
Abstract
Background
Pigeon pea (Cajanus cajan L.) is the sixth major legume crop widely cultivated in the Indian sub-continent, Africa, and South-east Asia. Cytoplasmic male-sterility (CMS) is the incompetence of flowering plants to produce viable pollens during anther development. CMS has been extensively utilized for commercial hybrid seeds production in pigeon pea. However, the molecular basis governing CMS in pigeon pea remains unclear and undetermined. In this study transcriptome analysis for exploring differentially expressed genes (DEGs) between cytoplasmic male-sterile line (AKCMS11) and its fertility restorer line (AKPR303) was performed using Illumina paired-end sequencing.
Results
A total of 3167 DEGs were identified, of which 1432 were up-regulated and 1390 were down-regulated in AKCMS11 in comparison to AKPR303. By querying, all the 3167 DEGs against TAIR database, 34 pigeon pea homologous genes were identified, few involved in pollen development (EMS1, MS1, ARF17) and encoding MYB and bHLH transcription factors with lower expression in the sterile buds, implying their possible role in pollen sterility. Many of these DEGs implicated in carbon metabolism, tricarboxylic acid cycle (TCA), oxidative phosphorylation and elimination of reactive oxygen species (ROS) showed reduced expression in the AKCMS11 (sterile) buds.
Conclusion
The comparative transcriptome findings suggest the potential role of these DEGs in pollen development or abortion, pointing towards their involvement in cytoplasmic male-sterility in pigeon pea. The candidate DEGs identified in this investigation will be highly significant for further research, as they could lend a comprehensive basis in unravelling the molecular mechanism governing CMS in pigeon pea.
Similar content being viewed by others

Background
Cytoplasmic male-sterility (CMS) is a maternally inherited trait in plants where they are unable to produce functional pollens [1]. It occurs due to unusual mitochondrial open reading frames (orfs) which are chimeric in structure and express proteins that restrict mitochondrial function and pollen development [2]. Cytoplasmic male-sterility phenotype is known to be restored by nuclear genes termed as the restorer of fertility (Rf) and hence result in the production of functional pollens in a plant with the aberrant mitochondrial genome [3]. CMS/Rf systems, therefore, give a clear view of the interaction and conflicts between nuclear and mitochondrial genome [4]. Cytoplasmic male-sterile systems along with the availability of restorer of fertility have emerged as a reliable tool for enhancing the yield of many crop plants by utilizing hybrid vigor or heterosis [5]. Hybrid seed production technology primarily involves three breeding systems: (a) CMS line, carrying male-sterile cytoplasm and devoid of functional nuclear Rf genes (b) Maintainer line, containing fertile cytoplasm along with similar Rf (c) Restorer line containing fertile cytoplasm along with functional dominant Rf gene(s). The F1 hybrids produced possess dominant Rf gene to restores the male-fertility and the blend of nuclear genes from the sterile and fertile lines results in hybrid vigor [2]. During the 1950s, the first CMS system used for hybrid corn production was maize CMS-Texas (CMS-T) system which substantially increased the hybrid seed production and remarkably enhanced the maize yield [6]. Till date, impressive advancement has been made in plants to comprehend the molecular basis of CMS and fertility restoration. A number of mitochondrial genes related to male-sterility have been reported such as urf13 from maize [7], pcf associated with atp9 gene from petunia [8, 9], and the radish atp8 of Ogura CMS [10]. However, in pigeon pea the gene associated with cytoplasmic male-sterility has not been much explored.
In flowering plants, anther development is a highly controlled mechanism which demands proper differentiation of the sporogenous tissue and timely programmed microsporogenesis [11]. The absence of male gametophyte (stamen) or defective development of anther, disruption of the pollen mother cells (PMC) or tapetum cells, abnormal development of pollen, and failure of anther dehiscence leads to pollen abortion [12, 13]. In all cases, CMS due to the dysfunctional mitochondrial genome is mainly responsible for the disruption of pollen development [14]. Previous studies on CMS provide definitive evidence that the organelle mitochondria are critical for the tapetum and microspores development [14]. During distinct stages of pollen development, the requirement of energy is extremely high, and even a slight defect in the mitochondrial function becomes deleterious and results in pollen sterility [15,16,17]. Previous studies have characterized many genes playing a pivotal role in anther development in Arabidopsis viz. SPOROCYTELESS (SPL)/NOZZEL (NZZ) [18,19,20,21], EXCESS MICROSPOROCYTES 1 (EMS1) [22, 23], TAPETUM DETERMINANT1 (TPD1) [24], ABORTED MICROSPORES (AMS) and MALE STERILITY1 (MS1) [25,26,27], MYB gene family (AtMYB103) [28, 29], RUPTURED POLLEN GRAIN1 (RPG1), CYTOCHROME P450 (CYP703A2), CYTOCHROME P450 (CYP704B1), acyl-coA synthetase5 (acos5), LAP6 and LAP5 [30,31,32,33,34,35,36,37] redundantly facilitate anther development and callose synthase5 (cals5) is essential for callose synthesis [38].
Pigeon pea (Cajanus cajan (L.) Millsp.) belongs to the family Fabaceae and regarded as the sixth major legume crop worldwide [39]. It is popularly known for its high protein content [40] and drought tolerance [41]. It is an important pulse crop worldwide and predominantly cultivated in the tropics and sub-tropics (http://faostat.fao.org). To meet the ever-growing demand for pigeon pea extensive research has been done during the last decade, a breakthrough has been accomplished in developing hybrid technology [42]. The pre-imperatives for large-scale hybrid seed production are pollen transfer method and advancement of stable cytoplasmic male-sterile (CMS) system. The male-sterility phenomenon could not be much explored in other legumes due to their self-pollinating nature. However, pigeon pea has a considerable natural out-crossing [43]. Till date, eight (A1 to A8) such CMS systems have been bred in pigeon pea by transferring the cytoplasm of the wild relatives in the nuclear background of the cultivated pigeon pea via interspecific-hybridization followed by several rounds of back-crossing [44]. Therefore, hybrid seed technology based on cytoplasmic male-sterility has given a chance to overcome the production constraints by efficiently increasing the yield potential in pigeon pea [45]. However, among the eight systems, A2 and A4 CMS system obtained from the wild-type Cajanus scarabaeoides and Cajanus cajanifolius respectively, are regarded as highly potential CMS systems [46,47,48]. The gene associated with CMS in A4 cytoplasm pigeon pea has been reported [49]. Recently, the genetics of fertility restoration in pigeon pea with A2 cytoplasm was studied in detailed [47]. Even though A2 cytoplasm has played a significant role in hybrid seed production, yet the underlying molecular processes governing CMS in A2 cytoplasm pigeon pea is still not known.
Presently, the next generation sequencing technology has revolutionized the field of life sciences and offers extraordinary speed and cost-effectiveness to study genomic and transcriptome data [50, 51]. RNA-Seq has emerged as an effective approach for transcriptome profiling which provides accurate measurement of gene expression level [52]. Transcriptome sequencing offers ease of identification and evaluation of thousands of genes in a single analysis [53]. Transcriptome analysis of the cytoplasmic male-sterile lines has been previously reported in many crops such as radish [54], watermelon [55], soybean [56], tomato [57], Brassica napus [58], chili pepper [59],cotton [60,61,62], sweet orange [63] leading to identification of several candidate genes in these systems. In this study, comparative transcriptome analysis of an A2 CMS system derived cytoplasmic male-sterile, and its fertility restorer line in pigeon pea was performed primarily to detect differentially expressed genes participating in pollen development which might help in understanding the CMS mechanism in this crop.
Results
Pollen fertility analysis and phenotypic characterization of sterile and fertile buds
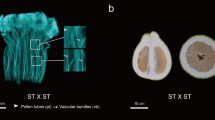
Pollens from male-sterile and fertile buds were observed under a microscope and were evaluated to determine fertility. Upon observation it was seen that the pollens grains showing red acetocarmine staining were fertile while sterile pollens remained unstained (Fig. 1a). During the phenotypic investigation of the sterile (AKCMS 11) and fertile (AKPR303) flower buds we observed that the filaments and anthers of the sterile flowers were smaller in contrast to the fertile flowers. It was clearly visible that the anthers of the fertile line were denser than the sterile line (Fig. 1b).

a Acetocarmine staining differentiating the pollens of AKCMS 11 (sterile line) and AKPR303 (fertile line). Fertile pollens are darkly stained whereas sterile pollens are almost colorless. b Phenotypic characterization of the sterile and fertile floral buds. A: Phenotype of sterile AKCMS11 buds; B: Phenotype of fertile AKPR303 buds
Transcriptome sequencing and de novo assembly
To create a reference transcriptome, we prepared cDNA libraries from the buds of male-sterile (CMS) and fertile (restorer) lines in pigeon pea (two replicates each) and subjected to Illumina paired-end sequencing. Workflow diagram of the analysis done is shown in Fig. 2. High throughput sequencing run generated a total of 109,600,365 and 72,575,988 raw paired-end reads of ~ 100 bp in the sterile and fertile buds, respectively. Also, the correlation between the replicate datasets of the sterile AKCMS11 and fertile restorer AKPR303 was analyzed by calculating Pearson’s correlation coefficient (r < 0.9) using an R studio package. Correlation analysis indicated that the replicate datasets of the sterile and fertile lines were positively correlated with Pearson’s coefficient value as r = 0.72 and r = 0.74, respectively (Additional file 1: Figure S1). The per base sequence quality and k-mer content of the raw paired-end reads was also determined (Additional file 1: Figure S2 and Table S1). After adapter trimming and removal of poor quality reads, 101,676,579 (49.5 Gb) and 69,481,226 (32.7 Gb) clean reads were attained for AKCMS11 (sterile) and AKPR303 (fertile) lines, respectively. Trinity software was further employed for de novo assembly of the pooled reads (171,157,805) from both the samples. There was a total of 1,98,587 transcripts with an average length of 641.34 bp, and the N50 value of 1009 bp (L50 = 6 contigs). We utilized Bowtie2 to align the clean reads to the assembled transcriptome, alignment score of 92.84 and 92.84% was obtained for sterile AKCMS11 (replicate 1 and replicate 2), respectively. Similarly, alignment score of 92.61 and 92.05% was obtained for fertile AKPR303 (replicate 1 and replicate 2), respectively. (Additional file 1: Table S2). An overall alignment rate of 92.58% was obtained indicative of the robust assembly. BUSCO analysis revealed that our assembly was relatively complete, with 98% (n = 297) of BUSCOs detected as complete sequences, just 2% (n = 6) as fragmented sequences, and none were missing in the assembly with eukaryotic lineage. Similarly, 93.5% (n = 402) of BUSCOs detected as complete sequences, 5.8% (n = 25) as fragmented sequences, and just 0.7% (n = 3) were missing in the assembly with viridiplantae lineage (Table 1). After the de novo assembly, 1,72,061 unigenes were obtained by using CD-HIT software. These unigenes were further BLASTN searched against the Rfam database to remove the non-coding RNAs (rRNA, tRNA, snoRNAs, snRNA etc.). Finally, a set of 1,71,095 unigenes were obtained with an average length and N50 of 561.66 bp and 757 bp, respectively (Table 2). The shortest and longest unigenes identified were 201 bp and 16,008 bp, respectively. We obtained 1,15,792 unigenes (67.7%) with a length range from 200 to 500 bp. 17,692 unigenes (10.3%) were greater than 1000 bp and 8700 (5.1%) were greater than 2000 bp (Fig. 3a). The average GC content of the unigenes was 45.8%. The RNA-Seq raw data generated from this investigation has been deposited in the NCBI Sequence Read Achieve (SRA) database (SRX3740150, SRX3740151, SRX3740153, SRX3740152) for sterile and fertile buds, respectively.

Systemic representation showing workflow of the analysis done

a Length distribution of the assembled unigenes. b Homology search and annotation statistics of pigeon pea unigenes. c) Similarity distribution of the BLASTX hits for each unigenes against nr database
Functional annotation and classification of the unigenes
Annotation results showed that, 1,02,463 (60%), 52,639 (30.6%), 28,812 (16.7%) and 7653 (4.4%) unigenes annotated in Nr, Virdiplantae, GO and KEGG databases, respectively (Fig. 3b). In total, 1,02,463 (60%) unigenes were annotated in the nr database, while the remaining 40% unigenes did not show any sequence similarity to the nr database (Additional file 2: Table S1). Sequence similarity distribution revealed that 61% of the aligned unigenes had sequence similarity more than 90%, on the other hand, 38.6% unigenes had similarity value between 50 to 90% and only 0.4% unigenes had lower than 50% (Fig. 3c). A total of 52,639 unigenes were uniquely mapped to the Virdiplantae database (Additional file 2: Table S2).
A total of 28,812 unigenes were assigned GO terms and were distributed into 53 GO categories including 26 biological processes, 14 molecular functions, and 13 cellular components. In the biological process category, “cellular process” (19,472 unigenes) was the most predominant functional group followed by “metabolic process” (17,365 unigenes) and “biological regulation” (9627 unigenes). Among the molecular function category, “binding” (19,531 unigenes), “catalytic activity” (14,757 unigenes) and “transcription regulator activity” were the main functional groups. In regard to the cellular component, “cell” (22,732 unigenes) and “cell part” (22,732 unigenes) were the largest categories followed by “organelle” (15,348 unigenes) and “macromolecular complex” respectively (Additional file 2: Table S3 and Additional file 3: Figure S1).
The unigenes were mapped to the KEGG pathway database, 7653 unigenes were assigned KEGG orthologos number involving 132 pathways. The result demonstrated that the five largest pathways were “metabolic pathways” (ko01100), “biosynthesis of secondary metabolites” (ko01110), “plant hormone signal transduction (ko04075), “carbon metabolism” (ko01200) and “biosynthesis of amino acids” (ko01230). The 7653 unigenes were allocated into six functional categories (Additional file 2: Table S4 and Additional file 3: Figure S2). A total of 5499 unigenes were found in “metabolism” with 1363 (24.8%) unigenes involved in “metabolic pathways”, 881 (16%) in “biosynthesis of secondary metabolites”, 870 (15.8%) in “carbohydrate metabolism”, 665 (12.1%) in “amino acid metabolism”, 458 (8.3%) in “lipid metabolism” and other sub-categories. 1222 unigenes accounted for the category of “genetic information and processing” in which 449 (36.7%) unigenes were engaged in “folding, sorting and degradation” followed by “translation” 446 (36.5%), “transcription” 181 (14.8%) and “replication and repair” 146 (11.9%).“Environmental information processing” was represented by a total of 462 unigenes with 416 (90%) involved in “signal transduction” and 46 (10%) in “membrane transport”. Additionally, 300 and 155 unigenes were classified in “cellular processes” and “organismal systems” categories, respectively.
Identification of transcription factors
PlantTFcat online tool [64] was utilized in the present study, for identification of transcription factors in pigeon pea. A total of 3803 unigenes (2.2% of the transcriptome) (Fig. 3b) were cataloged into 52 putative transcription factors (TF) families. Amid the 52 TF family, “C2H2” was found to be the most predominant category with 806 (21.2%) unigenes followed by “WD40-like” (540 unigenes, 14.2%), “MYB-HB-like” (344 unigenes, 9%), “bHLH” (200 unigenes, 5.3%), “AP2-EREBP” (163 unigenes, 4.3%), “WRKY” (161 unigenes, 4.2%) and “bZIP” (129 unigenes, 3.4%) transcription factors (Fig. 4a and Additional file 2: Table S5).

a Transcription factors identified in the transcriptome data of Cajanus cajan. b Number of differentially expressed genes (DEGs) in sterile AKCMS11 and restorer AKPR303
Identification of differentially expressed genes between lines AKCMS11 and AKPR303
We identified 3167 differentially expressed genes (DEGs) between the two lines containing 1432 up-regulated and 1390 down-regulated in the AKCMS11 sterile buds as compared to AKPR303 fertile buds (Additional file 2: Table S6). Additionally, 683 DEGs were uniquely expressed in the sterile genotype and 157 DEGs were distinctively expressed in the fertile restorer (Fig. 4b).
GO and KEGG enrichment analysis of the DEGs
To gain insight into the putative functions of the DEGs, we examined the significantly enriched Gene Ontology (GO) terms in DEGS. The annotated DEGs were assigned to 55 major groups covering three main ontologies: biological process, cellular component and molecular function (Fig. 5). The identified GO terms were further classified into down-regulated and up-regulated groups. In the down-regulated DEGs, a total of 221 GO terms were assigned, including 121, 59 and 41 GO terms in “biological process”, “molecular functions” and “cellular component”, respectively (Additional file 2: Table S7). In up-regulated DEGs, 174 GO terms were assigned, including 78 “biological process”, 53 “molecular functions” and 43“cellular component” (Additional file 2: Table S8). Overall, among the biological process category, the significantly over-represented GO terms were “cellular process”, followed by “metabolic process”, “primary metabolic process” and “cellular metabolic process”. “Catalytic activity”, “binding” and “transferase activity” were the most significantly expressed GO terms in the molecular functions category. Among the cellular component category, “cell part”, “cell” and “intracellular component” were the most significant GO terms (Figs. 6 and 7). Interestingly, many GO terms were specific to down-regulated DEGs and were involved in “pollen development” (10 DEGs), “reproductive structure development” (41 DEGs), “gametophyte development” (16 DEGs), and “oxidative phosphorylation” (7 DEGs), suggesting these may be related to male-sterility in AKCMS11. Hierarchical tree graph of over-represented GO terms in the biological process category of down-regulated and up-regulated DEGs are provided in Additional file 3: Figure S3 and S4.

GO enrichment analysis of the DEGs. X-axis represents the sub-categories; Y-axis represents number of genes in each sub-category. Blue and green indicates down-regulated and up-regulated genes in a sub-category

Significantly over-represented GO terms in down-regulated DEGs

Significantly over-represented GO terms in up-regulated DEGs
KEGG pathway enrichment analysis was performed to discern the metabolic pathways in which the DEGs were involved using the KEGG pathway database. In total, 110 enriched pathways were identified, of which 77 pathways were significantly enriched in both down and up-regulated DEGs whereas 15 and 19 pathways were specific to down and up-regulated DEGs, respectively. A total of 377 down-regulated and 396 up-regulated DEGs were assigned to 92 and 96 KEGG pathways, respectively (Additional file 2: Table S9). The top 25 significantly enriched pathways for down and up-regulated DEGs are mentioned in Fig. 8. The down-regulated DEGs were significantly over-represented in “metabolic pathways” (72 genes), “biosynthesis of secondary metabolites” (39 genes), “oxidative phosphorylation” (13 genes), “carbon metabolism” (11 genes), “ribosome” (9 genes) and glycerophospholipid metabolism (8 genes). Consistent with the GO analysis “oxidative phosphorylation was enriched with down-regulated DEGs. Several down-regulated DEGs participated in starch and sucrose metabolism, glycolysis, pentose phosphate pathway, reactive oxygen species (ROS) generation/scavenging and alpha-linolenic acid metabolism. Among the up-regulated DEGs, the most predominant pathway was “metabolic pathways” (66 genes) followed by “biosynthesis of secondary metabolites” (34 genes), “carbon metabolism” (14 genes), “protein processing in endoplasmic reticulum” (11 genes) and “plant hormone signal transduction” (11 genes). Also, some up-regulated genes were involved in “ascorbate and aldarate metabolism”, “linolenic acid metabolism” and “photosynthesis” etc. (Additional file 2: Table S9).

Top 25 significantly enriched KEGG pathways. a For down-regulated DEGs. b For up-regulated DEGs
Additionally, MapMan tool [65] was also used to visualize significant male-sterility related DEGs involved in different metabolic pathways. All the 3167 differentially expressed genes between sterile AKCMS11 and fertile AKPR303 were annotated to the TAIR database (http://www.arabidopsis.org) and finally, 951 DEGs were identified to be homologs of 930 Arabidopsis genes (Additional file 2: Table S10). To further explore, the potential functions of these DEGs in male-sterility, the Arabidopsis homologs genes were studied in MapMan to identify different metabolic processes in which these are involved (Fig. 9). In our network, the most significant differentially expressed genes were related to Energy/ATP synthesis, ROS metabolism, hormones, secondary metabolism and cell cycle. The expression of genes involved in ‘Energy (Glycolysis, TCA cycle, electron transport chain, transport p- and v-ATPase’s) was down-regulated in the sterile (AKCMS11) buds. Moreover, the genes related to ‘Redox’ (Ascorbate & Glutathione, Peroxiredoxin, Dismutase & Catalase) and cell cycle were also down-regulated in the sterile buds. Additionally, the genes implicated in ‘Auxin & Jasmonic acid synthesis’ were up-regulated in sterile buds, while the ones related to ‘Signaling pathway’ were down-regulated. Also, the expression of genes involved in ‘Lipid’ (Degradation & FA synthesis) and ‘Cell wall’ (degradation & modification) were mostly up-regulated, whereas genes related to ‘Secondary metabolism’ (Flavonoids) were down-regulated in the sterile buds compared to fertile buds. Among the transcription factors, NAC, WRKY (except one), CCAAT, SET, C2C2(Zn) GATA and AP2/EREBP (except one) were down-regulated in the sterile buds, whereas the majority of the MYB, MADS and C2C2(Zn) Dof transcription factors were up-regulated in comparison to the fertile buds.

Overview of differentially expressed genes (DEGs) involved in diverse metabolic pathways. DEGs were selected for the metabolic pathways using MapMan software. The colored red and green boxes indicate down-regulated and up-regulated genes, respectively. The scale bar represent fold change values
Candidate DEGs associated with male-sterility
The bioinformatics analysis gave us a deeper insight into the differentially expressed genes (DEGs) between the sterile (AKCMS11) and fertile genotypes (AKPR303). In this present investigation, several subsets of differentially expressed genes were identified which are potentially related to pollen development (Fig. 10).

Heat map of expression profiles of some of the focused genes related to cytoplasmic male-sterility. Color from red to green, indicated that the FPKM values were small to large, red color indicates low level of gene expression and green color indicates high level of gene expression
Putative gene related to pollen development
In flowering plants, pollen development is a complicated and sequential process of sexual reproduction. Arabidopsis thaliana is considered as the model plant to study putative genes related to pollen development [58]. To understand the mechanism related to male sterility, we queried all the 3167 DEGs in the TAIR database (http://www.arabidopsis.org). Additionally, all the 3167 DEGs were further compared with the Arabidopsis database (http://www.arabidopsis.org) to identify homologs using OrthoDB v10 [66] (Additional File 2: Table S11). In our study, 34 DEGs were found to be homologs of Arabidopsis genes associated with the development of pollen (Table 3). We found 10 DEGs involved in regulation of pollen development, among which 1 DEG encoding EXCESS MICROSPOROCYTES 1 (EMS1), 1 DEG encoding PHD-finger transcription factor MALE STERTILITY1 (MS1), 2 DEGs encoding callose synthase 7, 1 DEG encoding AUXIN-RESPONSIVE FACTOR (ARF17), 4 DEGs encoding CYTOCHROME P450-like, and 1 DEGs encoding aspartic protease. 5 DEGs were up-regulated and 5 were down-regulated in the sterile AKCMS11 in comparison to fertile AKPR303. 3 DEGs encoding polygalacturonase (PG) and endo-1,3- beta-glucosidase-like were involved in pollen cell wall remodeling. Total 5 DEGs encoding arabinogalactan glycoproteins (AGPs) and GPI-anchored like protein were involved in pollen tube growth. Single stress induced DEG was present, encoding late embryogenesis abundant protein (LEA) showing significant down-regulation in AKCMS11 with respect to AKPR303. We identified, 3 DEGs involved in the cell division process. Additionally, 12 DEGs with related functions were also identified in this study (Table 3).
Transcription factors (TFs) are of major importance as they regulate gene expression in plants. Alterations in the gene expression are associated with modifications in the expression of transcription factors [67]. In our study, there were 16 DEGs representing transcription factors, 9 DEGs were down-regulated and 7 were up-regulated in the AKCMS11. The 9 down-regulated DEGs included 2 bHLH family, 2 C2H2zinc-finger, 2 basic-leucine zipper transcription factor (bZIP), 1 dof zinc-finger and 2 NAC domain-containing transcription factors. Among the 7 up-regulated DEGs, 1 were bHLH, 2 MYB transcription factors, 2 basic-leucine zipper transcription factors (bZIP), 1 NAC domain-containing transcription factorsand 1 WRKY transcription factor. (Additional file 2: Table S5).
Metabolic pathways
Carbon fixation and energy metabolism are the most predominant metabolic pathways which are primarily responsible for furnishing the requirement of energy and carbon sources in the plants [68]. In the present study, 12 DEGs were observed to be participating in carbohydrate metabolism, 6 DEGs related to glycolysis/gluconeogenesis pathway and 6 DEGs related to pentose and glucuronate interconversions, respectively (Table 4). With respect to glycolysis, 4 DEGs were down-regulated and 2 DEGs were up-regulated in AKCMS11 in comparison to AKPR303. Additionally, among the 6 DEGs representing pentose and glucuronate interconversions, 3 DEGS were down-regulated and 3 DEGs were up-regulated in AKCMS11 (Table 4).
In this study, 2 DEGs participated in the Tricarboxylic acid cycle (TCA), 1 DEG encoding malate dehydrogenase and second encoding succinate-CoA synthetase (Table 4). Also, we detected 7 DEGs involved in oxidative phosphorylation, majority of the DEGs showed down-regulation with only 2 DEGs showing up-regulation in the AKCMS11 (Table 4). We examined a total of 15 DEGs involved in the elimination of reactive oxygen species (ROS), including 2 DEGs encoding peroxidase-like, 1 DEG encoding superoxide dismutase and 2 DEG encoding peroxiredoxin-2B-like, along with 1 DEG encoding glutathione S-transferase and 1 DEG encoding for glutathione peroxidase. Additionally, there were 5 DEGs encoding for monothiol glutaredoxin, and 1 DEG each encoding for ascorbate oxidase, thioredoxin and glutaredoxin. 10 of the DEGs showed down-regulation in AKCMS11 in comparison to AKPR303 (Table 4).
Confirmation of DEGs by qRT-PCR
We randomly selected 10 genes for qRT-PCR analysis with the aim to validate the expression profiles between AKCMS11 and AKPR303 obtained by RNA-Seq. The list of DEGs specific primers used for qRT-PCR analysis is listed in Additional file 2: Table S12. The DEGs selected for qRT-PCR confirmation were related to callose synthase 7-like, phosphoenolpyruvate carboxykinase, polygalacturonase, aspartic protease, late embryogenesis abundant (LEA), uncharacterized mitochondrial protein, glutathione S-transferase F10, calmodulin binding receptor-like protein, mitotic spindle assembly checkpoint protein (MAD1), and pentatricopeptide repeat-containing protein. Although the relative gene expression detected by RNA-Seq analysis was higher than those detected by qRT-PCR, the overall expression patterns were similar (Fig. 11 and Table 5). Linear regression analysis was performed and it revealed positive correlation (R2 = 0.9508) between the qRT-PCR and DGE fold change gene expression ratios, indicating that the expression of all the 10 genes depicted by qRT-PCR analysis was in agreement with the DGE data analysis (Fig. 12). These findings confirm the reliability of the Illumina RNA-Seq data and its analysis.

qRT-PCR validation of differentially expressed unigenes. S denotes the sterile AKCMS11 and F denotes the fertile AKPR303. The primary vertical axis represents qRT-PCR expression values and secondary vertical axis represents RNA-Seq expression values

Linear regression analysis of the fold change of the gene expression ratios between DEG data and qRT-PCR. 10 primers were selected for qRT-PCR analysis to confirm the accuracy and reproducibility of the Illumina expression. Scatterplots were generated by the log2 expression ratios from DGE sequencing data (x- axis) and qRT-PCR data (y-axis)
Discussion
Cytoplasmic male-sterility (CMS) is considered as an important tool for hybrid seeds production in many plant species. To elucidate the underlying mechanism governing CMS in pigeon pea, the present study was undertaken. Comparative transcriptome sequencing and analysis of the floral buds from sterile (AKCMS11) and fertile (AKPR303) pigeon pea were performed. In total, 3167 genes showed differential expression between the CMS and the fertile restorer. Based on the functional annotation, we identified some candidate DEGs related to pollen development and metabolism which were previously reported to be involved in male-sterility (Fig. 13).

Some of the candidate differentially expressed genes (DEGs) involved in cytoplasmic male-sterility sterility in pigeon pea. The upper arrows indicate up-regulation of the DEGs, and the down arrows represent down-regulation of the DEGs. Each color box depicts an individual metabolism
DEGs involved in pollen development potentially related to CMS
We identified 34 pollen development related genes which were found to have homologs in Arabidopsis (Table 3). Pollen formation is a critical developmental mechanism in plants which is highly dependent on co-ordinated metabolic pathways [69]. Male-sterility occurs due to the aberrant functioning of the genes responsible for tapetum development, pollen formation, callose degradation and pollen wall development [70]. In our findings, several DEGs with a potential role in pollen development were observed (Table 3). Pollen development is an intricate and programmed process which takes place within specialized structures called anthers from reproductive cells (microsporocytes). These microspores (pollens) are dependent on the sporophytic tissues for their development, finally leading to the release of functional pollens for fertilization in flowering plants [71]. In Arabidopsis, EXCESS MICROSPOROCYTES 1 (EMS1) proposes a leucine-rich repeat receptor protein kinase (LRR-RPK) which is actively involved in the determination and differentiation of the tapetal cells and the microsporocytes during plant reproduction. It was observed that the anthers of the EMS mutant (EMS1) exhibited excessive microsporocytes despite lacking the tapetum, producing non-functional pollens and leading to male-sterility [22]. We also observed 1 DEG (Ccajan_10186_c0_g1_i1) encoding leucine-rich repeat receptor protein kinase EMS1, down-regulated (4.53-fold) in the AKCMS11 respectively and the gene thus may also be related to pollen development and fertility in pigeon pea.
During the process of pollen development, tapetum is of great significance as it provides nourishment to the developing microspores leading to pollen wall formation [72]. In Arabidopsis, MALE STERILITY1 (MS1) and MALE SERILITY2 (MS2) genes regulate the stages of tapetum maturation and pollen wall biosynthesis and ending with the release of viable pollen [27, 73]. The MS1 gene exhibits homology to PHD-finger transcription factor critical for tapetum and pollen development [74]. A PHD-finger transcription factor MALE STERTILITY1 (MS1) conferring male-sterility was identified in barley [75]. In this study, 1 DEG (Ccajan_34303_c0_g2_i2) encoding PHD-finger transcription factor MALE STERTILITY1 (MS1) was detected with 4.27-fold down-regulation in the AKCMS11. In the MS1 mutant, immediately after the release of microspores from the tetrads, sudden preterm degeneration of the tapetum and pollen takes place leading to inviable pollen formation and cause male-sterility [27]. Similar results were reported in male-sterile tomato mutants ms3, ms15, ms5 and ms1035 due to dysfunctional regulation of the tapetum [57, 76, 77].
In flowering plants, pollens are secured within the pollen grains and physically protected by the complex pollen cell walls. The pollen mother cells (PMC) synthesize a polysaccharide, callose (β-1,3-glucan) and the outer protective callose wall segregates the newly developed microspore and restricts them from merging with the neighbouring tapetum tissues, giving the unique tetrad shape [78]. Finally, the callose wall surrounding the microspores degenerates, releasing them into the anther locule [79]. The callose synthase enzyme is required for the synthesis of a temporary callose wall around the developing microspore, twelve callose synthase (cals) genes, are reported in Arabidopsis [80, 81]. The callose synthase 5 (cals5) mutants showed the absence of callose deposition around the microspores, confirming the importance of this enzyme with reduced fertility [38]. Interestingly, we noticed 2 DEGs; Ccajan_44775c0_g1_i1 and Ccajan_52234_c0_g1_i1 representing callose synthase with 5.45 and 6.38-fold down-regulation in the AKCMS11 in comparison to the AKPR303 thus featuring as a prominent candidate in pollen development. These results are consistent with the earlier reports in watermelon and soybean [55, 56]. Additionally, primexine a microfibrillar polysaccharide matrix, is important for the normal development of functional pollen and is regulated by an AUXIN-RESPONSIVE FACTOR (ARF17). In Arabidopsis, ARF17 mutant displayed abnormal callose wall formation with no primexine and pollen abortion [82]. A single DEG Ccajan_41968_c0_g1_i1 for ARF72 displayed reduced expression of 6.21-fold in the AKCMS11 thus featuring as a prominent candidate in pollen development.
Sporopollenin is the key constituent of the outer rigid exine wall of the microspores and genes like CYTOCHROME P450 (CYP703A2), CYTOCHROME P450 (CYP704B1) and acyl-coA synthetase 5 (acos5) are responsible for regulating the biosynthesis of sporopollenin. Down-regulation of the CYP703A2 and CYP704B1 genes depicted the reduced level of sporopollenin, an absence of outer exine layer with unique stripped surface and impaired pollens in Arabidopsis [34, 35, 73]. Surprisingly, 4 genes corresponding to CYTOCHROME P450-like were drastically over-expressed in the AKCMS11 in comparison to AKPR303. For example, Ccajan_1528_c0_g3_i1 (8.30-fold), Ccajan_88538_c0_g1_i1 (8.23-fold), Ccajan_16351_c0_g1_i1 (8.81-fold) and Ccajan_55694_c0_g9_i1 (6.36-fold). This finding is supported by a previous study in cotton where they proposed that since these genes are involved in the synthesis of sporopollenin, their up-regulation must have ended up in immoderate accumulation sporopollenin and ultimately male-sterility [83]. Over-expression of this gene may be related to pollen abortion.
The developing microspores (pollen grains) within the anthers are enclosed by a layer of cells known as tapetum [84, 85], it is of considerable importance as it provides the necessary nourishment to the developing pollens and secretes components for the outer exine and regulates the pollen wall formation [86, 87]. During the subsequent later stages of pollen development, tapetum experiences a regulated disintegration by programmed cell death (PCD) and releases all the cellular components required for pollen wall formation into the anther locule [88,89,90]. Any delay in the timely regulation of tapetum PCD results in pollen lethality and male-sterility [87]. Previously, a PCS1 gene encoding an anti-cell-death factor known as an aspartic protease which governs PCD in Arabidopsis was reported [91]. The over-expression of the PCS1 gene produced excessive aspartic protease which further inhibited anther dehiscence and resulted in male-sterility. In this study, 1 DEG (Ccajan_24040_c0_g3_i1) was 10.42-fold over-expressed in the sterile AKCMS11 than the fertile AKPR303. The higher expression in the sterile line could have delayed the PCD of the tapetum, which is a must for anther dehiscence and ultimately lead to pollen sterility in pigeon pea. A similar observation was seen in soybean which therefore stands in support of the present finding [56].
In this study, 3 DEGs related to cell wall modification were identified and all were strongly down-regulated in the AKCMS11 as compared to the AKPR303. These genes encode cell wall-degrading enzymes such as polygalacturonase (PG), known for hydrolyzing pectin and polygalactouronic acid and endo-1,3-beta-glucosidase-like, cellulose- hydrolyzing enzyme. In an earlier report in Arabidopsis, QRT1 and QRT2 mutant produced non-functional pollen due to the inefficiency of polygalacturonase in pectin degradation around the microspores [92]. A single DEG (Ccajan_83966_c0_g1_i1) representing polygalacturonase (PG) showed 8.85-fold down-regulation with 2 DEGs (Ccajan_61890__c0_g1_i1 and Ccajan_7875_c0_g1_i1) encoding endo-1,3-beta-glucosidase-like were 4.87-fold and 2.20-fold down-regulated in the AKCMS11, respectively. The results were consistent with the findings in tomato and cotton [11, 93]. These under-expressed genes might be involved in pollen abortion.
Arabinogalactan glycoproteins (AGPs) are present in different cells and tissues of the higher plants and are actively involved in the growth and reproduction [94]. In Arabidopsis, AGP6, AGP11 and Fasciclin-like arabinogalactan proteins (FLA3) are the key genes involved in the synthesis of AGPs [95, 96] and are in primarily expressed in the pollen grains and pollen tube with their participation in the microspore development. Alterations in the genes result in obstruction in pollen tube growth and defective pollen release from the anthers leading to reduced fertility [95, 96]. In our results, we identified 3 differentially expressed transcripts encoding arabinogalactan proteins (AGPs) such as GPI-anchored like protein and Fasciclin-like arabinogalactan 11 (FLA11). Of the 2 DEGs encoding GPI-anchored like proteins, 1 was 5.0-fold down-regulated (Ccajan_38216_c1_g2_i3) in CMS genotype while the other gene showed up-regulation (7.25-fold). The DEG Ccajan_37653_c0_g4_i5 represents FLA11 with 9.14-fold down-regulation in CMS line with respect to the sterile line. Similar results were observed in watermelon, cotton and sesame [55, 61, 97].
Calcium ions (Ca2+) ions are known for their considerable physiological significance in plants [98]. They are actively involved in plant reproduction, specifically participating in pollen germination and pollen tube growth. It was earlier reported that sufficient Ca2+ ions result in normal pollen germination while any changes in the concentration (higher or lower) restrain pollen germination and tube elongation [99, 100]. In the present study, 2 DEGs Ccajan_47280_c0_g1_i1 and Ccajan_2910_c0_g1_i1 representing calmodulin-binding receptor-like proteins were identified, 1 showing 4.55-fold down-regulation and the other 7.53-fold down-regulation. Altered expression of these genes in the sterile AKCMS11 might have inhibited pollen germination and tube growth. Similar reports in watermelon, cotton and kenaf support the present results [55, 93, 101].
During the final stages of pollen maturation, the pollen dehydrates, and the mature pollen is ready for germination [102]. Late embryogenesis abundant (LEA) proteins are involved in conferring desiccation tolerance to the pollen. In lily, LP28 is a pollen-specific LEA-like protein is known which slowly accumulates in the developing pollen and generously present in the germinated pollen after hydration with their probable role in pollen maturation and pollen tube growth [103]. In this investigation, only 1 DEG was found related to LEA-like protein, for example, Ccajan_52697_c0_g1_i1 showing 4.57-fold down-regulation in the CMS line. Abnormal expression of this gene may be related to pollen development. The results are in accordance with the results in watermelon and soybean [55, 56].
Male gametogenesis is a complex process in flowering plants with rounds of mitotic and meiotic cell divisions leading to the development of male gametophyte. Any abnormality in the genes regulating the cell division can cause male-sterility [104]. In this study, 2 mitotic spindle assembly checkpoint protein (MAD1) genes and 1 cell division protein gene displayed abnormal expression in the sterile AKCMS11 in comparison to fertile restorer AKPR303. A similar finding was reported in watermelon [55] suggesting it might be involved in pollen development. The findings strongly indicate the potential role of these genes responsible for pollen development leading to CMS in pigeon pea.
DEGs encoding transcription factors potentially related to CMS
Transcription factors (TF’s) are proteins which bind to cis-regulatory specific sequences in the promoter region of the target genes and regulate the gene expression [105]. Any alterations in the expression of transcription factors lead to change in the gene expression causing substantial transformation during plant development [106]. In Arabidopsis, 608 transcription factors (TFs) belonging to 34 families have been described to be participating in pollen development [107]. In our study, 9 and 7 DEGs encoding transcription factors showed down-regulation and up-regulation in the sterile AKCMS11 in comparison to its fertile restorer, respectively. MYB proteins constitute a large and functionally diverse family with a major role in plant-specific processes [108]. In Arabidopsis, AtMYB32 and AtMYB103 are expressed in the tapetum and facilitate the development of pollen by controlling tapetum development, callose dissolution and exine formation [28, 29]. Any changes in their expression can lead to early tapetal degeneration, distorted pollens and male-sterility [28, 109]. In this study, 2 DEGs Ccajan_30780_c0_g1_i2 and Ccajan_36059_c0_g1_i1 encoding MYB transcription factors were identified, both up-regulated (3.59-fold and 3.11-fold) in the AKCMS11. Thus pointing towards the possible role of these differentially expressed genes in pollen development.
Transcription factors like bHLH are significant as they synchronize the process of normal tapetum development in the anthers. bHLH- type transcription factors such as DYT1 and AMS in Arabidopsis [110] and UNDEVELOPED TAPETUM (OsUDT1) in rice [111] are known to be crucial for proper tapetum development and finally leading pollen development. In our data, we observed 3 DEGs encoding bHLH transcription factors among those 2 DEGs showed down-regulation in the CMS line. For instance, Ccajan_47215_c0_g1_i1 was 7.14-fold down-regulated and Ccajan_68741_c0_g2_i1 was 2.08-fold down-regulated. Similar findings were seen in radish, sesame and kenaf [54, 97, 101].
In Arabidopsis, WRKY transcription factors like WRKY2, WRKY34 and WRKY 27 are known for their candidate role in pollen development during male gametogenesis [112]. Over-expression of WRKY 27 showed limited pollen viability leading to male-sterility [113]. In our study, there was 1 DEG which was over-represented in the AKCMS11; Ccajan_36686_c0_g1_i4 (10.05-fold up-regulated), this was in accordance with findings in soybean and sesame [56, 97]. These DEGS recognized in this study might have a possible role causing CMS in this crop.
DEGs involved in carbohydrate metabolism potentially related to CMS
In plants, carbohydrates in the form of sugars and starch are the substrate for energy supply and growth. Sufficient sugar level is highly important for the anther development and subsequently during pollen maturation sugars get converted into starch which later serves as the energy source for pollen germination [114, 115]. Dysfunctional sugar metabolism can significantly influence pollen development producing impaired pollens and results in male-sterility [116]. This condition was observed in pepper and cabbage [117, 118]. In this study, altered expression of genes involved in glycolysis, gluconeogenesis and pentose and glucuronate interconversions were detected in the AKCMS11 in comparison to the AKPR303 (Table 4). Total 6 DEGs were involved in glycolysis and gluconeogenesis, for instance Ccajan_36409_c0_g2_i1, Ccajan_34307_c0_g1_i1, Ccajan_37543_c0_g2_i4, Ccajan_34880_c0_g2_i1, Ccajan_39910_c0_g1_i1 and Ccajan_88886_c0_g1_i1, all of them were significantly down-regulated in the sterile AKCMS11 except for 1 DEG which showed up-regulation (Table 4). 6 DEGs were related to pentose and glucuronate interconversions, 3 of which were down-regulated and 3 DEGs were up-regulated in AKCMS11. These genes involved in carbohydrate metabolism exhibited lower expression suggesting the low availability of sugars during pollen development resulting in non-functional pollens and male-sterility. The results were in accordance with the previous report in soybean, chili pepper and cotton [56, 59, 62].
DEGs involved in the tricarboxylic acid cycle (TCA) and oxidative phosphorylation potentially related to CMS
Mitochondria are the main energy yielding-power houses of the cells with a number of important metabolic pathways, involving TCA cycle, respiratory electron transfer, and oxidative phosphorylation [119,120,121]. Pollen formation is an energy-utilizing process which is highly dependent on mitochondrial respiration and fermentation for satisfying their energy demands [102, 122]. We identified, 2 DEGs participating in the TCA cycle, Ccajan_64232_c0_g1_i1 encoding malate dehydrogenase and Ccajan_35485_c0_g1_i1 encoding succinate-CoA synthetase (Table 4). According to the previous reports in cotton, Brassica napus and cabbage lower expression of TCA related genes causes pollen abortion and are responsible for CMS [93, 123, 124]. These observations suggest that alterations in the energy-yielding metabolic pathways inhibits pollen development and may be the cause of CMS.
The mitochondria generate energy in the form of adenosine triphosphate (ATP) by utilizing two major pathways, the respiratory electron transfer chain and oxidative phosphorylation [125]. In this study, 7 genes related to respiratory electron transfer chain and oxidative phosphorylation were detected showing differential expression in the sterile AKCMS11 in comparison to the fertile AKPR303 (Table 4). Cytochrome c oxidase (cox) is the key enzyme involved in the last stage of the mitochondrial electron transport chain and considered as a major regulating site during ATP synthesis [126]. Previous literature on rice and cotton proposed the importance of cox genes in conferring CMS [127, 128]. Two DEGs Ccajan_13673_c0_g1_i1 and Ccajan_41295_c0_g1_i1 encoding cytochrome c oxidase subunit 6b and subunit 1 were 5.98-fold and 6.46-fold down-regulated in the AKCMS11 (Table 4). This was in accordance with the earlier reports in chili pepper, cotton, Brassica napus and beet [59, 93, 123, 129]. Hence, altered expression of cox 6 might have resulted in decreased ATP synthesis and disrupted the pollen formation in this crop.
DEGs involved in the elimination of reactive oxygen species (ROS) potentially related to CMS
In plants cells, reactive oxygen species (ROS) are the by-products of aerobic respiration, constantly being generated in the chloroplast, mitochondria, and peroxisomes and are maintained by ROS scavenging system [130]. The ROS are free radicals including superoxide (O2−), hydrogen peroxide (H2O2), and malondialdehyde (MDA) and their excessive accumulation in the cells can instigate cell apoptosis [131]. Previously, excessive accumulation of the ROS species and remarkable reduction of the antioxidative defense systems in the anthers of the cotton CMS line caused male-sterility [132]. In rice, the abnormal concentration of the ROS in the mitochondria induced acute oxidative stress during pollen development and leading to male-sterility [133]. In this study, 15 DEGs involved in the elimination of ROS were detected with 5 DEGs showing over-expression and 10 DEGs down-expression in the sterile AKCMS11 line in comparison to the fertile AKPR303 (Table 4). Down-regulation of the genes encoding oxygen scavenging enzymes in AKCMS11 triggered the excessive accumulation of ROS in the sterile buds, leading to pollen abortion [134]. In broccoli, higher-expression of glutathione S-transferase (GST) gene induced excessive ROS and resulted in male-sterility [70]. Interestingly, we came across 1 DEG encoding glutathione S-transferase (GST) for instance, Ccajan_36196_c0_g2_i3, showing significant higher-expression 2.41-fold in AKCMS11 in comparison to AKPR303. We presume, expression alterations of the genes involved in ROS scavenging probably induced pollen abortion in pigeon pea.
Conclusions
To our knowledge, this is the first reported attempt at transcriptome sequencing and comparative analysis of the floral buds of A2 CMS system (Cajanus scarabaeoides L.) derived cytoplasmic male-sterile (AKCMS11) and its fertility restorer (AKPR303) in pigeon pea. The differential gene expression analysis showed 3167 differentially expressed genes (DEGs) between AKCMS11 and AKPR303, among which 1432 were up-regulated and 1390 were down-regulated in the AKCMS11 when compared to the AKPR303. Based on the functional annotation of these DEGs in GO, KEGG databases and metabolic pathway analysis combined with previous studies, we hereby conclude that male-sterility of AKCMS11 is probably related to abnormalities of some of the putative DEGs in functional and metabolic pathways, such as involved in pollen development, encoding transcription factors, elimination of reactive oxygen species (ROS), carbon metabolism, oxidative phosphorylation, tricarboxylic acid cycle (TCA), etc. Further studies of these crucial genes will focus on clearly understanding their functions in conferring male-sterility. Therefore, this present report will be highly informative and provide support for future studies in elucidating the molecular basis related to cytoplasmic male-sterility in pigeon pea.
Methods
Plant material and RNA isolation
Cytoplasmic male-sterile AKCMS11 (Cajanus scarabaeoides cytoplasm) and fertility restorer line AKPR303 (Cajanus cajan cytoplasm) [47] of pigeon pea were used in this present investigation as described earlier [135]. The seeds for both the lines were procured from Pulse Research Unit, Dr. Panjabrao Deshmukh Krishi Vidyapeeth, Krishi Nagar, Akola, Maharashtra, India. The seeds were sown directly in the soil and the plants were maintained under natural conditions in the experimental greenhouse at ICAR- NIPB, New Delhi. Flower buds of different-sized were collected from two independent plants (representing replications) of each AKCMS11 and AKPR303 lines, respectively. The young buds were harvested, frozen in liquid nitrogen and stored at − 80 °C. Total RNA was extracted from the flower buds of both the lines (in replicates) using Spectrum Plant Total RNA Kit (Sigma-Aldrich, USA) following the manufacturer’s protocol.
Pollen fertility test
Pollen analysis was performed to evaluate pollen fertility in the male-sterile (AKCMS11) and fertile (AKPR303) buds. Anthers from the flower buds of AKCMS11 and AKPR303 were removed and squashed on a glass slide in 1% acetocarmine dye. Then the glass slide was examined under a microscope.
Library preparation and RNA sequencing
Total RNA was checked on 1% denaturing agarose gel and quantified using Nanodrop spectrophotometer (Thermo Fischer Scientific, USA). The RNA quality was analyzed with RNA 6000 NanoAssay kit (Agilent Technologies, USA) using Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Total four RNA libraries were constructed from the buds of sterile and fertile lines (two replicates each) using Truseq RNA Sample prep kit (Illumina, USA.) following the manufacturer’s protocol. The libraries were then sequenced by Illumina paired-end sequencing technology on Illumina HiSeq 1000 sequencer.
RNA-Seq data analysis and de novo transcriptome assembly
Sequencing raw data was received in the FASTQ format. The per base sequence quality of the raw reads from sterile (41,531,572; 68,068,793) and fertile (29,618,927; 42,957,061) lines respectively were determined by FastQC version 0.11.4 (http://www.bioinformatics.babraham.ac.uk/projects/) [136]. Rcorrector software was also employed for checking the k-mer content in the data [137]. Raw reads with PHRED quality score < 30 and shorter than 50 bp in length were filtered and not considered for further transcriptome assembly. Trimmomatic software version 0.36 [138] was used for trimming of the raw reads to remove the adaptor sequence followed by filtering of low-quality reads (quality score ≤ 5) and reads with the unknown ‘N’ base (‘N’ ratio ≥ 10%) to finally obtain the filtered clean reads. Next, following the read orientation (R1 and R2) the total clean reads from the sterile and fertile lines were concatenated together by using an in-house shell script. A de novo assembly of the pooled clean reads was achieved by Trinity software version v2.2.0 [139] using default parameters. Further, Bowtie2 aligner was used for the validation of the transcriptome assembly by mapping back the filtered reads onto the assembled transcriptome. Then, we evaluated the completeness of our final transcriptome assembly with Benchmarking Universal Single-Copy Orthologs tool version 3 (BUSCO) [140, 141]. BUSCO is ideal for assessing assembly completeness, because the expectation that these genes are found in a given genome as single copies is reasonable from an evolutionary point of view [140,141,142,143]. For the present analysis, we used the transcriptome assessment mode with the eukaryote lineage database (eukaryota_orthoDB9) and viridiplantae lineage database (viridiplantae_orthoDB10). Subsequently, non-redundant unigenes were obtained by employing the CD-HIT software version 4.6.1with identity parameter of 95% (http://weizhongli-lab.org/cd-hit/) [144]. Followed by removal of rRNAs and other housekeeping non-coding RNAs (tRNAs, snRNAs, snoRNAs etc.) by BLASTN search against Rfam database [145]. GC content of the total unigenes was calculated via GC-Profile tool (http://tubic.tju.edu.cn/GC-Profile/) [146].
Functional annotation of the unigenes
Functional annotation of the assembled unigenes was achieved by BLASTX search against Nr (NCBI non-redundant protein sequences), Virdiplantae, GO (Gene ontology) and KEGG (Kyoto encyclopedia of genes and genomes) databases with a stringent cut-off E-value of 1e− 3 [147,148,149]. Unigenes annotation against Nr database was performed with the aid of Blast2GO tool version 3.1.3 (http://www.blast2go.org) [150] up to 10 qualified blast hits for each unigene. Among the 10 blast hits the one with higher sequence similarity was accounted as a significant match for each unigene. Transcription factors were identified with the help of PlantTFcat online tool (http://plantgrn.noble.org/PlantTFcat) [73].
Differential gene expression analysis
Firstly, RSEM software [74] was used for calculating the read count of each uniquely mapped transcript on to the assembled transcriptome (for each sample) in terms of FPKM (fragments per kilo base per million). Then, differentially expressed genes (DEGs) between the sterile and fertile genotypes (two replications for each) were detected by DESeq2 [61]). Strict criteria of P-value and FDR (False discovery rate) ≤ 0.05 and Log2 FC (Fold change ratio sterile vs fertile) ≥ 2 and ≤ − 2 (for up-regulated and down-regulated genes) were used for determining significant differentially expressed genes (DEGs).
GO and KEGG enrichment analysis of the DEGs
Gene ontology (GO) annotation of all the differentially expressed genes (DEGs) was performed using Singular Enrichment Analysis Tool in AgriGO software version 2.0 (http://bioinfo.cau.edu.cn/agriGO/) with default parameters [151]. Based on GO enrichment results, the ones with P-value ≤0.05 were considered as significant GO terms. Then, the functional GO classification of all the DEGs was performed by WEGO software (wego.genomics.org.cn/) and the results were categorized into three independent hierarchies; biological processes, molecular functions and cellular components. KEGG (Kyoto encyclopedia of genes and genomes) pathway enrichment analysis of all the DEGs was conducted by KEGG Mapper (http://www.genome.jp/kegg/mapper.html) with default parameters. The DEGs were compared to the entire reference gene set by hypergeometric tests to identify the pathways enriched for DEGs and a P-value ≤ 0.05 indicated significant pathway enrichment. Additionally, MapMan version 3.3. 0 [65] was used for metabolic pathway analysis of the DEGs by searching against Arabidopsis thaliana TAIR database (http://www.arabidopsis.org).
Validation of DEGs by quantitative real time-PCR (qRT-PCR)
Total RNA was isolated from the buds of sterile (AKCMS11) and restorer (AKPR303) line as mentioned earlier. First-strand cDNA was prepared from 2 μg of total RNA using RevertAid First strand cDNA synthesis kit (Thermo Fischer Scientific, USA) according to manufacturer’s instructions. The first strand cDNA was then further diluted ten times for quantitative real-time PCR (qRT-PCR) reaction. qRT-PCR was performed using KAPA SYBR FAST qPCR Master Mix (2X) (KAPA Biosystems, USA) on ABI PRISM 7500 Real-Time PCR System (Applied Biosystems, USA). qRT-PCR was performed in three independent technical replicates for each genotype. The reaction mixture (20 μl) contained 3 μl of cDNA, 10 μl of 2X SYBR green qPCR master mix, 0.20 μl of primers (forward and reverse) and final volume was adjusted with nuclease-free water. PCR conditions were 94 °C for 3 min followed by 40 cycles of 94 °C for 3 s, 60 °C for 15 s and 72 °C for 15 s. α-tubulin was used as the internal reference gene. The relative level of gene expression was calculated using the 2(−ΔΔCt) algorithm, fertile restorer (AKPR303) was used as a calibrator.
Availability of data and materials
The dataset generated and/or analyzed during the current study are available in the NCBI SRA repository (SRX3740150, SRX3740151, SRX3740153, SRX3740152) for the sterile and fertile buds.(https://www.ncbi.nlm.nih.gov/sra/SRX3740153[accn]; https://www.ncbi.nlm.nih.gov/sra/SRX3740152[accn]; https://www.ncbi.nlm.nih.gov/sra/SRX3740151[accn]; https://www.ncbi.nlm.nih.gov/sra/SRX3740150[accn].
Abbreviations
- ACOS :
-
acyl-coA synthetase
- AGPs:
-
Arabinogalactan glycoproteins
- AMS :
-
ABORTED MICROSPORES
- AP2:
-
APETALA2
- ARF :
-
AUXIN RESPONSIVE FACTOR
- ATP:
-
Adenosine triphosphate
- bHLH:
-
Basic helix-loop-helix protein
- BLAST:
-
Basic Local Alignment Search Tool
- bp:
-
Base pair
- bZIP:
-
Basic leucine zipper
- C2H2 :
-
Cys2/His2
- Cals:
-
Callose synthase
- cDNA:
-
Complementary DNA
- CMS:
-
Cytoplasmic male-sterility
- cox:
-
Cytochrome c oxidase
- CYP703A2 :
-
CYTOCHROME P450
- CYP704B1 :
-
CYTOCHROME P450
- DEGs:
-
Differentially expressed genes
- DYT :
-
DYSFUNCTIONAL TAPETUM
- EMS :
-
EXCESS MICROSPOROCYTES
- ERF:
-
Ethylene-responsive factor
- FDR:
-
False discovery rate
- FLA:
-
Fasciclin-like arabinogalactan
- FPKM:
-
Fragments per kilo base per million
- GO:
-
Gene Ontology
- GST:
-
Glutathione S-transferase
- H2O2 :
-
Hydrogen peroxide
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LAP :
-
Polyketide synthase
- LEA:
-
Late embryogenesis abundant
- Log2 FC:
-
Fold change
- LRR-RPK:
-
Leucine-rich repeat receptor protein kinase
- MAD:
-
Mitotic spindle assembly checkpoint protein
- MDA:
-
Malondialdehyde
- MS :
-
MALE STERILITY
- NCBI:
-
National Center for Biotechnology Information
- Nr:
-
NCBI non-redundant protein
- NZZ :
-
NOZZEL
- O2 :
-
Superoxide
- orfs :
-
Open reading frames
- PCD:
-
Programmed cell death
- PCR:
-
Polymerase chain reaction
- PG:
-
Polygalacturonase
- PMC:
-
Pollen mother cells
- qRT-PCR:
-
Quantitative real-time PCR
- Rf :
-
Restorer of fertility
- RNA-Seq:
-
RNA Sequencing
- ROS:
-
Reactive oxygen species
- RPG :
-
RUPTURED POLLEN GRAIN
- SPL :
-
SPOROCYTELESS
- TCA:
-
Tricarboxylic acid cycle
- TF:
-
Transcription factors
- TPD :
-
TAPETUM DETERMINANT
- UDT :
-
UNDEVELOPED TAPETUM
References
Eckardt NA. Cytoplasmic male sterility and fertility restoration. Plant Cell. 2006;18:515–7.
Schnable PS, Wise RP. The molecular basis of male sterility and fertility restoration. Trends Plant Sci. 1998;3:175–80.
Bentolila S, Alfonso AA, Hanson MR. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. ProcNatlAcad Sci. 2002;99:10887–92.
Chen L, Liu YG. Male sterility and fertility restoration in crops. Annu Rev Plant Biol. 2014;65:579–606.
Mackenzie SA. The Influence of Mitochondrial Genetics on Crop Breeding Strategies. In: Janick J, editor. Plant Breeding Reviews: Wiley; 2010. p. 115–38.
Levings CS. The Texas cytoplasm of maize: cytoplasmic male sterility and disease susceptibility. Science. 1990;250:942–7.
Levings CS. Thoughts on cytoplasmic male sterility in cms-T maize. Plant Cell. 1993;5:1285–90.
Nivison HT, Sutton CA, Wilson RK, Hanson MR. Sequencing, processing, and localization of the petunia CMS-associated mitochondrial protein. Plant J. 1994;5:613–23.
Young EG, Hanson MR. A fused mitochondrial gene associated with cytoplasmic male sterility is developmentally regulated. Cell. 1987;50:41–9.
Budar F, Pelletier G. Male sterility in plants: occurrence, determinism, significance and use. CR AcadSci III. 2001;324:543–50.
Omidvar V, Mohorianu I, Dalmay T, Zheng Y, Fei Z, Pucci A, et al. Transcriptional regulation of male-sterility in 7B-1 male-sterile tomato mutant. PLoS One. 2017;12:1–19.
Shao LH, Zheng XW, Yi DX, Li C. Comparative sequence and expression analysis of tapetum specific male sterility related genes in Medicago truncatula. GenetMol Res. 2016;15:2.
Chase CD. Cytoplasmic male sterility: a window to the world of plant mitochondrial-nuclear interactions. Trends Genet. 2007;23:81–90.
Hanson MR, Bentolila S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell. 2004;16:154–69.
Bergman P, Edqvist J, Farbos I, Glimelius K. Male-sterile tobacco is plays abnormal mitochondrial atp1 transcript accumulation and reduced floral ATP/ADP ratio. Plant Mol Biol. 2000;42:531–44.
Sabar M, Gagliardi D, Balk J, Leaver CJ. ORFB is a subunit of F1F0-ATP synthase: insight into the basis of cytoplasmic male sterility in sunflower. EMBO Rep. 2003;4:1–6.
Yui R, Iketani S, Mikami T, Kubo T. Antisense inhibition of mitochondrial pyruvate dehydrogenase E1asubunit in anther tapetum causes male sterility. Plant J. 2003;34:57–66.
Liu X, Huang J, Parameswaran S, Ito T, Seubert B, Auer M, et al. The SPOROCYTELESS/NOZZLE gene is involved in controlling stamen identity in Arabidopsis. Plant Physiol. 2009;151:1401–11.
Ito T, Wellmer F, Yu H, Das P, Ito H, Alves-Ferreira M, et al. The homeotic protein AGAMOUS controls microsporogenesis by regulation of SPOROCYTELESS. Nature. 2004;430:356–60.
Schiefthaler U, Balasubramanian S, Sieber P, Chevalier D, Wisman E, Schneitz K. Molecular analysis of NOZZLE, a gene involved in pattern formation and early sporogenesis during sex organ development in Arabidopsis thaliana. ProcNatlAcad Sci. 1999;96:11664–9.
Yang WC, Ye D, Xu J, Sundaresan V. The SPOROCYTELESS gene of Arabidopsis is required for initiation of sporogenesis and encodes a novel nuclear protein. Genes Dev. 1999;13:2108–17.
Zhao DZ, Wang GF, Speal B, Ma H. The EXCESS MICROSPOROCYTES1 gene encodes a putative leucine-rich repeat receptor protein kinase that controls somatic and reproductive cell fates in the Arabidopsis anther. Genes Dev. 2002;16:2021–31.
Jia G, Liu X, Owen HA, Zhao D. Signaling of cell fate determination by the TPD1 small protein and EMS1 receptor kinase. ProcNatlAcad Sci. 2008;105:2220–5.
Yang SL, Xie LF, Mao HZ, Puah CS, Yang WC, Jiang L, et al. TAPETUM DETERMINANT1 is required for cell specialization in the Arabidopsis anther. Plant Cell. 2003;15:2792–04.
Xu J, Yang C, Yuan Z, Zhang D, Gondwe MY, Ding Z, et al. The ABORTED MICROSPORES regulatory network is required for post-meiotic male reproductive development in Arabidopsis thaliana. Plant Cell. 2010;22:91–107.
Sorensen AM, Kröber S, Unte US, Huijser P, Dekker K, Saedler H. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003;33:413–23.
Yang C, Vizcay-Barrena G, Conner K, Wilson ZA. MALE STERILITY1 is required for Tapetal development and Pollen Wall biosynthesis. Plant Cell. 2007;19:3530–48.
Preston J, Wheeler J, Heazlewood J, Li SF, Parish RW. AtMYB32 is required for normal pollen development in Arabidopsis thaliana. Plant J. 2004;40:979–95.
Zhang ZB, Zhu J, Gao JF, Wang C, Li H, Li H, et al. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007;52:528–38.
Aarts MGM, Keijzer CJ, Stiekema WJ, Pereira A. Molecular characterization of the CER7 gene of Arabidopsis lnvolved in Epicuticular wax biosynthesis and pollen fertility. Am SocPlant Physiol. 1995;7:2115–27.
Ariizumi T, Hatakeyama K, Hinata K, Sato S, Kato T, Tabata S, et al. A novel male-sterile mutant of Arabidopsis thaliana, faceless pollen-1, produces pollen with a smooth surface and an acetolysis-sensitive exine. Plant Mol Biol. 2003;53:107–16.
Guan YF, Huang XY, Zhu J, Gao JF, Zhang HX, Yang ZN. RUPTURED POLLEN GRAIN1, a member of the MtN3/saliva gene family, is crucial for Exine pattern formation and cell integrity of microspores in Arabidopsis. Plant Physiol. 2008;147:852–63.
Morant M, Jorgensen K, Schaller H, Pinot F, Moller BL, Werck-Reichhart D, et al. CYP703 is an ancient cytochrome P450 in land plants catalyzing in-chain hydroxylation of Lauric acid to provide building blocks for Sporopollenin synthesis in pollen. Plant Cell Online. 2007;19:1473–87.
Dobritsa AA, Shrestha J, Morant M, Pinot F, Matsuno M, Swanson R, et al. CYP704B1 is a long-chain fatty acid ω-hydroxylase essential for Sporopollenin synthesis in pollen of Arabidopsis. Plant Physiol. 2009;151:574–89.
de Azevedo SC, Kim SS, Koch S, Kienow L, Schneider K, McKim SM, et al. A novel fatty acyl-CoA Synthetase is required for pollen development and Sporopollenin biosynthesis in Arabidopsis. Plant Cell. 2009;21:507–25.
Kim SS, Grienenberger E, Lallemand B, Colpitts CC, Kim SY, Souza Cde A, et al. LAP6/POLYKETIDE SYNTHASE A and LAP5/POLYKETIDE SYNTHASE B Encode Hydroxyalkyl α-Pyrone Synthases Required for Pollen Development and Sporopollenin Biosynthesis in Arabidopsis thaliana. Plant Cell. 2010;22:4045–66.
Dobritsa AA, Lei Z, Nishikawa SI, Urbanczyk-Wochniak E, Huhman DV, Preuss D, et al. LAP5 and LAP6 encode anther-specific proteins with similarity to Chalcone synthase essential for pollen Exine development in Arabidopsis. Plant Physiol. 2010;153:937–55.
Dong X, Hong Z, Sivaramakrishnan M, Mahfouz M, Verma DPS. Callose synthase (CalS5) is required for exine formation during microgametogenesis and for pollen viability in Arabidopsis. Plant J. 2005;42:315–28.
Fu YJ, Liu W, Zu YG, Tong MH, Li SM, Yan MM, et al. Enzyme assisted extraction of luteolin and apigenin from pigeon pea [Cajanus cajan (L.) Millsp.] leaves. Food Chem. 2008;111:508–12.
Saxena KB, Kumar RV, Rao PV. Pigeon pea nutrition and its improvement. In: Basra AS, Randhawa IS, editors. Quality improvement in field crops. New York: Food Products Press; 2002. p. 227–60.
Odeny DA. The potential of pigeonpea (Cajanus cajan (L.) Millsp.) in Africa. Nat Resour Forum. 2007;31:297–05.
Nadarajan N, Ram SG, Petchiammal KI. Fertility restoration studies in short duration redgram (Cajanus cajan (L.) mill spp.) hybrids involving cgms system. Madras Agric J. 2008;95:320–7.
Saxena KB, Sharma D. Pigeonpea genetics. In: Nene YL, et al., editors. Pigeonpea. Wellingford: CAB International; 1990. p. 137–58.
Saxena KB. A novel source of CMS in pigeon pea derived from Cajanus reticulatus. Indian J of Genet Plant Breed. 2013;73:259–63.
Saxena KB, Sultana R, Mallikarjuna N, Saxena RK, Kumar RV, Sawargaonkar SL, et al. Male-sterility systems in pigeonpea and their role in enhancing yield. Plant Breed. 2010;129:125–34.
Saxena KB, Nadarajan N. Prospects of pigeonpea hybrids in Indian agriculture. Elec J Plant Breed. 2010;1:1107–17.
Meshram MP, Patil AN. Genetics of Fertility Restoration in A2 Cytoplasm based Hybrids of Pigeonpea (Cajanus cajan (L.) Millsp.). Int J CurrMicrobiol App Sci. 2018;6:565–71.
Choudhary AK, Singh IP. A study on comparative fertility restoration in A2 and A4 cytoplasm and its implication in breeding hybrid pigeonpea [Cajanus cajan (L.) Millspaugh]. Am J Plant Sci. 2015;6:385–91.
Sinha P, Saxena KB, Saxena RK, Singh VK, Suryanarayana V, SK SKCV, MAVS KK, Khan AW, Varshney R. Association of Gene with Cytoplasmic Male Sterility in Pigeonpea. Plant Genome. 2015;8. https://doi.org/10.3835/plantgenome2014.11.0084.
Schuster SC. Next-generation sequencing transforms today’s biology. Nat Methods. 2008;5:16–8.
Ansorge WJ. Next-generation DNA sequencing techniques. New Biotechnol. 2009;25:195–03.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, et al. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol. 2010;152:1787–95.
Mei S, Liu T, Wang Z. Comparative Transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int J Mol Sci. 2016;17:42.
Rhee SJ, Seo M, Jang YJ, Cho S, Lee GP. Transcriptome profiling of differentially expressed genes in floral buds and flowers of male sterile and fertile lines in watermelon. BMC Genomics. 2015;16:914.
Li J, Han S, Ding X, He T, Dai J, Yang S, et al. Comparative Transcriptome analysis between the cytoplasmic male sterile line NJCMS1A and its maintainer NJCMS1B in soybean (Glycine max (L.) Merr.). PLoS One. 2015;10:1–18.
Jeong HJ, Kang JH, Zhao M, Kwon JK, Choi HS, Bae JH, et al. Tomato Male sterile 1035 is essential for pollen development and meiosis in anthers. J Exp Bot. 2014;65:6693–09.
An H, Yang Z, Yi B, Wen J, Shen J, Tu J, et al. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genomics. 2014;15:2–11.
Liu C, Ma N, Wang PY, Fu N, Shen HL. Transcriptome sequencing and De novo analysis of a cytoplasmic male sterile line and its near-isogenic restorer line in chili pepper (Capsicum annuum L.). PLoS One. 2013;8:e65209. https://doi.org/10.1371/journal.pone.0065209
Wu J, Zhang M, Zhang B, Zhang X, Guo L, Qi T, et al. Genome-wide comparative transcriptome analysis of CMS-D2 and its maintainer and restorer lines in upland cotton. BMC Genomics. 2017;18:454.
Suzuki H, Rodriguez-Uribe L, Xu J, Zhang J. Transcriptome analysis of cytoplasmic male sterility and restoration in CMS-D8 cotton. Plant Cell Rep. 2013;32:1531–42.
Wei M, Song M, Fan S, Yu S. Transcriptomic analysis of differentially expressed genes during anther development in genetic male sterile and wild type cotton by digital gene-expression profiling. BMC Genomics. 2013;14:97.
Zheng BB, Wu XM, Ge XX, Deng XX, Grosser JW, Guo WW. Comparative transcript profiling of a male sterile Cybrid Pummelo and its fertile type revealed altered gene expression related to flower development. PLoS One. 2012;7:1–13.
Dai X, Sinharoy S, Udvardi M, Zhao PX. PlantTFcat: An online plant transcription factor and transcriptional regulator categorization and analysis tool. BMC Bioinformatics. 2013;14:321.
Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37:914–39.
Kriventseva EK, et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. NAR. 2018. https://doi.org/10.1093/nar/gky1053.
Yanagisawa S. Transcription factors in plants: physiological functions and regulation of expression. J Plant Res. 1998;111:363–71.
Wu Z, Cheng J, Qin C, Hu Z, Yin C, Hu K. Differential proteomic analysis of anthers between cytoplasmic male sterile and maintainer lines in Capsicum annuum L. Int J Mol Sci. 2013;14:22982–96.
Blackmore S, Wortley AH, Skvarla JJ, Rowley JR. Pollen wall development in flowering plants. New Phytol. 2007;174:483–98.
Pei X, Jing Z, Tang Z, Zhu Y. Comparative transcriptome analysis provides insight into differentially expressed genes related to cytoplasmic male sterility in broccoli (Brassica oleracea var. italica). SciHortic. 2017;217:234–42.
Gómez JM, Perfectti F, Abdelaziz M, Lorite J, Muñoz-Pajares AJ, Valverde J. Evolution of pollination niches in a generalist plant clade. New Phytol. 2015;205:440–53.
Heslop-Harrison J, Dickinson HG. Time relationships of sporopollenin synthesis associated with tapetum and microspores in Lilium. Planta. 1969;84:199–14.
Aarts MGM, Hodge R, Kalantidis K, Florack D, Wilson ZA, Mulligan BJ, et al. The Arabidopsis MALE STERILITY 2 protein shares similarity with reductases in elongation/condensation complexes. Plant J. 1997;12:615–23.
Wilson ZA, Morroll SM, Dawson J, Swarup R, Tighe PJ. The Arabidopsis MALE STERILITY1 (MS1) gene is a transcriptional regulator of male gametogenesis, with homology to the PHD-finger family of transcription factors. Plant J. 2001;28:27–39.
Gómez JF, Wilson ZA. A barley PHD finger transcription factor that confers male sterility by affecting tapetal development. Plant Biotechnol J. 2014;12:765–77.
Rick CM. Genetics and development of nine male-sterile tomato mutants. Hilgardia. 1948;18:599–33.
Gorman SW, McCormick S, Rick C. Male sterility in tomato. CRC Crit Rev Plant Sci. 1997;16:31–53.
Scott RJ, Spielman M, Dickinson HG. Stamen structure and function. Plant Cell. 2004;16:46–60.
Ariizumi T, Toriyama K. Genetic regulation of Sporopollenin synthesis and pollen Exine development. Annu Rev Plant Biol. 2011;62:437–60.
Hong Z, Delauney AJ, Verma DPS. A cell plate-specific Callose synthase and its interaction with Phragmoplastin. Plant Cell. 2001;13:755–68.
Enns LC, Kanaoka MM, Torii KU, Comai L, Okada K, Cleland RE. Two callose synthases, GSL1 and GSL5, play an essential and redundant role in plant and pollen development and in fertility. Plant Mol Biol. 2005;58:333–49.
Yang J, Tian L, Sun MX, Huang XY, Zhu J, Guan YF, et al. AUXIN RESPONSE FACTOR17 is essential for Pollen Wall pattern formation in Arabidopsis. Plant Physiol. 2013;162:720–31.
Wu Y, Min L, Wu Z, Yang L, Zhu L, Yang X, et al. Defective pollen wall contributes to male sterility in the male sterile line 1355A of cotton. Sci Rep. 2015;5:1–8.
Scott RJ. Pollen exine: the sporopollenin enigma and the physics of pattern. In: Scott RJ, Stead AD, editors. Molecular and cellular aspects of plant reproduction. Cambridge: Cambridge University Press; 1994. p. 49–81.
Mascarenhas JP. The male gametophyte of flowering plants. Plant Cell. 1989;1:657–64. https://doi.org/10.1105/tpc.1.7.657.
Maheshwari P. An introduction to the embryology of angiosperms. New York: McGraw-Hill Book Co Inc; 1950.
Kawanabe T, Ariizumi T, Kawai-Yamada M, Uchimiya H, Toriyama K. Abolition of the tapetum suicide program ruins microsporogenesis. Plant Cell Physiol. 2006;47:784–7.
Rogers LA, Dubos C, Surman C, Willment J, Cullis IF, Mansfield SD, et al. Comparison of lignin deposition in three ectopic lignification mutants. New Phytol. 2005;168:123–40.
Parish RW, Li SF. Death of a tapetum: a programme of developmental altruism. Plant Sci. 2010;178:73–89.
Vizcay-Barrena G, Wilson ZA. Altered tapetal PCD and pollen wall development in the Arabidopsis ms1 mutant. J Exp Bot. 2006;57:2709–17.
Ge X, Dietrich C, Matsuno M, Li G, Berg H, Xia Y. An Arabidopsis aspartic protease functions as an anti-cell-death component in reproduction and embryogenesis. EMBO Rep. 2005;6:282–8.
Rhee SY, Somerville CR. Tetrad pollen formation in quartet mutants of Arabidopsis thaliana is associated with persistence of pectic polysaccharides of the pollen mother cell wall. Plant J. 1998;15:79–88.
Kong X, Liu D, Liao X, Zheng J, Diao Y, Liu Y, et al. Comparative analysis of the cytology and transcriptomes of the cytoplasmic male sterility line H276A and its maintainer line H276B of cotton (Gossypium barbadense L.). Int J Mol Sci. 2017;18:2240.
Pennell RI, Roberts K. Sexual development in the pea is presaged by altered expression of arabinogalactan protein. Nature. 1990;334:547–9.
Levitin B, Richter D, Markovich I, Zik M. Arabinogalactan proteins 6 and 11 are required for stamen and pollen function in Arabidopsis. Plant J. 2008;56:351–63.
Li J, Yu M, Geng LL, Zhao J. The fasciclin-like arabinogalactan protein gene, FLA3, is involved in microspore development of Arabidopsis. Plant J. 2010;64:482–97.
Liu H, Tan M, Yu H, Li L, Zhou F, Yang M, et al. Comparative transcriptome profiling of the fertile and sterile flower buds of a dominant genic male sterile line in sesame (Sesamum indicum L.). BMC Plant Biol. 2016;16:250.
Bush DS. Calcium regulation in plant cells and its role in signaling. Annu Rev Plant Physiol Plant Mol Biol. 1995;46:95–122.
Brewbaker JL, Kwack BH. The essential role of calcium ion in pollen germination and pollen tube growth. Am J Bot. 1963;50:859–65.
Bednarska E. The effect of exogenous Ca2+ ions on pollen grain germination and pollen tube growth investigations with 45Ca2+ together with verapamil, La3+, and ruthenium red. Sex Plant Reprod. 1988;2:53–8.
Chen P, Ran S, Li R, Huang Z, Qian J, Yu M, et al. Transcriptome de novo assembly and differentially expressed genes related to cytoplasmic male sterility in kenaf (Hibiscus cannabinus L.). Mol Breed. 2014;34:1879–91.
Taylor LP, Hepler PK. Pollen germination and tube growth. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:461–91.
Mogami N, Shiota H, Tanaka I. LP28, a lily pollen-specific LEA-like protein, is located in the callosic cell wall during male gametogenesis. Sex Plant Reprod. 2002;15:57–63.
Chen C, Marcus A, Li W, Hu Y, Calzada JPV, Grossniklaus U, et al. The Arabidopsis ATK1 gene is required for spindle morphogenesis in male meiosis. Development. 2002;129:2401–9.
Guilfoyle TJ. The structure of plant gene promoters. In: Setlow JK, editor. Genetic engineering. New York: Plenum Press; 1997. p. 15–47.
Grotewold E, Chamberlin M, Snook M, Siame B, Butler L, Swenson J, et al. Engineering secondary metabolism in maize cells by ectopic expression of transcription factors. Plant Cell. 1998;10:721–40.
Honys D, Twell D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004;5:R85.
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15:573–81.
Higginson T, Li SF, Parish RW. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. 2003;35:177–92.
Zhang W, Sun Y, Timofejeva L, Chen C, Grossniklaus UMH. Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development. 2006;133:3085–95.
Jung KH, Han MH, Lee YS, Kim YW, Hwang I, Kim MJ, et al. Rice Undeveloped Tapetum1 is a major regulator of early Tapetum development. Plant Cell. 2005;17:2705–22.
Guan Y, Meng X, Khanna R, LaMontagne E, Liu Y, Zhang S. Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 2014;10:e1004384.
Mukhtar MS, Liu X, Somssich IE. Elucidating the role of WRKY27 in male sterility in Arabidopsis. Plant Signal Behav. 2017;12:e1363945.
Oliver SN, Dennis ES, Dolferus R. ABA regulates apoplastic sugar transport and is a potential signal for cold-induced pollen sterility in rice. Plant Cell Physiol. 2007;48:1319–30.
Datta R, Chamusco KC, Chourey PS. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant Physio. 2002;130:1645–56.
Mamun EA, Alfred S, Cantrill LC, Overall RL, Sutton BG. Effects of chilling on male gametophyte development in rice. Cell Biol Int. 2006;30:583–91.
Li YY, Wei YY, Zhang RH, Lu J. Studies on the metabolism of male sterile pepper in the development of microspore. ActaAgriculturaeBoreali- OccidentalisSinica. 2006;15:134–7.
Wang YF, Hu CQ, Lin ZG, Li FY, Jin JK, et al. ActaHort Sin. 1984;3:7.
Siedow J, Umbach A. Plant mitochondrial Electron transfer and molecular biology. Plant Cell. 1995;7:821–31.
Barrientos A. Yeast models of human mitochondrial diseases. IUBMB Life. 2003;55:83–95.
Logan DC. The mitochondrial compartment. J Exp Bot. 2006;57:1225–43.
Dickinson DB. Germination of lily pollen: respiration and tube growth. Science. 1965;150:30–1.
Du K, Liu Q, Wu X, Jiang J, Wu J, Fang Y, et al. Morphological structure and Transcriptome comparison of the cytoplasmic male sterility line in Brassica napus (SaNa-1A) derived from somatic hybridization and its maintainer line SaNa-1B. Front Plant Sci. 2016;7:1313.
Wang S, Wang C, Zhang XX, Chen X, Liu JJ, Jia XF, et al. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol Biochem. 2016;105:224–32.
Millar AH, Whelan J, Soole KL, Day DA. Organization and regulation of mitochondrial respiration in plants. Annu Rev Plant Biol. 2011;62:79–104.
Kadenbach B, Hüttemann M, Arnold S, Lee I, Bender E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free RadicBiol Med. 2000;29:211–21.
Liu Y, Wang X, Wang Y, Zhuo D. Structural variance analysis of mitochondria coI and coII genes from normal and cytoplasmic male-sterile varieties of rice Oryza sativa. ActaGenetica Sin. 1988;15:348–54.
Huang J, Yang P, Li B, Huang JL, Yang P, et al. Study on the activity of several enzymes of cytoplasmic male-sterile cotton line Jin A. Cotton Science. 2004;16:229–32.
Ducos E, Touzet P, Boutry M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001;26:171–80.
Sharma P, Jha AB, Dubey RS, Pessarakli M. Reactive oxygen species, oxidative damage, and Antioxidative defense mechanism in plants under stressful conditions. J Bot. 2012;2012:1–26.
Maxwell DP, Nickels R, McIntos L. Evidence of mitochondrial involvement in the transduction of signals required for the induction of genes associated with pathogen attack and senescence. Plant J. 2002;29:269–79.
Jiang P, Zhang X, Zhu Y, Zhu W, Xie H, Wang X. Metabolism of reactive oxygen species in cotton cytoplasmic male sterility and its restoration. Plant Cell Rep. 2007;26:1627–34.
Wan C, Li S, Wen L, Kong J, Wang K, Zhu Y. Damage of oxidative stress on mitochondria during microspores development in Honglian CMS line of rice. Plant Cell Rep. 2007;26:373–82.
Hanson MR. Plant mitochondrial mutations and male-sterility. Annu Rev Genet. 1991;25:461–86.
Junaid A, Kumar H, Rao AR, Patil AN, Singh NK, Gaikwad K. Unravelling the epigenomic interactions between parental inbreds resulting in an altered hybrid methylome in pigeonpea. DNA Res. 2018;25:361–73.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Song L, Liliana F. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. Giga Science. 2015;4:48.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat Biotechnol. 2011;29:644–52.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2017;35:543–8.
Waterhouse RM, Zdobnov EM, Kriventseva EV. Correlating traits of gene retention, sequence divergence, duplicability and essentiality in vertebrates, arthropods and fungi. Genome Biol Evol. 2011;3:75–86.
Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–D65.
Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9.
Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018;46:D335–42.
Gao F, Zhang CT. GC-profile: a web-based tool for visualizing and analyzing the variation of GC content in genomic sequences. Nucleic Acids Res. 2006;34:686–91.
Devani RS, Sinha S, Banerjee J, Sinha RK, Bendahmane A, Banerjee AK. De novo transcriptome assembly from flower buds of dioecious, gynomonoecious and chemically masculinized female Coccinia grandis reveals genes associated with sex expression and modification. BMC Plant Biol. 2017;17:241.
Fu Y, Esselink GD, Visser RGF, Tuyl JM, Arens P. Transcriptome analysis of Gerbera hybrida including in silico confirmation of defense genes found. Front Plant Sci. 2016;7:247.
Morandin C, Pulliainen U, Bos N, Schultner E. De novo transcriptome assembly and its annotation for the black ant Formica fusca at the larval stage. Sci Data. 2018;5:180282.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, et al. AgriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–9.
Acknowledgements
The authors are grateful to the Director, ICAR- National Institute for Plant Biotechnology (NIPB), New Delhi, India for providing facilities. The authors also thank the other members of our research groups and collaborators for technical assistance and discussions.
Funding
This work was supported by funds from Department of Science & Technology (DST), New Delhi, India and ICAR- Network Project on Transgenicsin Crops, New Delhi, India. The funders had no role in the design of the study, collection, data analysis, data interpretation and manuscript writing.
Author information
Authors and Affiliations
Contributions
SS1 carried out the experiment, prepared the cDNA library for sequencing, sequencing run, qRT-PCR validation, GO, KEGG pathway analysis, identification of transcription factors and wrote the manuscript. SS2 performed the read generation, de novo assembly, differential expression analysis, homology search and annotation. TK was involved in cDNA library preparation and sequencing run. DN and PKC assisted in the bioinformatics analysis. SS1, SS2, TK, DN and PKC were involved in result interpretation, analysis and finalization of the manuscript. NKS and ARR contributed in data analysis and manuscript finalization. SS3 contributed in manuscript finalization. KG conceived the study, designed the experiments, and coordinated the work. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Rcorrector ouput for k-mer content in the raw paired-end data. Table S2. Bowtie2 alignment statistics in sterile (AKCMS11) and fertile restorer (AKPR303). Figure S1. Pearson’s correlation coefficient between two replicates in sterile AKCMS11 (left) and fertile restorer AKPR303 (right). Figure S2. Graph representing per base sequence quality. a) Sterile AKCMS11 (replicates 1 and 2). b) Fertile AKPR303 (replicates 1 and 2).
Additional File 2: Table S1.
Sequences with significant BLASTX hits against nr protein database. Table S2. Sequences with significant BLASTX hits against Virdiplantae database. Table S3. Gene Ontology classification of the assembled unigenes. Table S4. KEGG classification of Cajanus cajan unigenes. Table S5. Transcription factors identified in transcriptome of Cajanus cajan. Table S6. List of differentially expressed genes between sterile AKCMS11 and fertile AKPR303 in pigeon pea. Table S7. List of significantly enriched GO terms in down-regulated DEGs. Table S8. List of significantly enriched GO terms in up-regulated DEGs. Table S9. Summary of KEGG annotations for down and up-regulated DEGs. Table S10. TAIR database BLASTX results of pigeon pea, 951 DEGs in MapMan pathway analysis. Table S11. OrthoDB homologs search of 3167 pigeon pea DEGs against TAIR database. Table S12. List of qRT-PCR primers used in this study.
Additional file 3: Figure S1.
Gene ontology classification of the assembled unigenes. The Y-axis indicates the number of unigenes and X-axis indicates the GO categories. Figure S2 Functional classification of KEGG pathways of the assembled unigenes. The KEGG pathways were classified into six functional categories: A- Metabolism; B- Genetic Information Processing; C- Environmental Information Processing; D- Cellular Processes; E- Organismal Systems; F- Human Diseases. The Y-axis represents the KEGG metabolic pathways. The X-axis represents number of unigenes annotated in that particular pathway. Figure S3 Hierarchical tree graph of over-represented GO terms in the biological process category of down-regulated DEGS. Boxes in the graph represent GO terms labeled according to their GO ID, term definition, and statistical information. Significant terms (adjusted P ≤ 0.05) are in color (red, orange, or yellow), while non-significant terms are shown as white boxes. In the diagram, the degree of color saturation of a box is positively correlated with the enrichment level of the term. Solid, dashed, and dotted lines represent two, one, and zero enriched terms at both ends connected by the line, respectively. The rank direction of the graph is set from top to bottom. Figure S4 Hierarchical tree graph of over-represented GO terms in the biological process category of up-regulated DEGS. Boxes in the graph represent GO terms labeled according to their GO ID, term definition, and statistical information. Significant terms (adjusted P ≤ 0.05) are in color (red, orange, or yellow), while non-significant terms are shown as white boxes. In the diagram, the degree of color saturation of a box is positively correlated with the enrichment level of the term. Solid, dashed, and dotted lines represent two, one, and zero enriched terms at both ends connected by the line, respectively. The rank direction of the graph is set from top to bottom.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Saxena, S., Sahu, S., Kaila, T. et al. Transcriptome profiling of differentially expressed genes in cytoplasmic male-sterile line and its fertility restorer line in pigeon pea (Cajanus cajan L.). BMC Plant Biol 20, 74 (2020). https://doi.org/10.1186/s12870-020-2284-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-2284-y