
Abstract
Background
Elytrigia Desv. is a genus with a varied array of morphology, cytology, ecology, and distribution in Triticeae. Classification and systematic position of Elytrigia remain controversial. We used nuclear internal-transcribed spacer (nrITS) sequences and chloroplast trnL-F region to study the relationships of phylogenetic and maternal genome donor of Elytrigia Desv. sensu lato.
Results
(1) E, F, P, St, and W genomes bear close relationship with one another and are distant from H and Ns genomes. Ee and Eb are homoeologous. (2) In ESt genome species, E genome is the origin of diploid Elytrigia species with E genome, St genome is the origin of Pseudoroegneria. (3) Diploid species Et. elongata were differentiated. (4) Et. stipifolia and Et. varnensis sequences are diverse based on nrITS data. (5) Et. lolioides contains St and H genomes and belongs to Elymus s. l. (6) E genome diploid species in Elytrigia serve as maternal donors of E genome for Et. nodosa (PI547344), Et. farcta, Et. pontica, Et. pycnantha, Et. scirpea, and Et. scythica. At least two species act as maternal donor of allopolyploids (ESt and EStP genomes).
Conclusions
Our results suggested that Elytrigia s. l. species contain different genomes, which should be divided into different genera. However, the genomes of Elytrigia species had close relationships with one another. Diploid species were differentiated, because of introgression and different geographical sources. The results also suggested that the same species and the same genomes of different species have different maternal donor. Further study of molecular biology and cytology could facilitate the evaluation of our results of phylogenetic in a more specific and accurate way.
Similar content being viewed by others


Background
Triticeae in Poaceae includes not only the most economically important cereal crops (wheat, barley, and rye) but also forage grasses and ecological species in grasslands. Approximately 450 Triticeae species exist worldwide [1,2,3]. Given the wide variety of biological mechanisms and genetic systems, this tribe represents an excellent group for research on plant systematics, genetic diversity, and speciation [4, 5].
As one of the most important perennial genera of Triticeae, Elytrigia Desv. includes 40 species, which are distributed in subtropical and warm temperate regions of both hemispheres [6]. Elytrigia Desv. was established by Desvaux [7], with Elytrigia repens (L.) Nevski as the type species. Morphologically, Elytrigia sensu lato is characterized by branched creeping rhizomes and caespitose, long anthers, lanceolate to liner glumes, lanceolate lemma, single spikelet per node, and cross-pollination, and most species were previously categorized under Agropyron Gaertner [1, 6, 8]. Cytogenetically, ploidy levels in Elytrigia s. l. vary from diploid (2n = 2× = 14) to decaploid (2n = 10× = 70) and contain Ee, Eb, St, ESt, StH, and NsXmStH genomes [1, 3, 8,9,10,11]. According to the proposed genomic system of classification, Löve [8] suggested that Elytrigia s. l. approximately includes 60 species and varieties and divided them into five genera, namely, Pseudoroegneria (Nevski) Á. Löve (St), Lophopyrum Á. Löve (E), Thinopyrum Á. Löve (J), Elytrigia (EJSt), and Elymus (StH). Dewey [1] considered Elytrigia s. l. into three independent genera: Pseudoroegneria (St), Thinopyrum (E or J), and Elytrigia (StX). Studies showed similarity of the E and J genomes [12,13,14,15,16,17,18,19]. Wang et al. [19] suggested that E and J should be considered as identical genomes and be distinguished from Ee and Eb. With the genomic system of classification in Triticeae taxonomy and systematics and genomic constitutions of increasing species identified, the definition of Elytrigia becomes narrower than that of traditional Elytrigia s. l. and only includes all polyploidy taxa with combination of Ee, Eb, and St genomes [3, 9, 11]. Ee genome originated from Lophopyrum elongatum (Host) Á. Löve, Eb genome from Thinoopyrum bessarabicum (Savul. and Rayss) Á. Löve, and St genome from diploid species in Pseudoroegneria (Nevski) Á. Löve. However, some studies reported that Et. repens, a type of Elytrigia, is a hexaploid species with StStStStHH genomes and was renamed as Elymus repens [3, 11, 20, 21]. Therefore, definition, precise taxonomic ranks, and number of Elytrigia species remain controversial.
Polyploidization and hybridization are the two main mechanisms in plant speciation and evolution [22, 23]. The changes in the cell size, genome size, gene expression, genomic repatterning, epigenetic effects and retrotransposon activation are caused by the polyploidization and chromosome doubling [5, 22,23,24,25,26]. As a result of these changes, stabilization of hybrid condition and full fertility may occur. And the establishment of phenotypes in nature could be enhanced. Therefore, polyploids could adjust to match with the new ecological niches or become more competitive than parental donors [5, 23, 26, 27]. The evolution of polyploidization alone and/or the combined effects of hybridization and polyploidization may lead to complex lineages, requiring an explanation of the phylogenetic relationship [27]. Molecular genetic analysis bears significance in elucidating phylogenetic relationships and genome evolution patterns in taxa for these kinds of plant groups [27, 28]. The analysis of Molecular phylogenetic exploits DNA sequences elucidated the history of revolution and origins of species in Triticeae. This illustrates their hybridization events and parental lineages contains their formation, and identifies the polyploidization mode [29,30,31,32,33,34,35,36,37,38]. Reproducibility and simplicity represent the main qualities that make DNA sequencing a suitable choice for identification of phylogenetic relationships among taxa and genomes [28, 39]. nrITS sequences were widely applied to explain genomic and phylogenetic relationship at a low taxonomic levels [40,41,42] and Triticeae species containing E, H, Ns, P, St and Xm genomes in Elymus, Hordeum, Psathyrostachys, Agropyron Pseudoroegneria and Leymus [5, 27, 33, 40,41,42,43]. Chloroplast DNA (cpDNA) sequences, including intron of trn-L and intergenic spacer of trnH-psbA, trnL-trnF, and trnS-trnG, are also widely used to identify maternal donors of polyploids with extra ability to analyze phylogenetic relationships among relevant species [41, 44,45,46,47].
The present study analyzed sequence data of one ITS region of nuclear DNA and one chloroplast gene (the intergenic region of trnL-trnF) from 18 species (subspecies) in Elytrigia s. l. with 21 species of related genera in Triticeae. The objectives are as follows: (1) to investigate phylogenetic relationships among species in Elytrigia s. l. and related genera; (2) to elucidate interspecific relationships among Elytrigia s. l. species; (3) to study phylogenetic relationships among species with different genomes and genome combinations; and (4) to discuss putative maternal donors for ESt genome species.
Methods
Plant materials
This study included 18 species (subspecies) of Elytrigia Desv. sensu lato and 21 species (subspecies) of related genera in Triticeae. Table 1 lists names, accession numbers, genomes, origins, and GenBank accession numbers. Bromus cartharticus Vahl. and Bromus tectorum L. were used as outgroup [5, 41, 48]. Seed materials with W6 and PI accession numbers were carefully offered by the American National Plant Germplasm System (Pullman, Washington, USA). We gathered seed materials with Y and ZY numbers. Voucher specimens and plants were deposited at the perennial nursery and herbarium of the Triticeae Research Institute, Sichuan Agricultural University, China.
DNA extraction, amplification, and sequencing
Total genomic DNA was extracted from leaves of single plants by slight modification of Cetyltrimethyl Ammonium Bromide (CTAB) procedure [49]. nrITS sequence and chloroplast trnL-F sequence were amplified with primers described in Table 2. A final volume of 20 μL of mixed reagents was obtained for each polymerase chain reaction (PCR); reagents included 2× Taq PCR MasterMix (10× ExTaq polymerase buffer, 3 mmol/L MgCl2, 500 μmol/L deoxynucleotide, 100 mmol/L KCl, and 20 mmol/L Tris–HCl), 1 μmol/L of each primer, 20–40 ng of template DNA, and distilled deionized water. PCR reactions were performed in GeneAmp T100 Thermal Cycler (Bio-Rad Inc., USA) employing protocols listed in Table 3. PCR products were electrophoresed on 1.0% agarose gels, and purified using EZNA™ gel extraction kit (Omega, GA, USA), and were cloned into pMD-19 T vector (TaKaRa) following the instructions of manufacturer. All sequences were derived from at least 3 independent clones for diploid species, and 5–8 independent clones for allopolyploid species. Sequencing was performed from both directions by Sunbiotech Company (Beijing, China) [36].
Phylogenetic analysis
Alignment of nrITS and trnL-F sequences were conducted by using Clustal W algorithm [50] with additional manual adjustment. Two data matrices with included nrITS were performed using Maximum likelihood (ML) in PAUP*4.0a (Swofford, D.L., Sinauer Associates, http://www.sinauer.com) and Bayesian inference (BI) in MrBayes version 3.1.2 [51]. Phylogenetic analyses based on trnL-F sequences were performed with MrBayes version 3.1.2. Evolutionary model employed for phylogenetic study was performed using Modeltest v3.7 with Akaike information criterion (AIC) [52]. Best-fit model was GTR + G for nrITS data. ML heuristic studies were carried out with 1000 random addition sequence replications and reconnection branch swapping algorithm and tree bisection [Dong 2013].
Similar to ML analysis, BI analyses of nrITS were perfomed with the alike evolutionary model. TVM + G was the optimal model for trnL-F data based on AIC in Modeltest v3.7. Observation of consistency and examined log likelihoods among all independent runs showed that burn-in periods very long enough for chains to become stationary [37]. Figures included nonsignificant bootstrap support (BS) of more than 50% and posterior probabilities of more than 70%.
Median-joining (MJ) network method was effectively employed to study detailed progenitor–descendant relationship among polyploidy species within tribe Triticeae [27, 35, 37, 53]. MJ network analysis was conducted by the Network 4.6.1.3 program (Fluxus Technology Ltd., Clare, Suffolk, UK). For the purpose of preventing single insertion/deletion events from being counted as multiple mutational stages in MJ network study, gaps in aligned nrITS and trnL-F sequences were not included in the calculation [37].
Results
nrITS data
Comparison of all species analysis suggested that DNA sequences for nrITS ranged from 596 bp to 605 bp in length. A TTTT insert at positions 58–61 in the nrITS sequence was detected for Et. caespitosa, Et. elongata (W6 21,859), Et. geniculata ssp. pruinifera, Et. intermedia (PI229917), Et. nodosa, Et. pontica, Et. rechingeri, Et. scirpea (PI 531750), Et. scythica, and Et. varnensis (Fig. 1).

Partial alignment of the amplified sequences of nrITS gene from the ten species of Elytrigia sensu lato. A TTTT insert at position 58–61
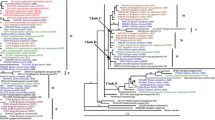
With assumed nucleotide frequencies A: 0.2286, C: 0.2966, G: 0.2794, and T: 0.1954, nrITS data yielded a single phylogenetic tree (−Lnlikelihood = 2553.6868). Proportion of invariable sites = 0, and gamma shape parameter = 0.4121. Likelihood settings from optimal model (GTR+ G) were selected by AIC in Modeltest v3.7. Similar to that of ML analysis, Bayesian study supposed the same topology. The tree demonstrated in Fig. 2 corresponds to the ML tree with posterior probabilities (PP) above and BS below branches [48].

Maximum-likelihood tree (−Lnlikelihood = 2553.6868, base frequencies A: 0.2286, C: 0.2966, G: 0.2794, T: 0.1954, pinvar = none, shape = 0.4121) inferred from the nrITS sequences of Elytrigia sensu lato and its affinitive species, under GTR+ G model. Numbers above and below branches indicate posterior probabilities (PP) ≥ 70% by BI analysis and bootstrap support (BS) ≥ 50% by ML, respectively
nrITS region from species were divided into five clades (Clades I–V). Clade I was divided into three groups, namely, A, B, and C. Group A (BS < 50 and PP < 70%) consisted of the St-genome sequence and included two Pseudoroegneria species (Pse. spicata PI 506259 and Pse. gracillima), two Elymus species (El. canadensis and El. caninus), and Elytrigia species, such as Et. caespitosa, Et. nodosa, Et. podperae, Et. pontica, and Et. scythica. Group B (PP = 81%) consisted of Et. bessarabica, Et. elongata (PI 531719), Et. farcta (PI 516555), Et. intermedia (PI 531725), and Et. scirpea (PI 531749); this group possesses an E-genome sequence. Group C (BS = 99% and PP = 100%) included 10 species with a TTTT insert at positions 58–61; this insert is a possible variation of E-genome sequence. This group comprised Et. caespitosa, Et. elongata (W6 21,859), Et. geniculata ssp. pruinifera, Et. intermedia (PI 229917), Et. nodosa, Et. pontica, Et. rechingeri, Et. scirpea (PI 531750), Et. scythica, and Et. varnensis. Clade II included St-genome sequences of Pse. spicata (PI 563870), Pse. tauri, and EStP genome species (Et. pungens and Et. pycnantha) and unknown genome sequences of Et. varnensis, P-genome sequences of Agropyron cristatum, W-genome sequences of Australopyrum pectinatum, Au. retrofractum, and F-genome sequences of Eremopyrum distans and Er. triticeum. Clade III consisted of St-genome sequences of Pse. libanotica, Pse. stipifolia, Pse. strigosa, Pse. strigosa ssp. aegilopoides, and three Elytrigia s. l. species (Et. geniculata ssp. pruinifera, Et. lolioides, and Et. repens). Clade IV comprised two Elytrigia s. l. species (Et. lolioides and Et. repens), two Elymus s. l. species (El. canadensis and El. caninus), and two Hordeum s. l. (H. bogdanii and H. chilense) (BS = 83%; PP = 100%). Clade V comprised Psathyrostachys juncea and Psa. huashanica (BS = 91% and PP = 100%).
In MJ analysis, each circular network node indicated a single sequence haplotype, and the node size is proportional to the number of isolates with that of haplotype [37]. Median vectors (standing for missing intermediates) present nodes that haven’t sampled deduced by MJ network study, and the numbers along branches illustrate the mutation positions. Distinguishing colors indicated various species species that share similar haplotype circular network node. Either alternative genealogies or true reticulation events are represented by network loops in closely related lineages [37]. The MJ network depicted genealogical relationships among 45 nrITS haplotypes from 49 taxa (Fig. 3) [48]. We found that MJ network represented a consistent phylogenetic reconstruction with ML tree. Then, we determined the names and group names of similar clusters to synchronize the MJ network. In nrITS MJ network analysis, five clusters (Clusters N-I to N-V) formed one star-like radiation. Three clusters (Clusters N-III to N-V) represented three different types of haplotypes (St, P, and Ns types) of Elytrigia s. l. and its related genera. Cluster N-I was divided into subclusters N-A, N-B, and N-C with E and St types, and Pse. spicata PI 506259 (PSP1) was placed at the central branching point. Cluster N-II included St type of Pse. spicata PI 563870 (PSP2) and Pse. tauri (PTAU), P type of Ag. cristatum (ACRI), Et. pungens (EPUN), and Et. pycnantha (EPYC), F type of Er. distans (EDIS) and Er. triticeum (ETRI), W type of Au. pectinatum (APEC) and Au. retrofractum (ARET), and unknown type of Et. varnensis (EVA1).

Median-joining networks based on nrITS locus haplotype of Elytrigia sensu lato and its affinitive species. Haplotypes are represented by circles. The numbers along the blanches indicate the frequency of mutations. Abbreviations of species names are listed in Table 1
trnL-F data
Comparison of all species studies showed that the length of trnL-F sequences ranged from 809 bp to 882 bp. Likelihood settings from optimal model (TVM + G) were chosed by AIC in Modeltest v3.7. Fig. 4 illustrates the BI tree with PP above branches. All trnL-F sequences from Elytrigia and its related genera species were similar.

Bayesian inference tree inferred from the trnL-F sequences of Elytrigia sensu lato and its affinitive species. Numbers above branches indicate posterior probabilities (PP) ≥ 70% by BI
Clade One involved 17 Elytrigia s. l. species [Et. bessarabica, Et. caespitosa, Et. elongata, Et. farcta, Et. geniculata, Et. geniculata ssp. pruinifera, Et. intermedia, Et. lolioides, Et. nodosa (PI 547344 and PI 547345), Et. podperae, Et. pontica (PI 383583 and PI 547313), Et. pycnantha, Et. rechingeri, Et. repens, Et. scirpea, Et. scythica, and Et. varnensis] and seven Pseudoroegneria species (Pse. gracillima, Pse. libanotica, Pse. spicata, Pse. stipifolia, Pse. strigosa, Pse. strigosa ssp. aegilopoides, and Pse. tauri). All diploid species with F, P, and W genomes were clustered together in Clade Two. Ns type trnL-F sequences from Psa. fragilis and Psa. juncea and H type trnL-F sequences from H. bogdanii and H. chilense were placed at Clusters Three and Four, respectively.
In the trnL-F MJ network analysis, 25 haplotypes were derived from 37 taxa. MJ network represented consistent phylogenetic reconstruction with BI tree. We determined clusters’ names following the name of groups shown in the ML tree. The trnL-F MJ network was divided into four clusters (Clusters N-One to N-Four). All species containing E or St genome were clustered together with E or St diploid species in Cluster N-One. Cluster N-Two included F, P, and W types of haplotypes. Ns type of Psathyrostachys haplotype species and H type of Hordeum haplotype species were grouped, respectively, in Clusters N-Three and N-Four (Fig. 5).

Median-joining networks based on trnL-F locus haplotype of Elytrigia sensu lato and its affinitive species. Haplotypes are represented by circles. The numbers along the blanches indicate the frequency of mutations. Abbreviations of species names are listed in Table 1
Discussion
Phylogenetic relationships among species in Elytrigia s. l.
Elytrigia s. l. is distributed in subtropical and warm temperate regions of both hemispheres [6]. Classification and systematic position of Elytrigia remain controversial [7, 8, 54,55,56]. Traditionally, the classification based on morphology and Elytrigia species contains Ee, Eb, EeEb, EeEeSt, EbEeSt, EeSt, StSt, StH, and EStP genomes. However, Dewey [1] and Löve [8] showed that taxonomic treatment of Triticeae species should be depended on genomic constitution. Therefore, Elytrigia species must be reclassified. Several current studies used molecular biology to study phylogenetic relationships of Elytrigia s. l. species and its related genera [43, 57, 58]. Hsiao et al. [59] estimated phylogenetic relationships of 30 diploid species of Triticeae (Poaceae) from the nrITS region of nuclear ribosomal DNA. Results illustrated that each genome group of species is monophyletic and consisten with cytogenetic evidence, and Australopyrum (Tzvelev) A. Löve (W) is closely concerned with Agropyron Gaertn. (P) [59]. Kim et al. [43] analyzed nrITS haplotypes, revealing close relationship of E, P, and St genomes. Cytologically, St and Y genomes and St, P, and W genomes are closely related [2, 60,61,62]. This finding indicates close relationship of E, P, St, and W genomes.
In this study, based on nrITS data, all Elytrigia s. l. species were classified in four groups (E, H, P, and St types) in the ML tree and MJ network. These results indicated that Elytrigia s. l. species contain different genomes. These findings also strongly support previous results. Genome species are not highly supported in Clades I-B, Ee, and Eb (BS < 50% and BI = 81%) (Fig. 2). This phenomenon provides evidence of close affinity between Ee and Eb genome species. Thus, these species are not homologous but homoeologous [63]. Our phylogenetic results also support previous cytological investigations reported by Löve [8, 64, 65], Yen and Yang [3], and Zhou [11].
Phylogenetic relationships between Elytrigia s. l. and related genera
In the present study, in the ML tree and MJ network based on nrITS data, seven types of nrITS region (E, F, H, P, W, Ns, and St types) were obtained from the Elytrigia s. l. species and its related genera. In polyploidy species Et. repens (StStH), H type was clustered with Hordeum species in Clade IV (83% BS, 100% PP), and St type was clustered with Pseudoroegneria (Fig. 2). In this study, we failed to obtain E and St type nrITS sequences from Et. pycnantha (EStP) and Et. pungens (EStP), whereas P type was clustered with Agropyron in Clade II (86% BS and 97% PP). In the phylogenetic tree, Clades II and IV formed a monophyletic group, and results support the distant relationship between H genome and other genomes (E, P, St, and W) (Fig. 2). This finding also indicated that St genome is the origin of Pseudoroegneria, whereas P genome is the origin of Agropyron.
Eremopyrum (Ledeb.) Jaub. et Spach and Agropyron. Gaertn. are highly similar based on one-keeled glumes and caryopsis morphology [66, 67]. According to molecular phylogenetic analysis, Er. triticeum and Er. distans were clustered with Agropyron based on rpoA, cpDNA, DMC1, and β-amylase data [44, 68,69,70]. Fan et al. [26] showed that Eremopyrum and Agropyron are closely related based on the presence of Acc1, Pgk1, and Acc1 + Pgk1. In the present nrITS gene data, allopolyploid species of Et. pycnantha and Et. pungens (EStP) were clustered with Er. triticeum, Er. distans, and Ag. cristatum with high statistical support (85% BS and 100% PP). Et. pycnantha, Et. pungens, Eremopyrum, and Agropyron species were grouped with St genome diploid species (Pse. spicata PI563870 and Pse. tauri) (57% BS and 100% PP) (Fig. 2). Estimates strongly support that Eremopyrum and Agropyron are closely related, and St, P, and F are very close to each other.
Sha et al. [48] studied phylogenetic relationships of Leymus based on trnH-psbA and indicated that Pseudoroegneria species are close to Lophopyrum bessarabicum. Evidence from meiotic chromosome pairing [71], morphological data [72], and DNA sequencing [44] suggested close relationship among Lo. bessarabicum, Lo. elongatum, and Pseudoroegneria. In the BI tree and MJ network based on trnL-F sequences, all Elytrigia species were categorized under Clade One with a number of zero-length branches; Elytrigia species are sisters with diploid species of Eremopyrum (F), Agropyron (P), and Australopyrum (W) (94% PP). H and Ns genome species formed the monophyletic group. These results indicated minimal differences in E and St genomes based on trnL-F sequence and close relationship of E, F, P, St, and W genomes, which are distant from H and Ns genomes. These findings support previous studies on morphology [66, 67], molecular biology [5, 44, 68,69,70], and cytogenetics [2, 60, 61].
Putative maternal donor and origin of Elytrigia species
cpDNA is mostly inherited from the female parent in tall plants. Therefore, it can be used to determine maternal donor in polyploids [37, 53, 73]. In the trnL-F ML, the phylogenetic tree showed high numbers of zero-length branches, which are mainly related to multifurcating relationships. Tree-based study methods are unable to represent multifurcating relationships and the coexistence of ancestors with their derivatives [53, 74]. Network approaches were designed to deal with such multifurcations [53, 73–76].
Previous studies based on cpDNA indicated that Pseudoroegneria (St genome donor) species are the maternal donor for species of Triticeae [41, 44, 77]. However, cytologically, Yen et al. [78] considered that rather than the St genome, the maternal donor of Kengyilia is the origin of P genome species. In this trnL-F-based BI tree, Et. bessarabica (2×) was clustered with polyploidy species Et. farcta, Et. nodosa (PI 547344), Et. pontica (PI 547313), Et. pycnantha, Et. scirpea, and Et. scythica (92% PP). MJ network analysis showed that diploid species Et. bessarabica (E), Et. pontica (PI 547313), Et. pycnantha (EStP), Et. scirpea (E), and Et. scythica (ESt) exhibit the same haplotype in Cluster N-One (Fig. 5). Combined with BI and MJ analyses, we can conclude that E genome-diploid species in Elytrigia served as maternal donor of E genome for Et. farcta, Et. nodosa (PI 547344), Et. pontica, Et. pycnantha, and Et. scythica. This conclusion agrees with results of Liao et al. [37]. Et. nodosa (P I547345 Ukraine) was not clustered with Et. bessarabica, and its haplotype differs from that of Et. nodosa (PI 547344 Turkey). Results showed that (1) at no less than two species served as maternal donor, indicating that formation of Et. nodosa occur multiple times. A similar conclusion was observed based on Et. caespitosa, Et. intermedia, Et. varnensis, and Kengyilia species [45, 58, 79]. (2) Different maternal donors in Et. nodosa are affected by altitude and climate conditions [80]. In the BI tree based on trnL-F sequence, we can conclude that Pseudoroegneria species (St genome donor) acted as maternal donor of Et. repens (StStH), whereas species of Agropyron Gaertn. (P genome donor) acted as maternal donor of Et. pungens (EStStP). However, E genome acted as maternal donor of Et. pycnantha (EStP). This result indicated that different species served as maternal donors that contributed to species containing the same genomes. Previous findings on Et. intermedia were similar to our results [58]. Other polyploidy species in Elytrigia and diploid species containing E or St genome formed zero-length branches in Clade One because of the close relation of E and St genomes (Fig. 4). Sources of maternal donor of these genomes remain to be identified.
Differentiation and relationship between E and St genomes
In Clade I, species containing E, St, and ESt genomes and those in Cluster N-I Pse. spicata appeared at the central part, indicating close relationship of St and E genome species (Figs. 2 and 3). These findings coincide with previous findings on morphology and molecular biology [44, 72, 81]. In the present study, E and St types were obtained from species containing ESt genome grouped with Elytrigia or Pseudoroegneria diploid species, respectively. This phenomenon showed that E genome was the origin of diploid Elytrigia species with the E genome. St genome was the origin of Pseudoroegneria. Results from morphology, genetics, and molecular biology indicate that species containing E, St, and ESt genomes are closely related with Elytrigia.
Taxonomy of species with ESt and EStP genomes
Polyploidization and hybridization are long recognized as prominent forces in evolution of plant species, which feature consequences of genomic changes [22, 23]. Genome relationship and differentiation are often vague in some species because of frequent introgression of alien genes, polyploidization and chromosome segments from wide hybridization [43]. Thus, classification is one of the most important issues that require understanding.
Previous studies indicated that Et. caespitosa, Et. intermedia, Et. nodosa, Et. scythica, and Et. varnensis contain ESt genomes, which belong to Trichopyrum [3, 17, 82, 83]. Comparison of partial sequences of nrITS gene showed that a TTTT insert at positions 58–61 in nrITS sequence was detected for 11 species (Fig. 1). This finding indicated that introgression of E genome during polyploidization or different independent hybridization events may create the variants in polyploidy ESt species. In the ML tree and MJ network based on nrITS sequence, one group is formed by ESt genome species (Et. caespitosa, Et. intermedia, Et. nodosa, Et. scythica, and Et. geniculata ssp. pruinifera) and unknown genome species of Et. varnensis. This result indicated that these species should be classified under the same genus (Trichopyrum). Species containing ESt genomes were grouped with diploid species Et. elongata (E genome), suggesting that E genome may be derived from Et. elongata. In this study, diploid species of Et. elongata were differentiated. We selected two Et. elongata (Iran, France) with different origins, which are divided into Clades I-B and C (Fig. 2). A TTTT insert at positions 58–61 in the sequence was also detected for Et. elongata (W6 21,859) (Fig. 1). This result indicates that ESt genome polyploid species and diploid Elytrigia species (E genome) displayed hybridization event, resulting in divided E genome.
Et. varnensis was reported by Löve to contain ESt genomes (2n = 12× = 84) [8]. Yang [84] showed that Et. varnensis is a tetraploid species. Diversity of species ploidy may be caused by chromosome variation under natural conditions. In this study, we discovered that Et. varnensis clustered with Et. pungens, Et. pycnantha, Ag. cristatum, Au. pectinatum, Au. retrofractum, Er. distans, and Er. triticeum (85% BS and 100% PP) (Fig. 2). We concluded that this species contains P or F genome. Another estimate indicated that St genome allopolyploid species possibly resulted from introgression of Eremopyrum or Agropyron during polyploidization. Results strongly support those of previous studies in cytogenetics [85].
Possible genome constitutions and taxonomic treatment of Et. lolioides
Cytologically, Et. repens comprised StH genome. Et. lolioides is a polyploid species, and its genomic constitutions remains unknown [8, 17, 62, 86]. In the present study, Et. lolioides was clustered with diploid Pse. libanotica (St genome); this result indicated that Et. lolioides possesses one St genome. Et. lolioides clustered with H. bogdanii, H. chilense (H), and Et. repens (H copy) with high statistical result (98% BS and 100% PP) (Fig. 2). Such finding also indicated that Et. lolioides contains H genome and is closely related to StH genome species Et. repens. Thus, we can conclude that genomic constitution of Et. lolioides includes St and H genomes and belongs to Elymus s. l.
Conclusion
This study analyzed the phylogenetic relationship among Elytrigia s. l. species on the basis of the nrITS sequence data. The results supported the conclusion that Elytrigia s. l. consists of various genomes (E, H, P, and St types), which should be classified as different genera. Analyses based on nrITS sequence data and chloroplast trnL-F region show that the E, F, St, P, and W genomes have a close intergroup relationship but are distant with the H and Ns genomes. This finding strongly supports previous studies on morphology, molecular biology, and cytogenetics. nrlTS sequence analysis demonstrated that the E genome of species Et. caespitosa, Et. caespitosa ssp. nodosa, Et. intermedia, Et. scythica and Et. geniculata ssp. pruinifera, which contains ESt genomes, originated from Et. elongata in Lophopyrum. However, differentiation was found in diploid species Et. elongata; this phenomenon was possibly due to diverse geographical origins or introgression. Et. lolioides, which is composed of unknown genomes, contains the H and St genomes and has a close genetic relationship with Et. repens and El. canadensis, which contain the St and H genomes. Accordingly, the genome of Et. lolioides is inferred to contain St and H. In this paper, polyploid species of Elytrigia s. l. was deduced based on trnL-F sequence, the female parent of Et. caespitosa ssp. nodosa (PI547344), Et. farcta, Et. pontica (PI547313), Et. pycnantha, Et. scirpea and Et. scythica is the diploid species of Elytrigia s. l. containing the E genome; the maternal donor of the polyploidy species Et. caespitosa ssp. nodosa (PI547345), Et. pontica (PI383583), Et. repens, Et. geniculata ssp. pruinifera is the St genome. Different maternal donors were also found in allopolyploid species. This result could be attributed to different growth environments, introgression, or incomplete separation of genome E lineage. Thus, different haplotypes were presented.
Abbreviations
- AIC:
-
Akaike information criterion
- BI:
-
Bayesian inference
- BS:
-
Bootstrap support
- CTAB:
-
Cetyltrimethyl ammonium bromide
- MJ:
-
Median-joining
- ML:
-
Maximum likelihood
- nrITS:
-
Nuclear ribosomal internal transcribed spacer
- PP:
-
Posterior probabilities
References
Dewey DR. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In: Gustafson JP, editor. Gene manipulation in plant improvement. New York: Plenum Publishing Corporation; 1984. p. 209–80.
Lu BR. The genus Elymus L. in Asia. Taxonomy and biosystematics with special reference to genomic relationships. In: RRC W, Jensen KB, Jaussi C, editors. Proc 2nd intern Triticeae Symp. Logan: The Utah State University Press; 1994. p. 219–30.
Yen C, Yang JL. Biosystamics on Triticeae (Poaceae), vol. 5. Beijing: Agriculture Press of China; 2013. p. 303–516.
von Bothmer R, Slomon B. Triticeae: a tribe for food, feed and fun. In: Wang RRC, Jensen KB, Jaussi C, editors. Proceedings of the 2nd international Triticeae symposium. Logan: Utah State University Press; 1994. p. 1–12.
Fan X, Sha LN, Dong ZZ, Zhang HQ, Kang HY, Wang Y, Wang XL, Zhang L, Ding CB, Yang RW, Zheng YL, Zhou YH. Phylogenetic relationships and Y genome origin in Elymus L. Sensu Lato (Triticeae: Poaceae) based on single-copy nuclear Acc1 and Pgk1 gene sequences. Mol Phylogenet Evol. 2013;69(3):919–28.
Tzvelev NN. Tribe 3. Trticeae dumort. Poaceae URSS. Leningrad: Nauka Publishers; 1976. p. 176–89.
Desvaux NB. Eustachys. Paris: Sci Soc Philom; 1810.
Löve Á. Conspectus of the Triticeae. Feddes Repertorium. 1984;95(7–8):425–521.
Yen C, Yang JL, Yen Y. Hitoshi Kihara, Áskell Löve and the modern genetic concept of the genera in the tribe Triticeae(Poaceae). Acta Phytotaxonomica Sin. 2005;43(1):82–93.
Mao PS, Huang Y, Wang XG, Meng L, Mao PC, Zhang GF. Cytological evaluation and karyotype analysis in plant germplasms of Elytrigia Desv. Agric Sci China. 2010;9(11):1553–60.
Zhou YH. The circumscription and genomic evolution of Elytrigia Sensu Lato (Poaceae: Triticeae). Proc 4nd wheat genomics Mol breed China. Nanjing; 2013. p. 105–6.
Cauderon Y, Saigne B. New interspecific and intergeneric hybrids involving Agropyron. Wheat Inffoem Serv. 1961;12:13–4.
Dvořák J. Chromosoma differentitiation in polyploidy species of Elytrigia, with special reference to the evolution of diploidlike chromosome pairing in polyploidy species. Can J Genet Cytol. 1981;23(2):287–303.
McGuire PE. Chromosome pairing in triploid and tetraploid hybrids in Elytrigia (Triticeae:Poaceae). Can J Genet Cytol. 1984;26(5):519–22.
Wang RRC. Genomic analysis of Thinopyrum bessarabicum and T. elongatum. Can J Genet Cytol. 1985;27(6):722–8.
Wang RRC, Hsiao CT. Genome relationship between Thinopyrum bessarabicum and T. elongatum: revisited. Genome. 1989;32(5):802–9.
Liu ZW, Wang RRC. Genome analysis of Elytrigia caespitosa, Lophopyrum nodosum, Pseudoroegneria eniculate ssp. scythica, and Thinopyrum intermedium (Triticeae: Gramineae). Genome. 1993;36(1):102–11.
Xu J, Conner RL. Intravarietal variation in satellites and C-banded chromosomes of Agropyron intermedium Ssp. trichophorum cv. Greenleaf. Genome. 1994;37(2):305–10.
Wang RRC, von Bothmer R, Dvorak J, Fedak G, Linde-Laursen I, Muramatsu M. Genome symbolsin the Triticeae (Poaceae). In: Wang RRC, Jensen KB, Jaussi C, editors. Proceedings of the 2nd international Triticeae symposium. Utah: Utah State University; 1994. p. 29–34.
Assadi M, Runemark H. Hybridization, genomic constitution, and generic delimitation in Elymus s. l. (Poaceae: Triticeae). Plant Syst Evol. 1995;194(3–4):189–205.
Vershinin A, Svitashev S, Gummesson PO, von Salomon B, Bothemer R, Bryngelsson T. Characterization of a family of tandemly repeated DNA sequences in Triticeae. Theor Appl Genet. 1994;89(2–3):217–25.
Soltis DE, Soltis PS, Tate JA. Advances in the study of polyploidy since plant speciation. New Phytol. 2003;161(1):173–91.
Otto SP. The evolutionary consequences of polyploidy. Cell. 2007;131(3):452–62.
Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42(1):225–49.
Cui L, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A, Albert VA, Ma H, de Pamphilis CW. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006;16(6):738–49.
Yan C, Sun GL. Multiple origins of allopolyploid wheatgrass Elymus caninus revealed by RPB2, PepC and TrnD/T genes. Mol Phylogenet Evol. 2012;64(3):441–51.
Dong ZZ, Fan X, Sha LN, Wang Y, Zeng J, Kang HY, Zhang HQ, Wang XL, Zhang L, Ding CB, Yang RW, Zhou YH. Phylogeny and differentiation of the St genome in Elymus L. Sensu Lato (Triticeae; Poaceae) based on one nuclear DNA and two chloroplast genes. BMC Plant Biol. 2015;15(1):179–93.
Yan C, Sun GL, Sun DF. Distinct origin of the Y and St genome in Elymus species: evidence from the analysis of a large sample of St genome species using two nuclear genes. PLoS One. 2011;6(10):e26853.
Sun GL, Daley T, Ni Y. Molecular evolution and genome divergence at RPB2 gene of the St and H genome in Elymus species. Plant Mol Biol. 2007;64(6):645–55.
Sun GL, Ni Y, Daley T. Molecular phylogeny of RPB2 gene reveals multiple origin, geographic differentiation of H genome, and the relationship of the Y genome to other genomes in Elymus species. Mol Phylogenet Evol. 2008;46(3):897–907.
Sun GL, Pourkheirandish M, Komatsuda T. Molecular evolution and phylogeny of the RPB2 gene in the genus Hordeum. Ann Bot. 2009;103(6):975–83.
Fan X, Sha LN, Yang RW, Zhang HQ, Kang HY, Ding CB, Zhang L, Zheng YL, Zhou YH. Phylogeny and evolutionary history of Leymus (Triticeae; Poaceae) based on a single-copy nuclear gene encoding plastid acetyl-CoA carboxylase. BMC Evol Biol. 2009;9(1):247–57.
Fan X, Liu J, Sha LN, Sun GL, Hu ZQ, Zeng J, Kang HY, Zhang HQ, Wang Y, Wang XL, Zhang L, Ding CB, Yang RW, Zheng YL, Zhou YH. Evolutionary pattern of rDNA following polyploidy in Leymus (Triticeae: Poaceae). Mol Phylogenet Evol. 2014;77(1):296–306.
Sha LN, Yang RW, Fan X, Wang XL, Zhou YH. Phylogenetic analysis of Leymus (Poaceae: Triticeae) inferred from nuclear rDNA ITS sequences. Biochem Genet. 2008;46(9–10):605–19.
Sha LN, Fan X, Yang RW, Kang HY, Ding CB, Zhang L, Zheng YL, Zhou YH. Phylogenetic relationships between Hystrix and its closely related genera (Triticeae: Poaceae) based on nuclear ACC1, DMC1 and chloroplast trnL-F sequences. Mol Phylogenet Evol. 2010;54(2):327–35.
Wang XL, Fan X, Zeng J, Sha LN, Kang HY, Kang HY, Zhang HQ, Yang RW, Zhang L, Ding CB, Zhou YH. Phylogeny and molecular evolution of the DMC1 gene within the StH genome species in Triticeae (Poaceae). Genes Genom. 2012;34:237–44.
Liao JQ, Loretta R, Fan X, Sha LN, Kang HY, Zhang HQ, Wang Y, Liu J, Wang XL, Yu XF, Yang RW, Ding CB, Zhang L, Zhou YH. Phylogeny and maternal donors of the tetraploid species with St genome (Poaceae: Triticeae) inferred from CoxII and ITS sequences. Biochem Syst Ecol. 2013;50(50):277–85.
Yang ZJ, Chen ZY, Peng ZS, Yu Y, Liao ML, Wei SH. Development of a high-density linkage map and mapping of the three-pistil gene (Pis1) in wheat using GBS markers. BMC Genomics. 2017;18(1):567–75.
Turktas M, Aslay M, Kaya E, Ertugrul F. Molecular characterization of phylogenetic relationships in Fritillaria species inferred from chloroplas trnL-trnF sequences. Turkish J Biol. 2012;36(5):552–60.
Wendel JF, Schnabel A, Seelanan T. An unusual ribosomal DNA sequence from Gossypium gossypioides reveals ancient, cryptic, intergenomic introgression. Mol Phylogenet Evol. 1995;4(3):298–313.
Liu QL, Ge S, Tang HB, Zhang XL, Zhu GF, Lu BR. Phylogenetic relationships in Elymus (Poaceae: Triticeae) based on the nuclear ribosomal internal transcribed spacer and chloroplast trnL-F sequences. New Phytol. 2006;170(2):411–20.
Mahelka V, Kopecký D. Gene capture from across the grass family in the allohexaploid Elymus repens (L.) Gould (Poaceae: Triticeae) as evidenced by ITS, GBSSI, and molecular cytogenetics. Mol Biol Evol. 2010;27(6):1370–90.
Kim TW, Kim JC, Fedak G, Son JH, Park KC, Kim NS. Sequence variation in ITS spacers and 5.8S rDNA and relationgship of E, St, P, ns, Xm, and H genomes in the genera of Agropyron, Elytrigia, Leymus, Pascopyrum, Psathyrostachys, and Hordeum. Genen Genom. 2010;32(5):477–85.
Mason-Gamer RJ, Orme NL, Anderson CM. Phylogenetic analysis of north American Elymus and the monogenomic (Poaceae: Triticeae) using three chloroplast DNA data sets. Genome. 2002;45(6):991–1002.
Zhang C, Fan X, Yu HQ, Zhang L, Wang XL, Zhou YH. Different maternal genome donor to Kengyi-lia (Poaceae: Triticeae) species inferred from chloroplast trnL-F sequences. Biol Plant. 2009;53(4):759–63.
Luo XM, Tinker NA, Fan X, Zhang HQ, Sha LN, Kang HY, Ding CB, Liu J, Zhang L, Yang RW, Zhou YH. Phylogeny and maternal donor of Kengyilia species (Poaceae: Triticeae) based on three cpDNA (matK, rbcL and trnH-psbA) sequences. Biochem Syst Ecol. 2012;44(10):61–9.
Mahmoud K, Alireza B, Fatemeh S, Mohamad-Reza N, Davood A, Daniel P. Phylogenetic relationship in Fritillaria spp of Iran inferred from ribosomal ITS and chloroplast trnL-trnF sequence data. Biochem Syst Ecol. 2014;57:451–7.
Sha LN, Fan X, Zhang HQ, Kang HY, Wang Y, Wang XL, Zhang L, Ding CB, Yang RW, Zhou YH. Phylogenetic relationships in Leymus (Triticeae; Poaceae): evidence from chloroplast trnH-psbA and mitochondrial COXII intron sequences. J Syst Evol. 2014;52(6):722–34.
Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1990;12:13–5.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80.
Huelsenbeck JP, Ronquist FR. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–5.
Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8.
Wang XL, Fan X, Zhang HQ, Zhang C, Sha LN, Kang HY, Yang RW, Zhang L, Ding CB, Zhou YH. Phylogeny of the StH genome species in (Triticeae:Poaceae) evidence from chloroplast trnL-F and mitochondria COX II intron sequences. Biochem Syst Ecol. 2011;39(4–6):758–65.
Nevski SA. Uber das system der tribe Hordeae Benth. Flora et Systematica Plantae vaeculares. Ser 1. Fasc 1, Leningrad. 1933;9–32.
Nevski SA. Tribe Hordeae Benth. In: Komarov VL, Roshevits RY, Shishkin BK, editors. Flora USSR, Leningrad, Acad Sci press USSR, vol. 2; 1934. p. 590–728.
Melderis A. Elymus. In: Tutin TG, et al., editors. Floa Eueopaea, vol. 5. Cambridge: Cambridge University Press; 1980.
Li D, Zhang X. Physical localization of the 18S-5.8S-26S rDNA and sequence analysis of ITS regions in Thinopyrum ponticum (Poaceae: Triticeae): implications for concerted evolution. Ann Bot. 2002;90(4):445–52.
Zeng J, Fan X, Sha LN, Kang HY, Zhang HQ, Liu J, Wang XL, Yang RW, Zhou YH. Nucleotide polymorphism pattern and multiple maternal origin in Thinopyrum intermedium inferred by trnH-psbA sequences. Biol Plant. 2012;56(2):254–60.
Hsiao C, Chatterton NJ, Asay KH, Jensen KB. Phylogenetic relationships of the monogenomic species of the wheat tribe, Triticeae (Poaceae), inferred from nuclear rDNA (internal transcribed spacer) sequences. Genome. 1995;38(2):221–3.
Hsiao C, Wang RRC, Dewey DR. Karyotype analysis and ganome relationships of 22 diploid species in the tribe Triticeae. Can J Genet Cytol. 1986;28(1):109–20.
Wang RRC. Genome relationships in the perennial Triticeae based on diploid hybrids and beyond. Hereditas. 1992;116(1–2):133–6.
Zhang XQ, Yang JL, Yen C. The genome constitution of Roegneria grandis Keng et S.L. Chen (Poaceae: Triticeae). Acta Agrestia Sin. 1996;4(3):207–12.
Jauhar PP. Multidisciplinary approach to genome analysisin the diploid species, Thinopyrum bassarabicum and T. elongatum (Lophoprum elongatum) of the Triticeae. Theor Appl Genet. 1990;80(4):523–36.
Löve Á. IOPB chromosome number reports LXVI. Poaceae- Triticeae- Americanae. Taxon. 1980;29(1):163–9.
Löve Á. Veröff. Geobot. Inst. Rübel. 1986;87:43–52.
Clayton WD, Renvoize SA. Genera Gramineum. Grasses of the world. London: Royal Botanical Gardens; 1986.
Frederiksen S. Taxonomic studies in Eremopyrum (Poaceae). Nord J Bot. 1991;11(3):271–85.
Petersen G, Seberg O. Phylogenetic analysis of the Triticeae (Poaceae) based on rpoA sequence data. Mol Phylogenet Evol. 1997;7(2):217–30.
Petersen G, Seberg O. Molecular evolutiom and phylogenetic application of DMC1. Mol Biol Evol. 2002;22(1):43–50.
Mason-Gamer RJ. The β-amylase genes of grasses and a phylogenetic analysis of the Triticeae (Poaceae). Am J Bot. 2005;92(6):1045–58.
Wang RRC. An assessment of genome analysis based on chromosome pairing in hybrids of perennial Triticeae. Genome. 1989;32(2):179–89.
Seberg O, Frederiksen S. A phylogenetic analysis of the monogenomic Triticeae (Poaceae) based on morphology. Bot J Linn Soc. 2001;136(1):75–97.
Cassens I, Mardulyn P, Milinkovitch MC. Evaluating intraspecific “network” construction methods using simulated sequence data: do existing algorithms outperform the global maximum parsimony apprpach? Syst Biol. 2005;54(3):363–72.
Posada D, Crandall KA. Selecting the best-fit model of nucleotide substitution. Syst Biol. 2001;50(4):580–601.
Jakob SS, Blanttner FR. A chloroplast genealogy of Hordeum (Poaceae): long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Mol Biol Evol. 2006;23(8):1602–12.
Kilian B, Özkan H, Deusch O, Effgen S, Brandolini A, Kohl J, Martin W, Salamini F. Independent wheat B and G genome origins in outcrossing Aegilops progenitor haplotypes. Mol Biol Evol. 2007;24(1):217–27.
Redinbaugh MG, Jones TA, Zhang Y. Ubiquity of the St chloroplast genome in St-containing Triticeae polyploids. Genome. 2000;43(5):846–52.
Yen C, Yang JL, Baum BR. Biosystematics of Triticeae (Vol.3). Beijing: Chinese Agriculture Press; 2006.
Zeng J, Fan X, Zhang L, Wang XL, Zhang HQ, Kang HY, Zhou YH. Molecular phylogeny and mate-rnal progenitor implication in the genus Kengyilia Triticeae (Poaceae): evidence from COX II intro-n sequences. Biochem Syst Ecol. 2010;38(2):202–9.
Liu HT, Gao AN, Yang XM, Liu WH, Li XQ, Li LH. Intergenomic rearrangements after polyploidization of Kengyilia thoroldiana (Poaceae: Triticeae) affected by environmental factors. PLoS One. 2012;7(2):e31033.
Yu HQ, Fan X, Zhang C, Ding CB, Wang XL, Zhou YH. Phylogenetic relationships of species in Pseudoroegneria (Poaceae: Triticeae) and related genera inferred from nuclear rDNA ITS (internal transcribed spacer) sequences. Biologia. 2008;63(4):498–505.
Chen Q, Conner RL, Larche A, Thomas JB. Genome analysis of Thinopyrum intermedium and Th. ponticum using genomic in situ hybridization. Genome. 1998;41(4):580–6.
Yu HQ, Zhang C, Ding CB, Wang XL, Zhang HQ, Zhou YH. Genome constitutions of Pseudoroegneria geniculata, P. geniculata Ssp. scythica and P. geniculata Ssp. pruinifera (Poaceae: Triticeae) revealed by genomic in-situ hybridization. Acta Physiol Pl. 2010;32(4):645–50.
Yang Y, Zhang HQ, Fan X, Sha LN, Kang HY, Wang Y, Zhou YH. Karyomorphological studies of twelve species in Elytrigia Desv. Sensu Lato (Poaceae: Triticeae). Caryologia. 2016;69(4):315–24.
Wang RRC, Dewey DR, Hsiao C. Intergeneric hybrids of Agropyron and Pseudoroegneria. Bot Gaz. 1985;146(2):268–74.
Dewey DR. The genome structure of intermediate wheatgrass. J Hered. 1962;53(6):282–90.
Acknowledgements
We thank anonymous reviewers for their very useful comments on this manuscript. In addition, we are very thankful to the US National Plant Germplasm System for providing the material seeds.
Funding
This work was supported by the National Natural Science Foundation of China [Grant Nos. 31,270,243, 31,470,305,31,670,331 and 31,200,252], the National Key Research and Development of China (2016YFD0102000), the fund from the Science and Technology Bureau and Education Bureau of Sichuan Province, China and the Meritocracy Research Funds of China West Normal Uniersity [Grant Nos. 17YC145]. Throughout the study the funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The sequencing data from our study was deposited in the National Center for Biotechnology Information (NCBI) under the accession number MF893146- MF893190.
Author information
Authors and Affiliations
Contributions
YY designed the study, and wrote the manuscript. YY, FX, ZJ and SLN acquisited, analyzed and interpred the data; YY and WL carried out of nrITS and trnL-F sequences.KHY and ZHQ participated in its design and coordination and helped to draft the manuscript. WL and ZJ participated in the language editing; WY and YXF gave the good suggestions in the experiments and manuscript. ZYH planned the study, participated in the design of the experiments, and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Y., Fan, X., Wang, L. et al. Phylogeny and maternal donors of Elytrigia Desv. sensu lato (Triticeae; Poaceae) inferred from nuclear internal-transcribed spacer and trnL-F sequences. BMC Plant Biol 17, 207 (2017). https://doi.org/10.1186/s12870-017-1163-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-017-1163-7