targeted, but two cis-positioned lox sites will be generated in the resulting SSI that would complicate subsequent use of Cre. However, excising one of the two cis-positioned sites, along with the marker gene, resolves the bottleneck without increasing the effort, and allows, in principle, unlimited rounds of gene stacking. The use of nucleases for marker excision is of strategic importance as the cut-repair site cannot be targeted by the same nuclease. Recently, a number of studies have described the use of nucleases in targeted mutagenesis [43], demonstrating their effectiveness and efficiency in plants. Excision of marker gene involves two concomitant breaks to delete a precise segment of DNA. Two studies have demonstrated the use of nucleases in marker gene excision. Antunes et al. [25] found ~15 % efficiency of PB1 in Arabidopsis for marker excision and inheritance of the excision locus. Petolino et al. [27] used CCR5-ZFN to delete transgenes from maize, and reported high rate of excision (35 %) in F1 plants, several of which stably transmitted the excision locus to F2 progeny. For gene stacking application, it is important that the sequences beyond the nuclease target site are preserved, and the integrity of the locus is not compromised. Using heat-inducible PB1, Antunes et al. found a fair number of clones containing very short indels or perfect re-ligation of the cut ends. A similar result was obtained by Petolino et al. in examining the site that were targeted by ZFN. These reports along with our data confirm the practical application of nucleases in deleting marker genes from plants. The use of inducible nucleases could streamline marker excision process and allow marker excision in the primary transgenic plants. The use of inducible nucleases could also allay the concern of off-target DSBs as we found that rice cells could tolerate heat-inducible I-SceI expression, but not the strong constitutive expression. Although, off-targeting by strong constitutive expression of I-SceI has not been reported in plants, and we did not find off-targeting at the predicted genomic sites in rice, off-targeting at non-canonical sites has been reported in human genome [44].
The gene stacking strategy presented here can be practiced with the current transformation platforms using dedicated target lines and designed donor constructs. SSR-mediated gene integrations have been practiced with both Agrobacterium and particle bombardment methods [12, 13, 23, 28]. Nuclease-mediated marker excisions could be done by genetic crosses or by deploying inducible nucleases as part of the donor construct (Fig. 1b, e: placed next to the promoter-less marker gene, upstream of nuclease target site) and integrating it into SSI structure for controlled ‘auto-excision’, an approach that has been demonstrated with Cre-lox [24, 45, 46]. More work is needed to determine the efficiency of the auto-excision approach with nucleases. More importantly, the two nucleases used in this study were effective in creating DSB or deleting marker gene, and, in case of I-SceI, preserving the integrity of the SSI locus. Hence, each step of the proposed gene stacking strategy was validated and found to be efficient, suggesting that this method is likely to function at 20 – 50 % efficiency in rice. Its efficiency in other plant species is also expected to be high as the efficiency of Cre-lox recombination and nucleases, individually, are reportedly high [27, 45, 47–49]; however species to species variation could occur, e.g., due to variation in DSB repair mechanisms [50].
Gene stacking methods that do not involve the use of nucleases have been reported. Two rounds of recombinase-mediated cassette exchange (RMCE) using 3 hetero-specific FRT sites was used for stacking 7 genes in soybean [51]. However, its application in subsequent rounds would be limited by the availability of new hetero-specific FRT sites. Currently, only a limited number of hetero-specific FRT sites are available, reactivity of which is variable [52]. Bxb1 system involves recombination between two non-identical sites (attP x attB), which cannot be reversed by Bxb1 recombinase without the helper protein [53]. This unidirectional recombination mechanism was used for gene stacking in tobacco through three rounds of transformation [19]. The resulting ‘stacked’ site, as expected, contained attL and attR between each construct, the number of which would increase with each round of site-specific integration. The use of nucleases, on the other hand, deletes all SSR footprints, seamlessly connecting gene constructs in the stacked locus.
Conclusions
We developed a practical approach of transgene stacking that would facilitate rapid introduction of multiple traits into crop varieties without complicating the downstream breeding process. The present work tested each component of the proposed gene stacking method that involves robust SSR systems for directing gene integrations and nucleases for marker excisions. The use of SSR allowed high rates of targeted integrations, which is a major bottleneck in gene targeting. The nucleases were instead used for excising marker genes that involves DSB repair through a more efficient process of joining chromosomal ends. By using the power of each reagent, this study developed an efficient approach of gene stacking that could integrate new genes to the specified site through unlimited rounds of plant transformations.
Methods
Plant line and transformation vectors
Transgenic rice line T5 (in variety Taipei 309) that contains a single lox76 site (right arm mutant) [9] within the construct as depicted in Fig. 2a and described earlier [28] was used as the founder line for gene stacking. Donor vectors, pNS27 and pNS35, for site-specific integration in lox76 site and FRT site, respectively, were constructed in pBluescript SK backbone. pNS27 (Fig. 2b) consists of a promoter-less Bar gene flanked by I-SceI target sites (Fig. 3d) followed by a GFP gene driven by a 35S promoter as a gene-of-interest, CCR5 site, the ZFN target, and a promoter-less neomycin phosphotransferase gene (NPT II) fused to FRT site. The whole construct is flanked by loxP upstream and lox75 downstream. pNS35 (Fig. 4b) contains maize ubiquitin-1 (Ubi) promoter [54] and GUS gene flanked by FRT sites. Sequences of lox and FRT are described by Albert et al. [9] and Senecoff et al. [55]. FLPe expression vector, pUbiFLPe, consists of FLPe gene [56] transcribed by Ubi promoter. I-SceI and ZFN expression vectors (pUbiISceI, pUbiZFN, pHSISceI, and pHSZFN) either contained Ubi promoter or soybean heat shock 17.5E (HS) promoter [57]. The optimized I-SceI coding sequence [58] and the CCR5-ZFN coding sequence was provided by Drs. Holger Puchta (Karlsruhe, Germany) and Joseph Petolino (Dow Agro Sciences, Inc.), respectively. Each gene in these vectors contain transcription termination sequence of nopaline synthase gene (nos 3′).
Rice transformation
Rice tissue culture media and protocols were essentially as described by Nishimura et al. [59]. All transformations were done by particle bombardment using PDS 1000/He gene gun (Bio-Rad, Inc.) as described earlier [13]. Scutellar calluses generated from mature seeds were bombarded with 1-μm gold particles coated with plasmid DNA. About 25 μg of particles were coated with 5 μg donor vectors (pNS27 or pNS35) or 1 – 2 μg of I-SceI/ZFN vectors and used for ten shots (plates). The pNS27-bombarded T5 callus were selected on bialaphos (5 mg/L) to isolate site-specific integration (SSI) lines. The selected SSI lines were bombarded with I-SceI or ZFN vectors along with a selection vector, pSS1, which contains 35S promoter driven NPT II gene, and selected on geneticin (100 mg/L).
Molecular analysis
Genomic DNA was isolated from the callus or leaves and subjected to PCR using primers (Table 2) and Taq polymerase following the manufacturer’s (Promega, Inc.) recommendations. All PCR reactions consisted of 40 cycles of 1 min denaturation at 94 °C, 1 min annealing at 56 °C and 1 min extension at 72 °C followed by final elongation step for 15 min at 72 °C. For sequencing, the PCR amplicon was extracted from the gel and sequenced by Eurofins MWG Operon Kentucky, and viewed using Sequence Scanner v1.0 (Applied Biosystems). For Southern hybridizations, ~5 μg of genomic DNA was digested overnight with appropriate restriction enzyme, fractionated on 0.8 % agarose gel, transferred to a nylon membrane, and hybridized with P32 labelled DNA probes.
Abbreviations
- DSB:
-
Double-stranded break
- indels:
-
Insertion-deletion
- ZFN:
-
Zinc finger nuclease
- SSR:
-
Site-specific recombination
- SSI:
-
Site-specific integration
- SMG:
-
Selectable marker gene
References
Que Q, Chilton MD, de Fontes CM, He C, Nuccio M, Zhu T, et al. Trait stacking in transgenic crops: Challenges and opportunities. GM Crops. 2010;1(4):220–9.
D’Halluin K, Ruiter R. Directed genome engineering for genome optimization. Int J Dev Biol. 2013;57(6–8):621–7.
Petolino JF. Genome editing in plants via designed zinc finger nucleases. In Vitro Cell Dev Biol Plant. 2015;51(1):1–8.
Puchta H, Fauser F. Gene targeting in plants: 25 years later. Int J Dev Biol. 2013;57(6–8):629–37.
Ow DW. Recombinase-mediated gene stacking as a transformation operating system. J Integr Plant Biol. 2011;53:512–9.
Srivastava V, Gidoni D. Site-specific gene integration technologies for crop improvement. In Vitro Cell Dev Biol - Plant. 2010;46:219–32.
Wang Y, Yau YY, Perkins-Balding D, Thomson JG. Recombinase technology: applications and possibilities. Plant Cell Rep. 2011;30:267–85.
Puchta H. The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot. 2005;56(409):1–14.
Albert H, Dale EC, Lee E, Ow DW. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995;7:649–59.
Srivastava V, Ariza-Nieto M, Wilson AJ. Cre-mediated site-specific gene integration for consistent transgene expression in rice. Plant Biotech J. 2004;2:169–79.
Louwerse JD, van Lier MC, van der Steen DM, de Vlaam CM, Hooykaas PJ, Vergunst AC. Stable recombinase-mediated cassette exchange in Arabidopsis using Agrobacterium tumefaciens. Plant Physiol. 2007;145(4):1282–93.
Li Z, Xing A, Moon BP, McCardell RP, Mills K, Falco SC. Site-specific integration of transgenes in soybean via recombinase-mediated DNA cassette exchange. Plant Physiol. 2009;151:1087–95.
Nandy S, Srivastava V. Site-specific gene integration in rice genome mediated by the FLP-FRT recombination system. Plant Biotechnol J. 2011;9:713–21.
Gidoni D, Srivastava V, Carmi N. Site-specific excisional recombination strategies for elimination of undesirable transgenes from crop plants. In Vitro Cell Dev Biol - Plant. 2008;44:457–67.
Gilbertson L. Cre-lox recombination: Cre-ative tools for plant biotechnology. Trends Biotech. 2003;21:550–5.
Ow DW. Recombinase-directed plant transformation for the post-genomic era. Plant Mol Biol. 2002;48:183–200.
Schnütgen F, Stewart AF, von Melchner H, Anastassiadis K. Engineering embryonic stem cells with recombinase systems. Methods Enzymol. 2006;420:100–36.
Nanto K, Yamada-Watanabe K, Ebinuma H. Agrobacterium-mediated RMCE approach for gene replacement. Plant Biotech J. 2005;3:203–14.
Hou L, Yau YY, Wei J, Han Z, Dong Z, Ow DW. An open-source system for in planta gene stacking by Bxb1 and Cre recombinases. Mol Plant. 2014;7(12):1756–65.
Day CD, Lee E, Kobayashi J, Holappa LD, Albert H, Ow DW. Transgene integration into the same chromosome location can produce alleles that express at a predictable level, or alleles that are differentially silenced. Genes Dev. 2000;14:2869–80.
Chawla R, Ariza-Nieto M, Wilson AJ, Moore SK, Srivastava V. Transgene expression produced by biolistic-mediated, site-specific gene integration is consistently inherited by the subsequent generations. Plant Biotech J. 2006;4:209–18.
Nanto K, Sato K, Katayama Y, Ebinuma H. Expression of a transgene exchanged by the recombinase-mediated cassette exchange (RMCE) method in plants. Plant Cell Rep. 2009;28:777–85.
Vergunst AC, Hooykaas PJ. Cre/lox-mediated site-specific integration of Agrobacterium T-DNA in Arabidopsis thaliana by transient expression of cre. Plant Mol Biol. 1998;38:393–406.
Nandy S, Srivastava V. Marker-free site-specific gene integration in rice based on the use of two recombination systems. Plant Biotech J. 2012;10:904–12.
Antunes MS, Smith JJ, Jantz D, Medford JI. Targeted DNA excision in Arabidopsis by a re-engineered homing endonuclease. BMC Biotech. 2012;12:86.
Fauser F, Roth N, Pacher M, Ilg G, Sánchez-Fernández R, Biesgen C, et al. In planta gene targeting. Proc Natl Acad Sci U S A. 2012;109:7535–40.
Petolino JF, Worden A, Curlee K, Connell J, Strange Moynahan TL, Larsen C, et al. Zinc finger nuclease-mediated transgene deletion. Plant Mol Biol. 2010;73:617–28.
Srivastava V, Ow DW. Biolistic mediated site specific integration in rice. Mol Breed. 2002;8:345–50.
Yu AM, McVey M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010;38:5706–17.
Vu GT, Cao HX, Watanabe K, Hensel G, Blattner FR, Kumlehn J, et al. Repair of Site-Specific DNA Double-Strand Breaks in Barley Occurs via Diverse Pathways Primarily Involving the Sister Chromatid. Plant Cell. 2014;26(5):2156–67.
Akbudak MA, Srivastava V. Improved FLP recombinase, FLPe, efficiently removes marker gene from transgene locus developed by Cre-lox mediated site-specific gene integration in rice. Mol Biotech. 2011;49:82–9.
Nguyen LD, Underwood JL, Nandy S, Akbudak MA, Srivastava V. Strong activity of FLPe recombinase in rice plants does not correlate with the transmission of the recombined locus to the progeny. Plant Biotech Rep. 2014;8(6):455–62.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–16.
Khattri A, Nandy S, Srivastava V. Heat-inducible Cre-lox system for marker excision in transgenic rice. J Biosci. 2011;36:37–42.
Ruiz-Lopez N, Haslam RP, Napier JA, Sayanova O. Successful high-level accumulation of fish oil omega-3 long-chain polyunsaturated fatty acids in a transgenic oilseed crop. Plant J. 2014;77:198–208.
Zhu C, Naqvi S, Breitenbach J, Sandmann G, Christou P, Capell T. Combinatorial genetic transformation generates a library of metabolic phenotypes for the carotenoid pathway in maize. Proc Natl Acad Sci U S A. 2009;105:18232–7.
Ainley WM, Sastry-Dent L, Welter ME, Murray MG, Zeitler B, Amora R, et al. Trait stacking via targeted genome editing. Plant Biotech J. 2013;11:1126–34.
D’Halluin K, Vanderstraeten C, Van Hulle J, Rosolowska J, Van Den Brande I, Pennewaert A, et al. Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant Biotech J. 2013;11:933–41.
de Pater S, Pinas JE, Hooykaas PJ, van der Zaal BJ. ZFN-mediated gene targeting of the Arabidopsis protoporphyrinogen oxidase gene through Agrobacterium-mediated floral dip transformation. Plant Biotech J. 2013;11:510–5.
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, et al. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature. 2009;2009(459):437–41.
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, et al. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature. 2009;459:442–5.
Kumar S, Al Abed D, Worden A, Novak S, Wu H, Ausmus C, et al. Modular gene targeting system for sequential transgene stacking in plants. J Biotechnol. 2015;207:12–20.
Voytas DF. Plant genome engineering with sequence-specific nucleases. Annu Rev Plant Biol. 2013;64:327–50.
Petek LM, Russell DW, Miller DG. Frequent endonuclease cleavage at off-target locations in vivo. Mol Ther. 2010;18(5):983–6.
Zhang W, Subbarao S, Addae P, Shen A, Armstrong C, Peschke V, et al. Cre/lox-mediated marker gene excision in transgenic maize (Zea mays L.) plants. Theor Appl Genet. 2003;107(7):1157–68.
Yau YY, Stewart Jr CN. Less is more: strategies to remove marker genes from transgenic plants. BMC Biotechnol. 2013;13:36.
Srivastava V, Anderson OD, Ow DW. Single-copy transgenic wheat generated through the resolution of complex integration patterns. Proc Natl Acad Sci U S A. 1999;96:11117–21.
Bala A, Roy A, Das A, Chakraborti D, Das S. Development of selectable marker free, insect resistant, transgenic mustard (Brassica juncea) plants using Cre/lox mediated recombination. BMC Biotechnol. 2013;13:88.
Lloyd AH, Wang D, Timmis JN. Single molecule PCR reveals similar patterns of non-homologous DSB repair in tobacco and Arabidopsis. PLoS ONE. 2012;7(2):e32255.
Kirik A, Salomon S, Puchta H. Species-specific double-strand break repair and genome evolution in plants. EMBO J. 2000;19(20):5562–6.
Li Z, Moon BP, Xing A, Liu ZB, McCardell RP, Damude HG, et al. Stacking multiple transgenes at a selected genomic site via repeated recombinase-mediated DNA cassette exchanges. Plant Physiol. 2010;154:622–31.
Turan S, Kuehle J, Schambach A, Baum C, Bode J. Multiplexing RMCE: versatile extensions of the Flp-recombinase-mediated cassette-exchange technology. J Mol Biol. 2010;2010(402):52–69.
Ghosh P, Kim AI, Hatfull GF. The orientation of Mycobacteriophage Bxb1 integration is solely dependent on the central dinucleotide of attP and attB. Mol Cell. 2003;2003(12):1101–11.
Christensen AH, Sharrock RA, Quail PH. Maize polyubiquitin genes: structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol Biol. 1992;18:675–89.
Senecoff JF, Rossmeissl PJ, Cox MM. DNA recognition by the FLP-recombinase of the yeast 2 lm plasmid. A mutational analysis of the FLP binding site. J Mol Biol. 1988;201:405–21.
Buchholz F, Angrand PO, Stewart AF. Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol. 1998;16:657–62.
Czarnecka E, Ingersoll JC, Gurley WB. AT-rich promoter elements of soybean heat shock gene Gmhsp17.5E bind two distinct sets of nuclear proteins in vitro. Plant Mol Biol. 1992;19:985–1000.
Hlubek A, Biesgen C, and Höffken H-W (2011) Chimeric endonucleases and uses therefore. Patent WO 2011/064750
Nishimura A, Aichi I, Matsuoka M. A protocol for Agrobacterium mediated transformation in rice. Nat Protoc. 2006;1:2796–802.
Acknowledgements
We thank Dr. Holger Puchta (Karlsruhe, Germany) for I-SceI gene, Dr. Joe Petolino for ZFN cDNA, Dr. James Thomson (USDA-ARS) for FLPe construct, and Jamie Underwood and Clinton Greub for technical help in the initial phase of this project.
Funding
This work was supported by USDA grant# 2010-33522-21715 and Arkansas Bioscience Institute-Division of Agriculture grant to VS; and USDA Capacity Building Grant # 2010-38821-21540 to MM and VS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
A patent related to the gene stacking approach presented in this paper has been filed by University of Arkansas.
Authors’ contributions
SN constructed vectors, carried out marker excision analysis and Southern analyses, SZ and BP carried out rice transformations and ZFN analysis, MM participated in data analysis and manuscript drafting, VS designed experiments, analyzed data, and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Availability of data and materials
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nandy, S., Zhao, S., Pathak, B.P. et al. Gene stacking in plant cell using recombinases for gene integration and nucleases for marker gene deletion. BMC Biotechnol 15, 93 (2015). https://doi.org/10.1186/s12896-015-0212-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-015-0212-2