
Abstract
Background
Triticum timopheevii (2n = 4x = 28; AtAtGG), is an important source for new genetic variation for wheat improvement with genes for potential disease resistance and salt tolerance. By generating a range of interspecific hybrid lines, T. timopheevii can contribute to wheat’s narrow gene-pool and be practically utilised in wheat breeding programmes. Previous studies that have generated such introgression lines between wheat and its wild relatives have been unable to use high-throughput methods to detect the presence of wild relative segments in such lines.
Results
A whole genome introgression approach, exploiting homoeologous recombination in the absence of the Ph1 locus, has resulted in the transfer of different chromosome segments from both the At and G genomes of T. timopheevii into wheat. These introgressions have been detected and characterised using single nucleotide polymorphism (SNP) markers present on a high-throughput Axiom® Genotyping Array. The analysis of these interspecific hybrid lines has resulted in the detection of 276 putative unique introgressions from T. timopheevii, thereby allowing the generation of a genetic map of T. timopheevii containing 1582 SNP markers, spread across 14 linkage groups representing each of the seven chromosomes of the At and G genomes of T. timopheevii. The genotyping of the hybrid lines was validated through fluorescence in situ hybridisation (FISH). Comparative analysis of the genetic map of T. timopheevii and the physical map of the hexaploid wheat genome showed that synteny between the two species is highly conserved at the macro-level and confirmed the presence of inter- and intra-genomic translocations within the At and G genomes of T. timopheevii that have been previously only detected through cytological techniques.
Conclusions
In this work, we report a set of SNP markers present on a high-throughput genotyping array, able to detect the presence of T. timopheevii in a hexaploid wheat background making it a potentially valuable tool for marker assisted selection (MAS) in wheat pre-breeding programs. These valuable resources of high-density molecular markers and wheat-T. timopheevii hybrid lines will greatly enhance the work being undertaken for wheat improvement through wild relative introgressions.
Similar content being viewed by others

Background
Wheat yields are plateauing in many countries at a time when production needs to be increased to feed the ever-growing population [3, 7]. A key factor in the plateauing of yields observed is the relatively small amount of genetic variation available in the gene pool of hexaploid wheat that can be used to develop new superior high yielding varieties adapted to the changing environment. However, unlike wheat its wild relatives provide a vast and almost untapped source of genetic variation that could be exploited to provide a step change in wheat breeding programmes (for reviews see [16, 29, 54]).
Triticum timopheevii is a tetraploid (2n = 4x = 28; AtAtGG) member of the triticeae. An allopolyploid, the progenitor species are thought to be the same as those for Triticum turgidum and Triticum aestivum, with Triticum urartu contributing the At genome [11] and an Aegilops speltoides-like species the G genome [12, 44]. A study on chromosome pairing by Rodriguez et al. [52] suggested that the G genome of T. timopheevii and the S genome of Ae. speltoides are closer together than either are to the B genome of T. aestivum. Meiotic analysis by Feldman [14] of F1 hybrids between T. timopheevii and T. aestivum showed that the chromosomes of the B and G genomes paired to form bivalents, etc., only 30% of the time. In contrast the A and At genomes, appear to be more closely related as in the same study by Feldman [14] the chromosomes from these genomes paired 70% of the time.
It is thought that the timopheevii wheats (including T. timopheevii) arose from a separate hybridisation event to T. turgidum and T. aestivum, based on the presence of different species-specific translocations. T. timopheevii contains the 4AtL/5AtL translocation found in T. turgidum and T. aestivum but also a 6At/1G/4G cyclic translocation [23]. However, like wheat, T. timopheevii carries a pairing control locus located on chromosome 5G, which acts to suppress homoeologous recombination. Chromosome 5B of wheat carries the Ph1 locus which also restricts pairing to homologous chromosomes. However, although this indicates a possible relationship, the Ph1 locus in wheat is thought to be stronger than that found on chromosome 5G of T. timopheevii [45].
T. timopheevii has been shown to be a valuable source of new disease resistance genes including leaf rust resistance [5, 31, 57, 59], stem rust resistance [1, 26, 47, 48, 63], powdery mildew resistance [21, 24, 49] and Fusarium head blast resistance [4, 6, 13]. In addition to the resistance genes, T. timopheevii has been shown to contain genetic variation for salt tolerance [67] and protein content [43, 69].
One of the most effective ways of introducing new genetic variation from wild relatives into wheat is by the generation of introgressions via homoeologous recombination and indeed introgressions previously produced between T. aestivum and T. timopheevii have shown the potential value of this approach [2, 17, 51]. A major bottleneck for the introduction of genetic variation into wheat from its wild relatives has been the difficulty in detecting and characterising introgressions. However, King et al. [27] reported the development of an Axiom® array, composed of circa 35 K single nucleotide polymorphism (SNP) markers, able to detect polymorphisms between wheat and 10 wild relatives including T. timopheevii. To date, this array has enabled the large-scale detection and characterisation of introgressions in wheat from Ambylopyrum muticum, Ae. speltoides, Thinopyrum bessarabicum and T. urartu [18, 19, 27, 28].
This paper reports the development of genome wide introgressions from T. timopheevii into wheat. At the Nottingham/BBSRC Wheat Research Centre (WRC) the Axiom® Wheat-Relative Genotyping Array was used to both detect and characterise the introgression lines produced, via the generation of a genetic linkage map of T. timopheevii consisting of 1582 SNP markers spread across both the At and G genomes. The genetic linkage map was also validated using fluorescence in situ hybridisation (FISH).
Results
Generation of wheat-T. timopheevii introgressions through homoeologous recombination in the absence of the Ph1 locus
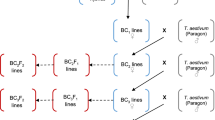
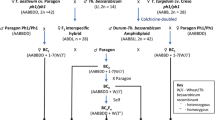
In order to generate wheat-T. timopheevii introgressions lines (Fig. 1) a total of 1947 crosses were made leading to the generation of 12,883 crossed seed and 7018 self-seed. The number of crosses made and seed set in each generation is shown in Table 1. In total, 150 interspecific F1 seeds were generated by crossing T. timopheevii with wheat having a mutation at the Ph1 locus resulting in homoeologous recombination between the chromosomes of the two species. Of these F1 hybrids, 86 were selected at random to produce the subsequent generations. Only 73 of these plants germinated, of which 25 (34%) produced seed when backcrossed to wild type Paragon in order to produce the BC1 generation. In contrast, the number of plants (again derived from randomly selected seed) that set seed in the BC1, BC2, BC3 and BC4 generations was close to or at 100%.

Wheat/T. timopheevii crossing programme undertaken in this work
The lowest frequencies of fertility were observed in the crosses between Paragon ph 1/ph1 x T. timopheevii and between the F1 x wild type Paragon with only 5 and 16% of crossed ears setting seed respectively. In contrast, the BC1, BC2, BC3 and BC4 generations showed much higher levels of fertility with 94, 93, 99 and 100% of crossed ears setting seed respectively. In addition, the average number of seed set per crossed ear was also considerably lower in the interspecific cross between Paragon ph 1/ph1 x T. timopheevii and the F1 x wild type, i.e. 0.3 and 0.4% respectively as compared to the BC1, BC2, BC3 and BC4 generations, i.e. 6.5, 10, 15 and 15% respectively. Self-fertilised seed was obtained from each generation with the exception of the F1.
Detection of introgressions using the Axiom® Wheat-Relative Genotyping Array
To detect introgressions from T. timopheevii in the interspecific hybrid lines, a 35 K Axiom® Wheat-Relative Genotyping Array was used. Of the SNPs on the array, 19,460 showed polymorphism between Paragon and T. timopheevii (Table 2). DNA from 344 individuals of BC1-BC4 populations were screened with the array. Genotype calls were generated, and the sample call rate ranged from 88.8 to 99.9% with an average of 99%. The lowest call rates were obtained for the three T. timopheevii samples with an average of 89.6%. The scores for each SNP was classified into one of six cluster patterns using Affymetrix software. Only those classified as Poly High Resolution (PHR; 3256) and thus, considered to be of optimum quality were used for genetic mapping. A majority of the polymorphic SNPs (10,537) were classified under the 'No minor homozygous allele' category. These were not considered to be ideal for genotyping a segregating backcross population due to the presence of heterozygous alleles, in at least one parent, for these SNPs.
Genetic mapping of T. timopheevii chromosomes
Joinmap [61] was used to analyse the PHR SNPS which resulted in the generation of 14 linkage groups with a total of 1528 SNPs. Each linkage group was assigned to either the At or the G genome of T. timopheevii depending on the BLAST analysis of the markers against the wheat genome. If the linkage group had most top hits in the A genome of wheat, it was assigned to the At genome of T. timopheevii. In contrast, if most top hits in the BLAST analysis were from the B genome of wheat then the linkage group was assigned to the G genome of T. timopheevii. Assuming synteny between wheat and T. timopheevii, the linkage groups were assigned to the same homoeologous group in the latter as indicated by the BLAST results in wheat, i.e. markers which produced a top hit on chromosome group 5 in wheat were assigned to linkage group 5 in T. timopheevii. The genetic map of the At genome of T. timopheevii was composed of 484 SNPs (Fig. 2a) and that of the G genome was composed of 1044 SNPs (Fig. 2b). The number of SNPs assigned to each of the 7 linkage groups of the At and G genomes varied between groups as is shown in Table 2. In both the At and G genomes, linkage group 5 had the highest number of SNPs (linkage group 5At = 20.5% and linkage group 5G = 20.9% of the SNPs on the genetic maps of the At and G genomes respectively) and linkage group 4 had the lowest number (linkage group 4At = 7.6% and linkage group 4G = 11.6% of the SNPs on the genetic maps of the At and G genomes respectively). The total length of the genetic map of the At genome was 384.5 cM and the total length of the map of the G genome was 290.7 cM. The length in cM of each linkage group varied considerably. For example, linkage group 4 of the At genome was only 15.3 cM in contrast to linkage group 5 which was 90.8 cM. For the G genome, linkage group 1 was 16.6 cM in contrast to linkage group 2 which was 76.5 cM (Table 2).

Genetic linkage map of (a) T. timopheevii At genome (b) T. timopheevii G genome
Genotyping indicated that, in total, 276 potential introgressions had been generated between wheat and T. timopheevii; 141 between wheat and the At genome and 135 between wheat and the G genome (Fig. 2). Marker analysis revealed that recombination between the genomes of wheat and those of T. timopheevii was not restricted to the gametes of the F1, i.e. recombination between the genomes of the two species also occurred during gametogenesis in the backcross progenies, e.g. linkage group 1At shows recombination between BC1 and BC2B and linkage group 5G shows recombination between BC2A and BC3B (Fig. 3).

Marker assisted selection of T. timopheevii introgressions across a family of back-cross lines. The red colour, in the GGT bar diagrams, is used to represent the presence of a T. timopheevii introgression, while the blue colour represents wheat (these diagrams cannot be used to assess which wheat chromosomes the T. timopheevii segments have recombined with). Recombination can be seen to have occurred in linkage group 1At between the BC1 and BC2 generations. Eventually the markers allow selection of a BC3F2 line containing a single T. timopheevii segment from linkage group 5G
The number of introgressions retained decreased considerably in each backcross population with one exception. For example, 71% of the BC1 genotypes carried introgressions from linkage group 1 of the At genome as compared to 50, 29 and 21% in the BC2, BC3 and BC4 generations respectively (Table 3). However, in contrast, linkage group 2 from the G genome which was present in 94% of the BC1 genotypes was still present in 75, 74 and 70% in the BC2, BC3 and BC4 generations, respectively (Table 3).
Fluorescence in situ hybridisation (FISH) validates the molecular markers
To confirm the presence of segments indicated by the genotyping, FISH was carried out on selected individuals with large T. timopheevii segments using oligos pSc119.2–1 [36] and the Afa family [39]. Firstly, it was confirmed that the FISH karyotype of the T. timopheevii accession used in this work was similar to a previously published karyotype for T. timopheevii (Fig. 4b; [37]). Differences between the FISH signals of wheat (Fig. 4a; [56]) and T. timopheevii were subsequently used to identify T. timopheevii segments in a wheat background (Figs. 4c-d) and validate the genotyping by molecular markers (Fig. 4e). While it was not possible to detect all the segments carried by an individual plant using this technique, either due to small size or no visible difference between the FISH signals for wheat and T. timopheevii, it was possible to confirm the expected larger segments.

FISH validation of the SNP genotyping of wheat-T. timopheevii introgression lines. Oligos pSc119.2–1 (green) and the Afa family (red) were used as FISH probes. a known FISH karyotype of wheat [56] (b) FISH karyotype of T. timopheevii accession P95–99.1-1 (based on that published by [37]) (c) FISH karyotype of line BC3F2–114-1 showing a homozygous introgression from T. timopheevii linkage group 5G into wheat chromosome 5B (d) FISH signals in the metaphase spread of line BC3F2–114-1 used to make the karyotype with white arrows indicating chromosomes T5GS.5GL-5BL (e) GGT bar diagram of line BC3F2–114-1 showing the T. timopheevii introgression from linkage group 5G in red markers while the wheat alleles are represented in blue
Comparative analysis between T. timopheevii and wheat genomes
Synteny analyses were carried out separately for both the At and G genomes with wheat using sequence information of the markers located on the genetic map of T. timopheevii. The sequences of the mapped markers were compared using BLAST (e-value cut-off of 1e-05) against the wheat genome sequence (Refseq v1; [20]) to obtain orthologous map positions of the best BLAST hits in the A, B and D genomes of wheat. 77.9% of the markers on the At genome had an overall top hit on the A genome of wheat and 72.2% of the markers on the G genome had an overall top hit on the B genome of wheat. This result was expected since the At and A genomes are closely related through a common progenitor T. urartu [11] and the G and B genomes are also thought to be closely related [12, 44].
The synteny analysis in Fig. 5 shows the comparison of the At and G genomes of T. timopheevii with the A and B genomes of wheat respectively. Figure 5 shows different coloured links, corresponding to the colour of the ideograms between T. timopheevii and wheat, which represent the physical positions of the markers on the wheat genome chromosomes as obtained through BLAST analysis. These results show that macro-synteny and collinearity is maintained between the At and G genomes of T. timopheevii and the A and B genomes of wheat respectively. Black links show where the BLAST hit of the marker sequence is on a non-homoeologous chromosome in wheat potentially indicating that there are chromosomal translocations within T. timopheevii. All chromosomal translocations are also shown as highlighted chromosome ideograms in the At and G genomes of T. timopheevii with part of the chromosome involved in the translocation highlighted with the colour of the chromosome that has been translocated onto it. Some of the inter-genome chromosomal translocations include 1GS/6AtS, 4GS/6AtS and 4GS/5AtL. The intra-genome chromosomal translocations include 4AtL/3AtL, 4AtL/6AtS and 4AtL/5AtL (the latter not indicated by a black link since this translocation is already present in wheat). Translocation 4AtL/5AtL, which the emmer wheat inherited from T. urartu [10, 40, 41] has been previously reported in T. timopheevii [23]. All the other translocations are potentially species-specific and have also been previously reported [22, 33] as having occurred potentially through three translocation events namely 6AtS/1GS/4GS, 4GS/4AtL, and 4AtL/3AtL, which most likely arose in that sequence. The 4AtL/5AtL translocation has been previously reported in wheat as part of a double translocation 5AL/4AL/7BS [32, 41], however, Fig. 5 shows that the 4AL/7BS translocation (or 4AtL/7GS in this case) does not exist in T. timopheevii matching previous reports [33]. These results also confirm previous reports that chromosomes 1At, 2At, 5At, 7At, 2G, 3G, 5G, and 6G of T. timopheevii do not differ structurally from their counterpart in the A and B genomes of wheat [2, 33]. Figure 6 shows a multi-colour GISH image of a metaphase spread showing some of these translocations.

Comparison of the At and G genomes of T. timopheevii and the A and B genomes of wheat, respectively, showing significant synteny and inter- and intra-genomic translocations. Chromosomes from both genome groups (At/A and G/B) are represented by differently coloured ideograms but chromosomes from the same homoeologous group in T. timopheevii and wheat are represented by the same colour. Ticks on the T. timopheevii genomes show the ideogram size in cM whereas those on the wheat genomes show the ideogram size in Mbp. BLAST results are represented by differently coloured links between the map positions of the markers on the genetic map of T. timopheevii and their corresponding physical positions on the wheat genome. Syntenic links are of the same colour as the homoeologous chromosomes which are linked whereas black links indicate where the BLAST hit was to a non-homoeologous chromosome

Multi-colour GISH of a metaphase spread of T. timopheevii accession P95–99.1-1. Chromosomes of the At genome are shown in green and chromosomes of the G genome in red. White arrows indicate the inter-genomic translocations 6AtS/1GS/4GS and 4GS/4AtL (the intra-genomic translocation 4AtL/3AtLcould not be detected using multi-colour GISH)
Discussion
T. timopheevii is a potentially important source of genetic variation for a range of agronomically important traits (see Introduction). In the past, one of the critical bottlenecks limiting the use of genetic variation from the wild relatives of wheat for crop improvement has been the inability to rapidly detect and characterise introgressions. However, the use of an Axiom® SNP genotyping array [18, 19, 27, 28], in combination with FISH, has led to the identification of 276 putative wheat-T. timopheevii introgressions.
The F1 interspecific hybrids generated between Paragon and T. timopheevii, which were backcrossed to Paragon (Fig. 1), were essentially haploid for the A, B and D genomes of wheat and also the At and G genomes of T. timopheevii. Therefore, intraspecific recombination between homologous chromosomes during meiosis was not possible. Thus, the rationale for using the strategy employed was to attempt to increase the frequency of recombination between 1) the A genome of wheat and the At genome of T. timopheevii and 2) the B genome of wheat and the G genome of T. timopheevii.
It has previously been shown that the A and At genomes and the B and the G genomes recombine, at a frequency of 70 and 30% respectively, in the presence of the Ph1 locus located on the long arm of chromosome 5B of wheat [14] (T. timopheevii also carries a pairing control locus/gene although it has been reported to have a weaker affect than Ph1 in wheat, [45]). This chromosome pairing study indicates that the A and B genomes of wheat do not show complete homology with the At and G genomes of T. timopheevii. In an attempt to increase recombination between the genomes of wheat and T. timopheevii, a ph 1 mutant wheat was used to generate F1 hybrids, i.e. the resulting interspecific hybrids thus lacked the 5B Ph1 locus but carried its weaker putative T. timopheevii allelic variant [45]. Since a control experiment was not undertaken as this was not the object of this research (i.e. the object of this research was purely to generate large numbers of introgressions for exploitation in breeding programmes) it was not possible to determine if removal of the 5B Ph1 locus increased recombination between wheat and T. timopheevii chromosomes. However, we note that significant numbers of wheat/T. timopheevii introgressions, i.e. 276, were generated. The fact that some linkage groups of T. timopheevii, derived from introgressions, had much lower frequencies of recombination than others, e.g. for the G genome 16.6 cM for linkage group 1 in contrast to linkage group 2 which was 76.5 cM (Fig. 2 and Table 2), indicates that the level of homology between these two species varies across the genome.
Large numbers of wheat/wild relative introgressions were also observed in recent work on Am. muticum (218 – [27]), Ae. speltoides (294 – [28]) and T. urartu (176 – [19]). In contrast, the numbers of introgressions obtained between wheat and Th. bessarabicum was extremely low, i.e. only 12 [18]. The lower frequency of wheat/Th. bessarabicum introgression observed may result from the fact there was considerable disruption in synteny between this species and the genomes of wheat as compared to the other species described.
The F1 hybrids contained three genomes from wheat (A, B and D) and two genomes from T. timopheevii (At and G). Thus, even though a degree of recombination was expected to occur between the A/At and B/G genomes, it was expected that a large proportion of the gametes produced would be unbalanced. As a result, it was predicted that the fertility of the F1 hybrids would be low. This was found to be the case with only 16% of crossed F1 hybrid ears setting seed (Table 1). A very similar level of infertility was observed in F1 hybrids between wheat and Am. muticum (A, B, D and T) [27]. However, wheat/Ae. speltoides F1 hybrids (A, B, D and S) exhibit higher fertility, i.e. 29% [28]. As a result of the infertility of the wheat/T. timopheevii F1 hybrids, only 152 BC1 seed were generated from 351 crossed ears with Paragon. Twenty-five F1 seeds gave rise to the BC1, BC2, BC3 and BC4 populations in this work. The level of interspecific recombination detected by genetic mapping was such that it was possible to assemble 14 linkage groups of T. timopheevii. Furthermore, the use of blast analysis enabled us to determine which linkage groups were derived from which two genomes of T. timopheevii. We were able to characterise these introgressions and track them through the backcross generations (Fig. 3). However, while we used the genetic map to identify and characterise introgressions, it is important to note that the maps were not produced using proper mapping families and thus the cM distances should be treated with caution.
All of the backcross populations were derived from gametes of 25 F1 plants. Recombination between the chromosomes of wheat and T. timopheevii was not limited to the gametes of the F1 population, i.e. genotyping showed that interspecific chromosome recombination had occurred in the gametes of individuals of the backcross populations in the presence of the wild type Ph1 locus. However, SNP analysis from each of the backcross generations showed that the majority occurred due to recombinant events in the F1 gametes rather than in later generations (Fig. 3).
In total, out of 276 introgressions, 141 involved the At genome and 135 involved the G genome of T. timopheevii. Slightly more At introgressions were observed than G introgressions. The difference in the number of introgressions involving the At chromosomes may result from a higher frequency of recombination of this genome with the genomes of wheat (presumably the A genome). However, the difference in the number of introgressions could reflect marker coverage, i.e. there may be insufficient markers to accurately access the actual levels of recombination occurring with each of the T. timopheevii genomes with those of wheat. Thus, it is therefore not possible to reliably compare and contrast the frequencies of recombination of the At and G genomes of T. timopheevii with wheat in this work.
Validation of introgressions identified via the Axiom® array was performed on selected individuals, potentially carrying large T. timopheevii segments, using FISH. Unlike GISH [27, 28], FISH could only be used to detect introgressions when the resulting hybridisation signals of oligos pSc119.2–1 and the Afa family could distinguish wheat chromosomes from T. timopheevii chromosomes. However, in each case where FISH analysis was undertaken it confirmed the SNP analysis (Fig. 4).
In this work it was found that chromosome 2G was transmitted at a much higher frequency to each of the backcross generations than other chromosomes from T. timopheevii (Table 3). This work confirms similar findings for the elevated transmission of either a complete chromosome 2G [42, 57] or a 2G introgression in wheat [9]. Chromosome 2S from Ae. speltoides has also been shown to carry a gametocidal gene which results in its preferential transmission to the next generation through both the male and female gametes [28, 34, 35, 58]. However, while the level of preferential transmission exhibited by chromosome 2S is at, or close to, 100% [28, 34], this was not found to be the case with chromosome 2G. Thus, the gene(s) responsible for the preferential transmission of chromosome 2G from T. timopheevii appear to be weaker than the gene for preferential transmission on chromosome 2S from Ae. speltoides.
One of the implications of preferentially transmitted chromosomes is that they need to be removed if introgressions without them are to be developed. In the case of chromosome 2S from Ae. speltoides this is extremely difficult as it is transmitted exclusively to the next generation. Removal of these genes thus requires the use of additional strategies [15]. However, since chromosome 2G is not transmitted exclusively, it can be removed via further backcrossing combined with selection, e.g. molecular markers, FISH.
One of the major bottle-necks preventing the exploitation of the vast reservoirs of genetic variation in the wild relatives for wheat improvement has been the lack of a high-throughput system to detect and characterise introgressions. The Axiom® Wheat-Relative Genotyping Array was selected as the technology to identify wheat/wild relative introgressions for the work being undertaken on multiple species at the Nottingham BBSRC Wheat Research Centre. A key factor in choosing this technology was that a single array is able to identify introgressions from all of the ten species being analysed in the programme within the wheat genotypes being used. To date it has been used to identify large numbers of introgressions into wheat from Am. muticum, Ae. speltoides, Th. bessarabicum, T. urartu and Th. intermedium ([27] and 2018; [19] and b; [8]).
While the Axiom® Wheat-Relative Genotyping Array has proved invaluable in detecting and characterising introgressions, in our hands, we were only able to utilise 1582 of the markers to identify wheat/T. timopheevii introgressions. However, it should be noted that since the 35 K SNPs on the array were selected to be polymorphic with all ten wild relatives used in the programme, only a subset of the SNPs (~ 19.5 K) were polymorphic with T. timopheevii to begin with. In addition, for a majority of the polymorphic SNPs (~ 10.5 K), at least one parent showed a heterozygous call. Therefore, a smaller group of markers showing a typical cluster pattern for a codominant marker were selected for genetic mapping.
In addition to the array, other methods for the detection of wheat/wild relative introgressions could be exploited in the future, including next generation sequencing technologies such as genotyping by sequencing (GBS; [53]) and Specific Locus Amplified Fragments (SLAF-seq; [55]). Population-specific SNPs can be discovered through the GBS procedure through sequencing of DNA libraries obtained after restriction digest of samples. Unlike the Axiom array, GBS is free of ascertainment bias [50] but the utility of GBS for this type of work in the future will depend on the cost of sequencing, bioinformatics, etc. SLAF-seq is an alternative method that could be used to identify introgressions. However, as many of the SNPs generated by this technology are likely to be from non-genic regions, only a limited number are likely to be transferable between species. Thus, it could be necessary to undertake the SLAF-seq protocol for each of the wild relatives and the wheat genotype in each introgression programme. Whichever technologies are used, they will need to take into account that some of the wild relatives are out-breeders and some are polyploids (i.e. the technologies will need to be able to distinguish polymorphisms that are between a wild relative and wheat versus polymorphisms between alleles within a wild relative).
Irrespective of the detection platform used the technologies already available, and those that will become available in the future, are resulting in a step change in our ability to detect wheat/wild relative introgressions that will allow us, for the first time, to begin to systematically exploit the vast reserve of genetic variation available in the wild relatives for wheat improvement.
In the present work we are currently unable to use SNP markers on the Axiom® array or FISH to determine which chromosomes of wheat are involved in each of the introgressions generated in this work. However, we are developing wheat chromosome-specific Kompetitive Allele Specific PCR (KASP™) markers which allow the high- throughput analysis of large numbers of introgressions enabling us to track introgressions in future derivative material and also to determine which wheat chromosomes have introgressions from T. timopheevii.
Conclusions
In this work, we have used the Axiom® Wheat-Relative Genotyping Array for high-throughput genotyping of wheat-T. timopheevii introgression lines. The characterisation of these interspecific hybrid lines has resulted in the development of a set of SNP markers, spread across all 7 chromosomes of both the At and G genomes, that can detect the presence of T. timopheevii in a hexaploid wheat background making it a potentially valuable tool for marker assisted selection (MAS) in wheat pre-breeding programs. These valuable resources of high-density molecular markers and wheat-T. timopheevii hybrid lines will greatly enhance the work being undertaken for wheat improvement through wild relative introgressions.
Methods
Generation of introgressions
It has previously been shown that even in the presence of the Ph1 locus, the At and G genomes of T. timopheevii are sufficiently closely related to the A and B genomes of wheat for a level of recombination, and hence genetic exchange, to occur at meiosis in hybrids between the two species, with the resulting generation of interspecific recombinant chromosomes or introgressions. In order to generate introgressions, hexaploid wheat cv. Paragon ph 1/ph1 mutant (2n = 2x = 14) (obtained from the Germplasm Resource Unit (GRU) at the John Innes Centre) was pollinated with T. timopheevii, (accession P95–99.1-1, obtained from the United States Department of Agriculture, USDA; 2n = 4x = 28), to produce F1 interspecific hybrids. These hybrids were then grown to maturity and backcrossed as the female parent with Paragon to generate BC1 populations. The BC1 individuals and their resulting progenies were recurrently pollinated to produce BC2, BC3 and BC4 populations (Fig. 1).
Genotyping of introgression lines
A 35 K Axiom® Wheat-Relative Genotyping Array (Affymetrix, Santa Clara, California) was used to detect the presence of putative wheat/T. timopheevii introgressions in each of the backcross generations [18, 19, 27, 28]. This array is composed of SNPs between various wild relatives and wheat genotypes, including T. timopheevii [27]. All the SNPs incorporated in this array formed part of the Axiom® 820 K SNP array [66], the data-set for which is available from www.cerealsdb.uk.net [64, 65]. Table 2 shows the number of putative SNPs between T. timopheevii and each of the wheat genotypes included on the array. The array allows 384 lines to be screened at one time. Genotyping was performed as described by King et al. [27] with slight modifications (see below).
DNA was extracted according to the Somers and Chao protocol (http://maswheat.ucdavis.edu/PDF/DNA0003.pdf, verified 21 January 2019, original reference in [46]) from 344 individuals of the back-crossed populations, BC1, BC2, BC3, and BC4, derived from the wheat/T. timopheevii F1 hybrids and control samples which included three replicates of each of the parental lines, i.e. wheat cv. Paragon and T. timopheevii. These populations were genotyped with the Axiom® Wheat-Relative Genotyping Array. Only Poly High Resolution (PHR) SNP markers, which were co-dominant and polymorphic, with at least two examples of the minor allele were used for genetic mapping [27]. Call rate for a sample was calculated as the percentage of the number of SNP probes on the array that resulted in a definitive genotype call (AA, AB, BB) for that sample. The equipment, software, procedures and criteria used for this genotyping are as described by King et al. [27].
Genetic mapping
SNP markers which showed: 1) heterozygous calls for either parent(s) 2) no polymorphism between the wheat parents and T. timopheevii and/or 3) no calls for either parent(s) were removed using Flapjack™ ([38]; v.1.14.09.24). The resulting markers were sorted into linkage groups (Fig. 2) in JoinMap® 4.0 [61] with a LOD score of 50. All markers that did not show any heterozygous calls or were unlinked were ignored and only the highest-ranking linkage groups with more than 30 markers were selected for map construction. Linkage groups were assigned to one of the 14 chromosomes of T. timopheevii through a BLAST analysis against the wheat genome reference sequence (RefSeq v1.0; [20]). Markers from each linkage group were used in a BLAST function (e-value cut-off of 1e-05) against the wheat genome to obtain the orthologous map positions of the top hits in the A, B and D genomes of wheat. If majority of the markers from a linkage group had a top hit in the A genome of wheat, they were assigned to the At genome of T. timopheevii. Similarly, if most markers from a linkage group had a top hit on the B genome of wheat then the group was mapped to the G genome of T. timopheevii. The linkage groups were assigned to the same homoeologous group in T. timopheevii as indicated by the BLAST results in wheat. Linkage group data was used to produce two genetic maps (one each for the At and the G genome of T. timopheevii – Fig. 2a and b) using MapChart 2.3 [62]. Markers at the same genetic map position were ordered according to their physical positions on the wheat genome (RefSeq v1.0; [20]) but only the first two markers were selected as anchors and represented on the map for that position. All markers and their order in the linkage group are shown in Additional file 1 Graphical genotype visualization was performed using Graphical GenoTypes 2.0 (GGT; [60]).
Cytogenetic analysis
Preparation of metaphase spreads
Preparation of chromosome spreads was as described in Zhang et al. [68] and King et al. [27] but briefly: Roots were excised from germinated seeds, treated with nitrous oxide gas at 10 bar for 2 h, fixed in 90% acetic acid for 10 min and then washed three times in water on ice. Root tips were dissected and digested in 20 μl of 1% pectolyase Y23 and 2% cellulase Onozuka R-10 (Yakult Pharmaceutical, Tokyo) solution for 50 min at 37 °C and then washed three times in 70% ethanol. Root tips were crushed in 70% ethanol, cells collected by centrifugation at 5000 rpm for 1 min, briefly dried and then re-suspended in 30–40 μl of 100% acetic acid prior to being placed on ice. The cell suspension was dropped onto glass slides (6–7 μl per slide) in a moist box and dried slowly under cover.
Fluorescence in situ hybridisation (FISH)
Slides were initially probed for multi-colour fluorescence in situ hybridization (FISH). Two repetitive DNA sequences pSc119.2 [36] and the Afa family [39] were labelled with Alexa Fluor 488–5-dUTP and Alexa Fluor 594–5-dUTP, respectively, and hybridized to the slides.
Genomic in situ hybridization (GISH)
The protocol for GISH was as described in Zhang et al. [68], Kato et al. [25] and King et al. [27]. Genomic DNAs was isolated from T. urartu (A genome) and Ae. speltoides (B genome) and labelled by nick translation with Chroma Tide Alexa Fluor 488–5-dUTP (Invitrogen, Carlsbad, California; C11400) and Alexa Fluor 594–5-dUTP (Invitrogen; C11397), respectively.
Slides of T. timopheevii (accession P95–99.1-1) were probed with labelled DNAs of T. urartu (100 ng) and Ae. speltoides (200 ng) in a ratio of 1:2 per slide to detect the AtAtGG genomes. Slides were counterstained with Vectashield mounting medium with DAPI, and analysed using a Zeiss Axio ImagerZ2 upright epifluorescence microscope (Carl Zeiss Ltd., Oberkochen, Germany) with filters for Alexa Fluor 488 and Alexa Fluor 594. Photographs were taken using a MetaSystems Coolcube 1 m CCD camera. Image analysis was carried out using Meta Systems ISIS and Metafer software (Metasystems GmbH, Altlussheim, Germany).
Comparative analysis
Synteny analysis was carried out using sequence information of the markers located on the genetic map of T. timopheevii. The sequences of the mapped markers were compared using BLAST (e-value cut-off of 1e-05) against the wheat genome reference sequence (RefSeq v1.0; [20]) to obtain the orthologous map positions of the top hits in the A, B and D genomes of wheat (Fig. 5). To generate the figures, cM distances on the linkage groups of the present map of T. timopheevii were scaled up by a factor of 100,000 to match similar base pair lengths of the chromosomes of the wheat genome. Figure 5 was visualized using Circos (v. 0.67; [30]) with chromosomes from both genome groups (At/A and G/B) being represented by differently coloured ideograms. Ideograms from the same homoeologous group between T. timopheeevii and wheat are represented by the same colour, e.g. chromosomes 1At and 1A are represented by the same colour. Corresponding genetic and physical positions of the markers on T. timopheevii and wheat, respectively, are shown in Additional file 1.
Abbreviations
- BLAST:
-
basic local alignment search tool
- FISH:
-
fluorescence in situ hybridisation
- GISH:
-
genomic in situ hybridisation
- PHR:
-
poly high resolution
- SNP:
-
single nucleotide polymorphism
References
Allard RW, Shands RG. Inheritance of resistance to stem rust and powdery mildew in cytogentetically stable spring wheats derived from Triticum timopheevii. Phytopathology. 1954;44:266–74.
Badaeva ED, Budashkina EB, Bilinskaya EN, Pukhalskiy VA. Intergenomic chromosome substitutions in wheat interspecific hybrids and their use in the development of a genetic nomenclature of Triticum timopheevii chromosomes. Russ J Genet. 2010;46:769–85.
Brisson N, Gate P, Gouache D, Charmet G, Oury F-X, Huard F. Why are wheat yields stagnating in Europe? A comprehensive data analysis for France. Crops Res. 2010;119:201–12.
Brown-Guedira GL, Bockus WW, Davis MA, Gill BS, Van Sanford DA, Murphy JP. Notice of release of KS04WGRC46 Fusarium head blight-resistant hard red winter wheat germ plasm. Ann. Wheat Newsl. 2005; p. 189.
Brown-Guedira GL, Singh S, Fritz AK. Performance and mapping of leaf rust resistance transferred to wheat from Triticum timopheevii subsp armeniacum. Phytopathology. 2003;93:784–9.
Cao W, Fedak G, Armstrong K, Xue A, Savard ME. Registration of spring wheat germplasm TC 67 resistant to Fusarium head blight. J Plant Regist. 2009;3:104–6.
Charmet G. Wheat domestication: lessons for the future. C.R. Biol. 2011;334:212–20.
Cseh A, Yang C, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge AJ, et al. Development and validation of an exome-based SNP marker set for identification of the St, Jr and Jvs genomes of Thinopyrum intermedium in a wheat background. Theor Appl Genet. 2019. https://doi.org/10.1007/s00122-019-03300-9.
Devos KM, Atkinson MD, Chinoy CN, Francis HA, Harcourt RL, Koebner RMD, et al. Chromosomal rearrangements in the rye genome relative to wheat. Theor Appl Genet. 1993;85:673–80.
Devos KM, Dubcovsky J, Dvorak J, Chinoy CN, Gale MD. Structural evolution of wheat chromosomes 4A, 5A and 7B and its impact on recombination. Theor Appl Genet. 1995;91:282–8.
Dvorák J, di Terlizzi P, Zhang HB, Resta P. The evolution of polyploid wheats: identification of the a genome donor species. Genome. 1993;36:21–31.
Dvorák J, Zhang HB, Kota RS, Lassner M. Organisation and evolution of the 5s ribosomal RNA gene family in wheat and related species. Genome. 1989;32:1003–16.
Fedak G. Potential of wide crosses to improve the resistance to vomitoxin accumulation in wheat following infection by Fusarium head blight, wheat improvement, management and utilization, Ruth Wanyera, James Owuoche, editors. InTechOpen. 2017. https://doi.org/10.5772/67272.
Feldman M. Identification of unpaired chromosomes in F1 hybrids involving T. aestivum and T. timopheevii. Can. J. Genet. Cytol. 1966;8:144–51.
Friebe B, Zhang P, Nasuda S, Gill BS. Characterization of a knock-out mutation in the Gc2 locus in wheat. Chromosoma. 2003;111:509–17.
Gill BS, Friebe BR, White FF. Alien introgressions represent a rich source of genes for crop improvement. Proc Natl Acad Sci U S A. 2011;108:7657–8.
Gordeeva EI, Leonova IN, Kalinina NP, Salina EA, Budashkina EB. Comparative cytological and molecular analysis of common wheat introgression lines containing genetic material of Triticum timopheevii Zhuk. Russ J Genet. 2009;45:1428–37.
Grewal S, Hubbart-Edwards S, Yang C, Scholefield D, Ashling S, Burridge A, et al. Detection of Triticum urartu introgressions in wheat and development of a panel of interspecific introgression lines. Front Plant Sci. 2018b. https://doi.org/10.3389/fpls.2018.01565.
Grewal S, Yang C, Hubbart-Edwards S, Scholefield D, Ashling S, Burridge AJ, et al. Characterisation of Thinopyrum bessarabicum chromosomes through genome-wide introgressions into wheat. Theor Appl Genet. 2018a;131:389–406.
International Wheat Genome Sequencing Consortium (IWGSC), Appels R, Eversole K, Feuillet C, Keller B, Rogers J, et al. Shifting the limits in wheat research and breeding using a fully annotated reference sequence. Science. 2018;361:eaar7191.
Järve K, Peusha HO, Tsymbalova J, Tamm S, Devos KM, Enno T. Chromosomal location of a T timopheevii-derived powdery mildew resistance gene transferred to common wheat. Genome. 2000;43:377–81.
Jiang J, Gill BS. Different species-specific chromosome translocations in T. timopheevii and T. turgidum support the diphyletic origin of polyploid wheats. Chromosom Res. 1994a;2:59–64.
Jiang J, Gill BS. New 18S-26S ribosomal RNA gene loci: chromosomal landmarks for the evolution of polyploid wheats. Chromosoma. 1994b;103:179–85.
Jorgensen JH, Jensen CJ. Gene Pm6 for resistance to powdery mildew. Euphytica. 1973;22:423.
Kato A, Lamb JC, Birchler JA. Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc Natl Acad Sci U S A. 2004;101:13554–9.
Khlebova IP, Barysheva NV. Genetic control of resistance to stem rust in durum wheat introgressive lines derived from Triticum timopheevii Zhuk. Biol. Bull. Bogdan Chmelnitskiy Melitopol State Pedagogical University. 2016;6:121–31.
King J, Grewal S, Yang C, Hubbart S, Scholefield D, Ashling S, et al. A step change in the transfer of interspecific variation into wheat from Amblyopyrum muticum. Plant Biotech J. 2017;15:217–26.
King J, Grewal S, Yang C, Hubbart-Edwards S, Scholefield D, Ashling S, et al. Introgression of Aegilops speltoides segments in Triticum aestivum and the effect of the gametocidal genes. Ann Bot. 2018;121:229–40.
King J, Gustafson P, Allen A, King IP. Exploitation of interspecific diversity in wheat. In: Bonjean AP, Angus WJ, van Ginkel M, editors. The world wheat book: a history of wheat breeding. Vol. 3. Paris: Lavoisier; 2016. p. 1125–39.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Leonova I, Börner A, Budashkina E, Kalinina N, Unger O, Röder M, et al. Identification of microsatellite markers for a leaf rust resistance gene introgressed into common wheat from Triticum timopheevii. Plant Breed. 2004;123:93–5.
Liu CJ, Atkinson MD, Chinoy CN, Devos KM, Gale MD. Non-homoeologous translocations between group 4, 5 and 7 chromosomes within wheat and rye. Theor Appl Genet. 1992;83:305–12.
Maestra B, Naranjo T. Structural chromosome differentiation between Triticum timopheevii and T. turgidum and T. aestivum. Theor. Appl. Genet. 1999;98:744–50.
Marais GF, Pretorius ZA. Gametocidal effects and resistance to wheat leaf rust and stem rust in derivatives of Triticum turgidum ssp. durum/Aegilops speltoides hybrids. Euphytica. 1996;88:117–24.
McIntosh RA. Breeding wheat for resistance to biotic stress. Euphytica. 1998;100:19–34.
McIntyre CL, Pereira S, Moran LB, Appels R. New Secale cereale (rye) DNA derivatives for the detection of rye chromosome segments in wheat. Genome. 1990;33:635–40.
Miko P, Megyeri M, Farkas A, Molnar I, Molnar-Lang M. Molecular cytogenetic identification and phenotypic description of a new synthetic amphiploid, Triticum timococcum (AtAtGGAmAm). Genet Resour Crop Ev. 2015;62:55–66.
Milne I, Shaw P, Stephen G, Bayer M, Cardle L, Thomas WTB, et al. Flapjack – graphical genotype visualisation. Bioinformatics. 2010;26:3133–4.
Nagaki K, Tsujimoto H, Isono K, Sasakuma T. Molecular characterisation of a tandem repeat, Afa family, and its distribution among Triticeae. Genome. 1995;38:479–86.
Naranjo T. Chromosome structure of durum wheat. Theor Appl Genet. 1990;82:577–86.
Naranjo T, Roca A, Goicoechea PG, Gira H, Ldez R. Arm homoeology of wheat and rye chromosomes. Genome. 1987;29:873–882.
Nyquist WE. Differential fertilisation in the inheritance of stem rust resistance in hybrids involving a common wheat strain derived from Triticum timopheevii. Genetics. 1962;47:1109–24.
Obukhova LV, Budashkina EB, Ermakova MF, Kalinina NP, Shumnyi VK. The quality of grain and flour in introgression spring wheat lines with resistance genes to leaf rust from Triticum timopheevii Zhuk. Ag Biol. 2008;5:38–42.
Ogihara Y, Tsunewaki K. Diversity and evolution of chloroplast DNA in Triticum and Aegilops species as revealed by restriction fragment analysis. Theor Appl Genet. 1988;76:321–32.
Ozkan H, Feldman M. Genotypic variation in tetraploid wheat affecting homoeologous pairing in hybrids with Aegilops peregrine. Genome. 2001;44:1000–6.
Pallotta MA, Warner P, Fox RL, Kuchel H, Jefferies SJ, Langridge P. Marker assisted wheat breeding in the southern region of Australia. Proc. 10th Inter. Wheat Genetics Symp. Paestum, Italy; 2003. P. 789–791.
Patterson FL, Gallun RL, Roberts JJ. Registration of Arthur wheat. Crop Sci. 1974;14:910.
Patterson FL, Gallun RL, Roberts JJ, Finney RE. Registration of Arthur 71 and Abe wheat. Crop Sci. 1975;15:736.
Peusha H, Enno T, Priilinn O. Chromosomal location of powdery mildew resistance genes and cytogenetic analysis of meiosis in common wheat cultivar Meri. Hereditas. 2000;132:29–34.
Poland JA, Brown PJ, Sorrells ME, Jannink J-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;7:E32253.
Pshenichnikova TA, Permyakov AV, Osipova SV, Permyakova MD, Rudikovskaya EG, Verchoturov VV. Effects of limited introgressions from Triticum timopheevii Tausch. into the genome of bread wheat (Triticum aestivum L.) on physiological and biochemical traits under normal watering and drought. Russ. J. Genet: Appl. Res. 2016;6:553–9.
Rodriguez S, Maestra B, Perera E, Diez M, Naranjo T. Pairing affinities of the B- and G-genome chromosomes of polyploid wheats with those of Aegilops speltoides. Genome. 2000;43:814–9.
Saintenac C, Jiang D, Wang S, Akhunov E. Sequence-based mapping of the polyploid wheat genome. G3: Genes genomes Genet. 2013;3:1105–14.
Schneider A, Molnar I, Molnar-Lang M. Utilisation of Aegilops (goatgrass) species to widen the genetic diversity of cultivated wheat. Euphytica. 2008;163:1–19.
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One. 2013;8:e58700.
Tang Z, Yang Z, Fu S. Oligonucleotides replacing the roles of repetitive sequences pAs1, pSc119.2, pTa-535, pTa71, CCS1, and pAWRC.1 for FISH analysis. J Appl Genet. 2014;55:313–8.
Timinova EM, Leonova IN, Belan IA, Rosseeva LP, Salina EA. The influence of particular chromosome regions of Triticum timopheevii on the formation of resistance to diseases and quantitative traits in common wheat. Russ J Genet: Appl Res. 2012;2:330–43.
Tsujimoto H, Tsunewaki K. Gametocidal genes in wheat and its relatives I. genetic analyses in wheat of a gametocidal gene derived from Aegilops speltoides. Can J Genet Cytol. 1984;26:78–84.
Uhrin A, Szakács E, Láng L, Bedö Z, Molnár-Láng M. Molecular cytogenetic characterization and SSR marker analysis of a leaf rust resistant wheat line carrying a 6G(6B) substitution from Triticum timopheevii (Zhuk.). Euphytica. 2012;186:45–55.
van Berloo R. GGT 2.0: versatile software for visualization and analysis of genetic data. J. Hered. 2008;99:232–6.
van Ooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res. 2011;93:343–9.
Voorrips RE. Mapchart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93:77–8.
Watson IA, Ling NH. Timvera – a Steinwedel x Triticum timopheevii derivative. Agron J. 1958;50:644.
Wilkinson PA, Winfield MO, Barker GLA, Allen AM, Burridge A, Coghill J, et al. CerealsDB 2.0: an integrated resource for plant breeders and scientists. BMC Bioinformatics. 2012;13:219.
Wilkinson PA, Winfield MO, Barker GLA, Tyrell S, Bian X, Allen AM, et al. CerealsDB 3.0: expansion of resources and data integration. BMC Bioinform. 2016;17:256. 441.
Winfield MO, Allen AM, Burridge AJ, Barker GLA, Benbow HR, Wilkinson PA, et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotech. J. 2016;14:1195–206.
Yudina RS, Leonova IN, Salina EA, Khlestkina EK. Change in salt tolerance of bread wheat as a result of the introgression of the genetic material of Aegilops speltoides and Triticum timopheevii. Russ. J. Genet: Appl. Res. 2016;6:244–8.
Zhang H, Bian Y, Gou X, Zhu B, Xu C, Qi B, et al. Persistent whole chromosome aneuploidy is usually associated with nascent allohexaploid wheat. Proc Natl Acad Sci U S A. 2013;110:3447–52.
Zhukovsky PM. Cultivated plants and their wild relatives. Systematics, geography, cytogenetics, immunity, origin and use. Kolos: Leningrad; 1971. p. 121.
Acknowledgements
The authors would like to thank Professor Keith Edwards and Dr. Sacha Allen for their expertise and support in the use of the array.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council [grant number BB/P016855/1] as part of the Designing Future Wheat programme (DFW). The funding body played no role in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
All data analysed during this study is included in this published article (and its Additional file 1). All raw genotyping data can be made available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
UD made the back-cross populations. SG, CY, IPK and JK made the initial crosses. CY, SA and DS prepared plant material for genotyping. AB genotyped the samples on the array. UD, SG and JK analysed the genotyping data. UD and SG did the BLAST analysis. CY and JK performed the FISH analysis. CY and UD performed the GISH work. IPK, JK and UD conceived the experimental design. SG, JK and IPK wrote the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Physical positions on the A, B and D genomes of wheat of all markers present on the genetic maps of the At and G genomes of T. timopheevii, as obtained through BLAST against the wheat genome (RefSeq v1; [20]). Positions highlighted in yellow were the top hits in the BLAST search and markers highlighted in green represent a translocated region of the T. timopheevii chromosomes (XLSX 98 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Devi, U., Grewal, S., Yang, Cy. et al. Development and characterisation of interspecific hybrid lines with genome-wide introgressions from Triticum timopheevii in a hexaploid wheat background. BMC Plant Biol 19, 183 (2019). https://doi.org/10.1186/s12870-019-1785-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1785-z