
Abstract
Background
Although sleep deprivation is associated with neurobehavioral impairment that may underlie significant risks to performance and safety, there is no reliable biomarker test to detect dangerous levels of impairment from sleep loss in humans. This study employs microarrays and bioinformatics analyses to explore candidate gene expression biomarkers associated with total sleep deprivation (TSD), and more specifically, the phenotype of neurobehavioral impairment from TSD. Healthy adult volunteers were recruited to a sleep laboratory for seven consecutive days (six nights). After two Baseline nights of 10 h time in bed, 11 subjects underwent an Experimental phase of 62 h of continuous wakefulness, followed by two Recovery nights of 10 h time in bed. Another six subjects underwent a well-rested Control condition of 10 h time in bed for all six nights. Blood was drawn for measuring gene expression on days two, four, and six at 4 h intervals from 08:00 to 20:00 h, corresponding to 12 timepoints across one Baseline, one Experimental, and one Recovery day.
Results
Altogether 212 genes changed expression in response to the TSD Treatment, with most genes exhibiting down-regulation during TSD. Also, 28 genes were associated with neurobehavioral impairment as measured by the Psychomotor Vigilance Test. The results support previous findings associating TSD with the immune response and ion signaling, and reveal novel candidate biomarkers such as the Speedy/RINGO family of cell cycle regulators.
Conclusions
This study serves as an important step toward understanding gene expression changes during sleep deprivation. In addition to exploring potential biomarkers for TSD, this report presents novel candidate biomarkers associated with lapses of attention during TSD. Although further work is required for biomarker validation, analysis of these genes may aid fundamental understanding of the impact of TSD on neurobehavioral performance.
Similar content being viewed by others

Background
Sleep plays a key role in health, performance, and cognition [1,2,3,4,5]. Yet sleep deficiencies, be they from mistimed sleep, insufficient sleep, or sleep disorders, are widespread [4]. Neurobehavioral tests have revealed assorted forms of performance deficits from sleep loss, including impairment of learning and of responses to feedback in decision making [3, 6, 7]. Since its introduction over 30 years ago, the Psychomotor Vigilance Test (PVT) is one of the most commonly applied neurobehavioral assays of performance impairment due to sleep loss [8, 9]. This test assays stimulus-response time, with failure to respond within 500 ms recorded as a lapse. Sleep deprivation is associated with increased variability in stimulus-response times, and more lapses, on the PVT [10].
Besides neurobehavioral testing, efforts have been made to identify molecular biomarkers such as differentially expressed genes or metabolites affected by sleep loss [11,12,13,14,15,16,17]. A biomarker has been defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [18]. Beyond identifying the mere presence of a process or response, many biomarkers such as differentially expressed genes can provide mechanistic insights. Humans are known to differ in their sensitivity to sleep loss [19,20,21], and recent work has sought to identify biomarkers distinguishing individuals as susceptible or resistant to sleep deprivation [12, 22,23,24]. Yet surprisingly little effort has been made to synthesize molecular biomarker research with results from neurobehavioral assays.
This study tests the hypothesis that gene expression not only responds to total sleep deprivation (TSD), but also can be related to neurobehavioral impairment measured by PVT lapses. Healthy human adults acclimated to the laboratory with two nights of Baseline sleep consisting of 10 h time in bed (TIB). Then subjects in the TSD group underwent 62 h continuous wakefulness during an Experimental phase, followed by two Recovery nights of 10 h TIB. In contrast, individuals in the Control (C) group received 10 h TIB all six nights. Neurobehavioral impairment was assessed with the PVT during scheduled wakefulness, and blood was drawn for whole-transcriptome microarrays during day two, day four (hours 24–36 of wakefulness for the TSD group), and day six. The analytical approach largely focused on biomarker discovery, as defined by Mullington and colleagues [16]. Another aim was advancing mechanistic insights into the impact of sleep deprivation, as seen through a bioinformatics lens of the predicted function and molecular networks associated with the proposed biomarkers.
Methods
Sample collection and neurobehavioral data
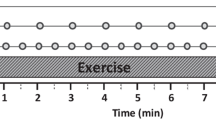
Study design and population demographics, including subjects’ prior sleep history, have been reported previously [7]. All individuals provided written informed consent, and Institutional Review Board approval was obtained at both Washington State University (WSU), and the Civil Aerospace Medical Institute (CAMI) of the Federal Aviation Administration. In brief, blood samples were obtained from 17 healthy adults (ages 22–37, 7 females) who were not using drugs. Subjects remained in the sleep laboratory at the Sleep and Performance Research Center of WSU (Spokane, WA) for six consecutive nights. Meals were semi-standardized with selection from among a limited number of menu options; blood draws were performed immediately prior to meals. A two-night Baseline phase for laboratory acclimation was followed by a two-night Experimental phase, and the study concluded with two Recovery nights. During both the Baseline and Recovery phases all subjects received 10 h TIB for sleep each night (22:00–08:00 h). For the Experimental phase, 11 randomly selected subjects underwent TSD, consisting of 62 h of continued wakefulness. The remaining 6 C subjects received the usual 10 h TIB nightly. Blood samples were collected with an intravenous catheter approximately every 4 h during time awake on days two, four, and six (Fig. 1), corresponding to 1 day each during the Baseline, Experimental, and Recovery phases. At each of the 12 timepoints (Fig. 1, Additional file 1: Table S1), 2.5 mL blood was collected in a PAXgene™ Blood RNA tube, and the number of lapses per test bout was recorded from a 10 min PVT assay (Additional file 2: Supplementary text).

Overview of the study design, consisting of 7 days (six nights) in the sleep laboratory. During the Baseline and Recovery phases, all 17 subjects received 10 h Time in Bed (TIB) for sleep each night. During the Experimental phase, the 11 Total Sleep Deprivation (TSD) persons underwent a 62 h period of continuous wakefulness, whereas the 6 Control subjects continued to have 10 h TIB each night. Vertical lines indicate the 12 timepoints with blood collection and Psychomotor Vigilance Test data analyzed here
RNA isolation and microarray data collection
Blood samples in PAXgene™ tubes were shipped to CAMI for RNA extraction on a QIAcube robot. Each total RNA sample was prepared for hybridization to Affymetrix GeneChip Human Gene 1.0 ST arrays, and scanned on an Affymetrix Scanner (Additional file 2: Supplementary text).
Differential gene expression: Treatment and PVT effects
Quality assessment was performed and Transcript Cluster-level expression values from microarrays were filtered for low expression (Additional file 2: Supplementary text). Affymetrix expression data are archived at the NCBI GEO online repository, accession GSE98582. Tests for differential expression were conducted for biomarker discovery with R/limma v. 3.24.15 using linear models [25]. Visual examination of the PVT data indicated that three of the 11 TSD subjects were fatigue resistant, in terms of neurobehavioral impairment detectable by the PVT (Additional file 2: Supplementary text – Figure S1), and these were excluded from the RMA expression file for Treatment effect analyses. Models with all 17 subjects were explored but found substantially fewer genes (Additional file 2: Supplementary text). Thus the reported Treatment effect analysis tested for a significant difference in gene expression between the 8 TSD (not fatigue resistant) and 6 C subjects at the Experimental or Recovery phase, but not at Baseline. Linear models also were run in limma to test for a significant relation of PVT lapses to gene expression, with inclusion of data for all 17 subjects. Finally, Ingenuity Pathway Analysis® biomarker filtering was done to test whether Transcript Clusters significantly related to Treatment and PVT have been found in human blood previously.
Co-expression and temporal networks
Weighted Gene Co-expression Network Analysis (WGCNA) was performed using data from all 17 subjects with the software R/WGCNA v. 1.47 [26], which grouped Transcript Clusters based on similarity of expression across samples (Additional file 2: Supplementary text). Each group of co-expressed Transcript Clusters was termed a module, and designated by a color. Pearson correlations were computed separately between each module and each of three variables: Treatment, PVT lapses, and Time of Day. The WGCNA software also allowed identification of the top hub Transcript Cluster for each module, namely, the most highly connected Transcript Cluster within the module.
A second, temporal clustering approach was performed using R/Mfuzz v. 2.28.0 [27] to visualize gene expression shifts over time in response to TSD, for those not fatigue resistant. Thus, Mfuzz was used to cluster and plot the mean expression of differentially expressed Treatment Transcript Clusters in the 8 TSD subjects across the 12 timepoints (Additional file 2: Supplementary text). The same Mfuzz clustering strategy was employed a second time on the Transcript Clusters associated with PVT lapses, again based on the average data from the 8 TSD subjects. Subsequently, Mfuzz clustering was re-run on the Treatment and PVT Transcript Clusters based on data from the Control subjects.
Transcription factor regulators
Regulatory Impact Factor (RIF) scores were assigned to known human transcription factors as a means of ranking potential regulators of the differentially expressed genes. The RIF analysis tests for a difference between the correlation of expression levels of known transcription factors, to the expression levels of a given gene list in each of two conditions (Additional file 2: Supplementary text) [28, 29]. Results are reported as z-scores, and a larger absolute value of the z-score is interpreted as greater evidence that the transcription factor has a regulatory role distinguishing the conditions. The RIF analysis was run twice, once using the Treatment effect Transcript Clusters as the input gene list, and once with the PVT effect Transcript Clusters. In each run the two conditions consisted of C and TSD.
In addition to the RIF differential co-expression analysis, the BIOBASE F-match tool (http://www.biobase-international.com) was utilized to search for regulatory transcription factors based on the promoter sequence of differentially expressed genes. The tool scanned for over-represented binding sites in the Treatment effect and in the PVT Transcript Clusters (Additional file 2: Supplementary text).
Functional enrichment and pathway analysis
Affymetrix’s online tool NetAffx™ [30] was used to annotate the Transcript Clusters, and functional enrichment of Transcript Clusters was assessed with the DAVID v. 6.7 bioinformatics tool [31]. The Ingenuity Pathway Analysis® (IPA®, QIAGEN Redwood City, http://www.qiagen.com/ingenuity) Core Analysis tool was used to explore molecular pathways and networks based on previously published interactions among genes. The IPA® Causal Network pathways [32] were reviewed for connections between differentially expressed genes and upstream regulatory molecules (Additional file 2: Supplementary text).
microRNA quantitative PCR
A 300 ng aliquot of total RNA from each sample was used for the analysis of microRNA (miRNA) expression with 30 TaqMan® assays according to the recommendations from Fluidigm® for miRNA analysis on the BioMark system. Normalized, efficiency-corrected gene expression values were analyzed via linear mixed-effects models with the R package nlme v. 3.1–126 (Additional file 2: Supplementary text).
Results
Statistical models confirmed a significant effect of the TSD Treatment on PVT lapses (Figs. 1 and 2, Additional file 2: Supplementary text). Model selection by the lowest AIC and BIC scores preferred models including a Treatment by Phase interaction (χ2 = 21, df = 2, P = 2.58E-05; AIC =783 and BIC =819) over models with just the individual terms of Treatment, Time of Day, and Phase (AIC =800 and BIC = 830), where Phase distinguishes Baseline, Experimental, and Recovery periods.

Mean (± 1 SE) Psychomotor Vigilance Test (PVT) lapses. Results represent the 11 Total Sleep Deprivation and 6 Control subjects from three of the seven consecutive study days, consisting of 1 day each during the Baseline, Experimental, and Recovery phases
Differential gene expression: Treatment and PVT effects
In the Treatment effect analysis, 225 Transcript Clusters, corresponding to 212 genes with annotated gene symbols or mRNA assignments (Additional file 3: Table S2; Additional file 2: Supplementary text – Figure S2), exhibited a significant difference between TSD and C subjects in the Experimental but not Baseline phase. Of these, 91 genes passed the IPA® human blood biomarker filter (Additional file 3: Table S2). Most of the Treatment Transcript Clusters (~ 88%) were down-regulated in TSD relative to C subjects during the Experimental phase. Only two Transcript Clusters, Cathelicidin Antimicrobial Peptide (CAMP, with False Discovery Rate (FDR) =3.56E-03 and log2 fold change of − 0.57), as well as Defensin, Alpha 4, Corticostatin (DEFA4, FDR =0.033, log2 fold change of − 1.00), were differentially expressed between TSD and C subjects in the Recovery phase but not Baseline. These Transcript Clusters also were differentially expressed in the Experimental phase (Additional file 3: Table S2). Overlap of Transcript Clusters from this study with lists of genes responding to TSD in a handful of published datasets [15, 17, 22] was low, but this was not entirely unexpected considering differences in study design, models, and even microarray platforms (Additional file 2: Supplementary text, Additional file 4: Table S3).
Thirty-four Transcript Clusters, representing 28 annotated genes (Additional file 5: Table S4, Additional file 2: Supplementary Text – Figure S3), were associated with PVT lapses. Of these, 13 genes passed the IPA® human blood biomarker filter (Additional file 5: Table S4). There were 29 Transcript Clusters down-regulated and five up-regulated as PVT lapses increased during TSD. One of the up-regulated Transcript Clusters was Outer Dense Fiber Of Sperm Tails 2-Like (ODF2L), and the remaining four belonged to the Speedy/RINGO (SPDY) cell cycle regulator gene family. There were 15 Transcript Clusters corresponding to 13 distinct genes in the PVT list not found in the Treatment list (Additional file 3: Table S2, Additional file 5: Table S4), including EF-Hand Domain Family, Member D2 (EFHD2); Ankyrin Domain Family (POTE); Growth Factor Receptor-Bound Protein 2 (GRB2); Potassium Inwardly-Rectifying Channel, Subfamily J, Member 15 (KCNJ15); and Flotillin 1 (FLOT1). Specificity of these 15 Transcript Clusters to the PVT list was robust to inclusion or exclusion of the three fatigue resistant subjects in tests of differential gene expression (Additional file 2: Supplementary text).
Co-expression and temporal networks
Weighted Gene Co-expression Network Analysis (WGCNA) was used to group Transcript Clusters with similar expression levels across samples into modules. Analyses focused on modules associated with PVT lapses. A total of 23 modules of co-expressed Transcript Clusters were constructed, along with a group of three remaining Transcript Clusters not exhibiting co-expression (Grey “module”). Each module was tested for an association with each of the key variables (Time of Day, Treatment, PVT). Some modules correlated with Time of Day, but analyses focused on the association of modules with PVT. At a Bonferroni-corrected Type I error threshold of 0.0167 to account for multiple comparisons in the Pearson tests on the three variables, the White and Darkturquoise modules were significantly correlated with PVT lapses (Fig. 3, Additional file 6: Table S5), and Darkturquoise also was significantly correlated with Treatment. The genes represented in the Darkturquoise module primarily were involved in the immune response, with a large number of immunoglobulins including its top hub Transcript Cluster Immunoglobulin Kappa Constant (IGKC) (Table 1). In the White module, several Transcript Clusters represented members of the SPDY gene family, including the top hub Transcript Cluster (Table 1). The tests of correlations of modules to key variables were run separately in contrast to the multi-factor models in limma, and the multiple testing correction was less stringent as compared to a FDR for testing thousands of Transcript Clusters. Thus, results of WGCNA were anticipated to complement limma models of differential expression, but not always be confirmatory.

Matrix of Pearson correlations from Weighted Gene Co-expression Network Analysis. Co-expression modules (rows) are separately correlated with each of three variables (columns): Treatment, Psychomotor Vigilance Test (PVT) lapses, and Time of Day. Positive correlations are shown in red, and occur when gene expression was higher in samples from Total Sleep Deprivation relative to Control subjects, in samples with higher PVT lapses, and in samples at later times of day. Negative correlations are blue. Correlation coefficients are depicted for the correlation of each module and variable, with P-values in parentheses
A second clustering approach with Mfuzz aided visualization of changes across timepoints. Running Mfuzz clustering of the Treatment effect Transcript Clusters in TSD subjects resulted in a single group of 26 Transcript Clusters up-regulated during the Experimental phase (Mfuzz Treatment Group 2), whereas Transcript Clusters in the other two Mfuzz groups were down-regulated (Fig. 4, Additional file 3: Table S2, Additional file 2: Supplementary text). Down-regulated Mfuzz Treatment Group 1 contained 104 Transcript Clusters, and DAVID analysis revealed functional enrichment of ion binding and cell adhesion. Genes represented in Mfuzz Treatment Group 1 included Argonaute RISC Catalytic Component 4 (AGO4), Prostaglandin-Endoperoxide Synthase 2 (PTGS2), Casein Kinase 1, Alpha 1-Like CSNK1A1L, Protein Kinase C, Beta (PRKCB), Lipopolysaccharide-Induced TNF Factor (LITAF), Interleukin 1B (IL1B), Eukaryotic Translation Initiation Factor 4E Family Member 3 (EIF4E3), and Glycogen Synthase Kinase 3 Beta (GSK3B). The up-regulated Mfuzz Treatment Group 2 was smaller and lacked significantly enriched functional clusters in DAVID; members included Transcript Clusters associated with B cell signaling, and genes in the SPDY family. Down-regulated Mfuzz Treatment Group 3 contained 94 Transcript Clusters, with functional enrichment of immunoglobulins, cell motility, and the inflammatory response. Among the genes in this group were Hypoxia Inducible Factor 1, Alpha Subunit (Basic Helix-Loop-Helix Transcription Factor) (HIF1A), Chemokine (C-X-C motif) Receptor 2 (CXCR2), Solute Carrier Family 11 (Proton-Coupled Divalent Metal Ion Transporter), Member 1 (SLC11A1), Chemokine (C-X-C motif) Receptor 1 (CXCR1), Interleukin 17 Receptor A (IL17A), Cytoplasmic Polyadenylation Element Binding Protein 4 (CPEB4), and Immediate Early Response 3 (IER3).

Temporal expression of microarray Transcript Clusters that are significant for the Treatment effect. Gray lines represent individual Transcript Clusters, and the thick black line represents the group center. Values represent normalized expression based on data from Total Sleep Deprivation (TSD) subjects during three of the seven consecutive study days, consisting of 1 day each during the Baseline, Experimental, and Recovery phases
Although members of both Mfuzz Treatment Groups 1 and 3 based on TSD subject data exhibited down-regulation during the Experimental phase, the temporal patterns differed slightly. Mfuzz Treatment Group 1 showed highest expression at midday in Recovery and a weak trend toward highest expression around noon in Baseline, which was disrupted by down-regulation in the Experimental phase. Overall, Mfuzz Treatment Group 3 Transcript Clusters had their highest expression during Baseline at 08:00 h and decreased expression to 20:00 h. Not only was this pattern disrupted during the Experimental phase similar to Mfuzz Treatment Group 1, but also there was a delay in the maximum expression until 12:00 h in the Recovery phase.
When Transcript Clusters associated with PVT lapses were temporally clustered based on data from TSD subjects, there again were two Mfuzz down-regulated groups and one up-regulated group during the Experimental phase (Fig. 5, Additional file 5: Table S4, Additional file 2: Supplementary text). For down-regulated Mfuzz PVT Group 1 with 15 Transcript Clusters, cell motility was the only significantly enriched functional cluster found in DAVID. Members of Mfuzz PVT Group 1 included Aquaporin 9 (AQP9), chemokine receptors CXCR1 and CXCR2, and HIF1A. In down-regulated Mfuzz PVT Group 2 with 14 Transcript Clusters, only an ion transport group was significantly enriched. Included in Mfuzz PVT Cluster 2 were LITAF, KCNJ15, and FLOT1. There were no significant enrichment terms for Mfuzz PVT Group 3, which consisted of four Transcript Clusters representing the SPDY family (red and pink lines in Fig. 5), plus OD2FL.

Temporal expression of microarray Transcript Clusters significant for the Psychomotor Vigilance Test effect. Gray lines represent individual Transcript Clusters, and the thick black line represents the group center. Values represent normalized expression based on data from Total Sleep Deprivation (TSD) subjects during three of the seven consecutive study days, consisting of 1 day each during the Baseline, Experimental, and Recovery phases
Similar to Group 3 of the Mfuzz Treatment results on TSD subjects, the Mfuzz PVT Group 1 for TSD subjects showed a pattern of decreasing expression from 08:00 to 20:00 at Baseline that shifted to overall low expression in the Experimental phase, with continuing distortion during the Recovery phase. The Mfuzz PVT Group 2 was roughly similar in expression pattern to Mfuzz Treatment Group 1 in having a circadian maximum midday in the Baseline and Recovery phases, which was depressed during the Experimental phase. Mfuzz PVT Group 3 exhibited a roughly similar expression pattern to Treatment Group 2, with up-regulation during the Experimental phase.
Overall, Mfuzz clustering of Treatment and PVT Transcript Clusters based on expression values from Control subjects showed differences from plots made on TSD subject data (Additional file 2: Supplementary text, Additional file 2: Supplementary text – Figs. S4 and S5). However, clustering resulted in Transcript Clusters sorting themselves out differently in runs of TSD and C subject data. Individual Transcript Clusters in Group 1 of the Mfuzz cluster from TSD subjects’ data are not all found in Group 1 of the Mfuzz results for Control subjects, and so on. There was roughly 75% overlap of Transcript Clusters associating with the same group in runs of Mfuzz on the Treatment list for C and TSD subjects. For PVT there was a reasonable amount of overlap for Mfuzz Group 3, but very poor relation of Transcript Cluster membership for the other two groups between C and TSD plots. Transcript Clusters in Mfuzz Group 2 for Control subjects, were roughly evenly split between Mfuzz Group 1 and 2 for the TSD subjects. Therefore, caution is needed when comparing specific groups between plots of TSD and C subjects’ data.
Transcription factor regulators
Regulatory Impact Factor z-scores were used to rank human transcription factors as potential regulators of the differentially expressed Transcript Clusters, first for the Treatment list and second for PVT Transcript Clusters. For the Treatment list, no transcription factors had RIF z-scores > 2, whereas 41 had z-scores <− 2. The smallest RIF value corresponded to the ELK3, ETS-Domain Protein - SRF Accessory Protein 2 (ELK3), with RIF z-score = − 7.40. From the PVT list, 7 Transcript Clusters had z-scores > 2 including the Basic Helix-Loop-Helix Family Member 40 (BHLHE40, RIF z-score = 2.64), and 25 had z-scores <− 2. The largest absolute value of the RIF score again was for ELK3, with RIF z-score = − 6.36.
To further characterize potential regulators of the response to TSD, the BIOBASE F-match algorithm was used to assess potential transcription factor binding sites in the promoters of Transcript Clusters from the Treatment and PVT lists. Seven transcription factor matrices were predicted to regulate genes that respond to Treatment: c-Myb, E2A, Ets, GKLF, E-box, GLI, and myogenin. Only Hic1 met the screening criteria for the PVT list.
Two matrices, Ets and E-box, were identified by F-match for the Treatment list, and associated with transcription factors having an RIF z-score > 2 or < − 2. The Ets and E-box binding matrices were found in many Treatment Transcript Clusters by F-match, and among transcription factors known to bind these matrices were four genes with high RIF z-scores: Upstream Transcription Factor 1 (USF1) with an E-box binding site; Transcription Factor 4 (TCF4) with an E-box binding site; GA Binding Protein Transcription Factor, Alpha Subunit 60 kDa (GABPA) with an Ets binding site; and ELK3 with an Ets binding site (Table 2). Three of these transcription factors, ELK3, TCF4, and USF1, had an RIF z-score < − 2 in the PVT list; however, F-match PVT results only identified the Ets matrix as over-represented against three background sets, and the E-box motif against two backgrounds sets. Expression levels of the gene Hypermethylated in Cancer 1 (HIC1) did not meet the low-expression filter (Methods), and thus it was excluded from the RIF analysis.
Functional enrichment and pathway analysis
Functional enrichment in DAVID was similar for both the Treatment and PVT effects, albeit with fewer findings for the much smaller list of differentially expressed PVT Transcript Clusters (Tables 3 and 4). Several enriched functional categories were related to the immune response and motility, including immunoglobulins, components of the inflammatory response, cell adhesion, and calcium ion binding. There were no significantly enriched functional clusters for the up-regulated Transcript Clusters of the Treatment list (Mfuzz Treatment Group 2, Fig. 4) or PVT list (Mfuzz PVT Group 3, Fig. 5), likely due to the small number of up-regulated Transcript Clusters. Most Transcript Clusters were down-regulated.
The 28 genes associated with PVT lapses were part of multiple upstream Causal Networks in IPA®, 25 of which were connected to at least 10 of the differentially expressed PVT genes. One had similarity to a network from the Treatment list in that the Bradykinin Receptor (BDKR) was a master regulator in both (Additional file 2: Supplementary text – Figure S6, Network B). This network was significantly enriched for both the Treatment analysis (P = 8.75E-04) and PVT analysis (P = 4.22E-03). Both Treatment and PVT Causal Networks provided evidence for purinergic activity, but with different purine type receptors as the master regulator: P2RX4 for Treatment (P = 9.16E-04), and P2RX7 for PVT (P = 2.46E-03) (Additional file 2: Supplementary text – Figure S7, Network P). A PVT Causal Network of interest (P = 1.72E-03) that contained evidence of ion channel activity had Adenylate Cyclase (ADCY) as its master regulator (Fig. 6, Network A). Also, a PVT Causal Network pathway with the master regulator DnaJ Heat Shock Protein Family (HSP40), known as DNAJ (Additional file 2: Supplementary text – Figure S8, Network D), predicted up-regulation of several heat shock proteins (P = 4.09E-03). These include DNAJ, Heat Shock Protein 70 (HSP70) and Heat Shock Protein 90 (HSP90). However, there were no corresponding Treatment Causal Networks with ADCY or DNAJ as the master regulator. The P-values reported above for causal networks are raw P-values of overlap; the network bias-corrected P-values likewise fall below a P-value threshold of 0.05 for the BDKR and P2RX4 networks from the Treatment dataset, as do those for ADCY, BDKR, P2RX7, and DNAJ networks from the PVT dataset (Additional file 7: Table S6, Additional File 8: Table S7).

Ingenuity Pathway Analysis® Causal Network pathway A with master regulator Adenylate Cyclase (ADCY). Pathway A was predicted from the Psychomotor Vigilance Test effect gene list. Lighter molecule color suggests less confidence in prediction of the direction of expression; darker indicates more confidence
microRNA qPCR
None of the tested miRNAs were significantly (FDR < 0.05) associated with Treatment, but three were significantly related to PVT lapses: microRNA 152 (MIR152), microRNA 27b (MIR27B), and microRNA 24 (MIR24) (Additional file 9: Table S8). Evidence is slightly weaker for MIR27B based on visual inspection of plots, with separation during the final Baseline timepoint of mean (±1 SE) expression between C and TSD fatigue susceptible subjects (Additional file 2: Supplementary text – Figure S9).
Discussion
This study identifies candidate biomarkers for acute total sleep deprivation in humans, as well as promising candidates for a biomarker test of neurobehavioral impairment caused by TSD. Moreover, functional enrichment analyses and prediction of molecular networks advanced mechanistic insights into the impact of sleep deprivation. Some of the difficulty identifying biomarkers for sleep deprivation [16] may be caused by the large inter-individual variability in responses to sleep loss. In particular, the greater ability of some persons to resist performance degradation during sleep loss has been recognized for over a decade [19]. In the present study three out of 11 TSD subjects were identified as fatigue resistant in terms of PVT lapses (Additional file 2: Supplementary Text – Figure S1). By testing for the relationship of gene expression in blood to PVT lapses that encompass some of this variability, additional biomarkers were found that were not identified by assessment of a simple sleep deprivation Treatment effect. Of course the same was true in reverse, as we identified 212 Treatment effect genes in blood (Additional file 3: Table S2) and a mere 28 genes associated with PVT lapses (Additional file 5: Table S4). Detecting a relationship of gene expression with PVT lapses may be more difficult, considering the added complexity, greater outcome specificity [33], and perhaps narrower suite of associated genes for neurobehavioral traits.
While identifying genes associated with a sleep loss Treatment has value, biomarkers for neurobehavioral impairment such as our list of genes associated with PVT may aid fundamental understanding of the relationship between sleep and cognition. Shifting the focus from sleep deprivation biomarkers, to biomarkers for impairment from sleep deprivation, strengthens characterization of the molecular basis of the phenotype. By directly assaying the molecular changes associated with neurobehavioral performance, and drawing predictions of associated impacts on function, this research enhances understanding of the relation between sleep loss and capacity for sustained attention.
Most genes identified in this study exhibited down-regulation in TSD relative to C during the Experimental phase, a pattern consistent with prior studies in humans such as [15]. As reviewed by Mackiewicz et al. [34], sleep is associated with macromolecule biosynthesis, and prolonged wakefulness leads to down-regulation of genes associated with multiple metabolic processes. The current study indicates potential effects on translation in the down-regulation of Cytoplasmic Polyadenylation Element Binding Protein 4 (CPEB4, Treatment and PVT effect lists) and Eukaryotic Translation Initiation Factor 4E Family Member 3 (EIF4E3, Treatment list only). The CPEB4 gene is one of four vertebrate cytoplasmic polyadenylation binding proteins that regulates translation via effects on poly(A) elongation [35, 36]. The phosphorylated form of the CPEB protein promotes translation of mRNAs with roles in learning, memory, and synaptic plasticity [36, 37], which may explain its relation to PVT lapses. Grønli and colleagues report that sleep deprivation leads to reduced phosphorylation of Cpeb in the hippocampus and Eif4e in the dentate gyrus of rats [37]. Although the EIF4E gene was not significantly related to PVT lapses in the present study, it was down-regulated in response to the TSD Treatment. The protein EIF4E is a component of the translation initiation complex [38]; decreased levels of this protein would be detrimental to synthesis of new protein and could contribute to the known effects of sleep loss on macromolecular biosynthesis.
Sleep deprivation biomarkers and immunity
Cytokine and stress-associated networks frequently are associated with sleep deficits [15, 39], and results here further support the association of TSD with the immune system. Ingenuity Pathway Analysis® Causal Networks detected in both Treatment and PVT analyses have BDKR as the master regulator (Additional file 2: Supplementary Text – Figure S6, Network B). Bradykinin receptors are mediators of the inflammatory response [40], as indicated by inclusion of differentially expressed genes such as the chemokine CXCR1 and transcription factor LITAF in the PVT network. The LITAF gene is a key mediator of the inflammatory cytokine response to lipopolysaccharides [41]. Multiple genes related to the immune system were down-regulated in both Treatment and PVT lists, including LITAF, CXCR1, and CXCR2. The genes IL17RA and IL1B were down-regulated for Treatment only.
In contrast, several reviews suggest that sleep loss results in increased levels of cytokines such as IL1 [39, 42,43,44]. While many reports are based on protein assays, studies reviewed by Krueger [42] have shown that in brain, IL1 mRNA increases during sleep deprivation. However, results in the present study are based on blood rather than brain samples. Also, much of the IL1 data in the reviews are derived from studies of animals, particularly rodents, and results may differ in humans. Details of the experimental design such as the time of measurement also may influence results. For example, in human blood higher mRNA levels of IL1B are found in day vs. nighttime samples [17].
Nonetheless, findings in the current study do indicate that specific aspects of the immune system were up-regulated. For example, an up-regulated group of Treatment effect genes (Mfuzz Treatment Group 2 – TSD subjects’ data, Fig. 4) contains members associated with B cell signaling. This is consistent with the study by Aho and colleagues [45] of leukocyte gene expression in humans following partial sleep restriction, in which B cell activation is among the top up-regulated Gene Ontology pathways. Besides cytokines, the immune genes CAMP and DEFA4 are of interest. These molecules were significantly down-regulated in TSD subjects in both Experimental and Recovery phases. Their continuing down-regulation suggest the need for more than one Recovery night of sleep to restore molecular homeostasis. This idea is further supported by the Mfuzz plots for TSD subjects showing potential circadian disruption in the Recovery phase, with gene expression patterns from morning to evening differing from the temporal patterns observed at Baseline (Figs. 4 and 5). Nevertheless, there appear to be some potential differences during Baseline between Mfuzz TSD and C clusters (Additional file 2: Supplementary text – Figs. S4–S5), warranting some caution in over-interpreting circadian trends from Mfuzz. Yet as aforementioned, care is needed in comparing results of Mfuzz for C and TSD subjects due to differences in Transcript Cluster membership among Mfuzz groups (Results, Additional file 2: Supplementary text).
Besides immunity, sleep deprivation typically is associated with evidence of a stress response including induction of heat shock proteins [34, 39, 46, 47]. One of the PVT Causal Networks predicts up-regulation of stress response genes including HSP70 and HSP90 (Network D, Additional file 2: Supplementary Text – Figure S8). Differentially expressed genes in this network included cytokine receptors CXCR1 and CXCR2, as well as the transcription factors HIF1A and LITAF. While HIF1A is known for its role in activating hypoxic response genes, recent work suggests that HIF1A induction from hypoxia caused by obstructive sleep apnea may disrupt circadian rhythms [48]. Overall stress response networks and cytokines may eventually contribute to a larger biomarker panel for diagnosing TSD, but by themselves such genes may be too variable or too pleiotropic to discriminate between sleep loss and other phenotypes such as illness.
Homeostatic and circadian clock genes
Overlap between the circadian and homeostatic sleep processes is increasingly documented [49, 50], and results here further suggest a link with neurobehavioral function. Among the high-scoring transcription factors in the RIF analysis was BHLHE40 (also known as DEC1), which together with Basic Helix-Loop-Helix Family Member 41 (BHLHE41, also known as DEC2) acts as a transcriptional repressor of the Circadian Locomoter Output Cycles Kaput (CLOCK)/Brain and Muscle ARNT-Like 1 (BMAL1) promoter [51, 52]. Mutations of BHLHE41 have been associated with resisting effects of sleep loss in humans [53]. Interestingly 62 of the genes identified by Möller-Levet et al. as rhythmic in a well-rested condition [17], were identified in our Treatment list, and six were found in the PVT list (Additional file 4: Table S3).
Additionally, expression profiles of three miRNAs were significantly related to PVT lapses: MIR24, MIR27 B, and MIR152 (Additional file 9: Table S8). MicroRNAs are known for their roles in regulating gene expression [54], and have been associated with sleep and neurodegenerative disease [55]. Due to their relation to PVT lapses in the current study, these three miRNAs are intriguing candidates for regulating the molecular mechanism linking sleep deprivation and sustained attention. In mice Mir27 b regulates the clock gene Bmal1 at the posttranscriptional level [56]. Although not part of the ADCY Causal Network A generated with IPA® (Fig. 6), bioinformatics analyses suggest that Mir27 b interacts with the Adenylate cyclase 6 (Adcy6) gene [57]. It has been proposed that Mir24 plays a role in regulating the period genes in mice [58], and based on sequence analysis in humans, MIR24 is predicted to interact with Cryptochrome Circadian Clock 2 (CRY2) and Period Circadian Regulator 2 (PER2) [59]. Finally we note that microRNAs themselves can exhibit circadian rhythm in their expression. For example, Mir152 exhibits diurnal oscillations in mice [60]. Plasma samples in humans revealed diurnal oscillations in MIR24 [59], although evidence is mixed for such rhythmicity in MIR27 B [57, 59].
Transcription factor analyses highlight further regulators with possible roles in both homeostatic and circadian processes, such as USF1. In mammals, the CLOCK/BMAL1 protein heterodimer binds E-boxes in the promoters of the Period (PER1 and PER2) and Cryptochrome (CRY1 and CRY2) genes leading to their activation, and the protein products of these genes repress the CLOCK/BMAL1 complex and in turn their own expression, until degradation of PER and CRY products releases CLOCK/BMAL1 [61,62,63]. Oscillations in this molecular clock contribute to initiating circadian rhythms. USF1, like the CLOCK/BMAL1 heterodimer, binds E-box regulatory sites but with peak binding at night, antiphase to CLOCK/BMAL1 [61]. It has been proposed that USF1 may help generate circadian rhythms by maintaining an open chromatin state, enhancing the ability of CLOCK/BMAL1 binding to the E-boxes on the next circadian cycle [61]. In the current study, not only was there a strong prediction of a regulatory role for USF1 in the RIF analysis, but also the Biobase F-match tool revealed over-representation of E-box binding sites in the differentially expressed genes.
Other genes with regulatory roles supported by both RIF and F-match analyses were GABPA, TCF4, and ELK3. The protein encoded by the GABPA gene is a transcription factor that may function in human cognition [64]. Previous research on chronic sleep restriction in humans suggested a possible association between GABPA and gene down-regulation [45], but more work is needed to elucidate the relation of TCF4 and ELK3 to sleep deprivation. It is noted that RIF and F-match test for regulatory effects in distinctly different ways (Additional file 2: Supplementary text) and should be considered complementary, not necessarily confirmatory.
Novel biomarkers and genes specific to neurobehavioral impairment
Altogether 13 genes represented by 15 Transcript Clusters were associated with PVT lapses but not with Treatment (Additional file 2: Supplementary text; Additional file 5: Table S4), including FLOT1. In mice, flotillins are up-regulated with sleep and down-regulated with sleep deprivation [47], which in the present study would be seen as a Treatment effect. Due to their association with lipid rafts, flotillins may have a role in neurotransmitter signaling [34, 47]. In contrast to the results of Mackiewicz et al. [47], lack of a Treatment effect in the current study could reflect a difference between mice and humans. Confirming the absence of a Treatment effect for the 13 genes specific to the PVT analysis will require additional studies with more individuals. Nevertheless, a tantalizing hypothesis is that these 13 genes are specifically related to the mechanism by which TSD affects the capacity for sustained attention.
Another down-regulated gene specifically associated with PVT was KCNJ15, an inward rectifying potassium channel proposed to be a key component of the potassium circadian cycle [65]. It has been suggested that cycling of sodium and potassium currents is an evolutionarily conserved mechanism of governing clock neurons in the brain [66]. Recent work points to the role of neuromodulators influencing extracellular ion concentrations in the brain, in turn impacting sleep/wake activity [67]. Further evidence linking PVT lapses and ion channels exists in the IPA® Causal Network A (Fig. 6). The direction of change of the differentially expressed genes within this network was consistent with inhibition of an L-type calcium channel complex and activation of Potassium Calcium-Activated Channel Subfamily N Member 4 (KCCN4). In mice, knockouts of Kccn4 lead to reduced sleep duration [68].
Beyond suggesting a role of ion signaling in TSD and the resulting neurobehavioral deficits, Network A (Fig. 6) was intriguing due to the implications for cyclic adenosine monophosphate (cAMP) signaling. In general, activation of adenylate cyclase leads to production of cAMP [69]. Here it was predicted that ADCY was down-regulated, which in turn would reduce cAMP levels. Other components of Network A include predicted down-regulation of complexes for protein kinase A (PKA) and phospho-cAMP response element binding protein (CREB), which are implicated in memory storage [70]. A study in mice demonstrated that increasing cAMP in hippocampal neurons can rescue the typical memory consolidation impairment caused by sleep deprivation [71]. Via its impact on cAMP, this network also implicates the immune system.
Narasimamurthy and colleagues [72] proposed a model in which Cryptochrome 1 (CRY1) inhibits adenylate cyclase, reducing levels of cAMP and ultimately of IL6. In this study the IL6 gene was excluded from analyses due to its low expression across multiple samples, but reduction of this cytokine would be consistent with the predictions of inhibition of the immune system. Additional Causal Networks contained purine type 2X7 (PVT) and 2X4 (Treatment) receptors (Additional file 2: Supplementary text – Figure S7, Network P). Binding of ATP to P2X4 receptors is known to promote REM sleep, whereas binding to P2X7 receptors promotes non-REM sleep [43]. As reviewed previously [73, 74], adenosine binding to P2X7 receptors has been implicated in effecting the cumulative deficits in PVT performance due to chronic sleep loss, but these receptors also can act independently of adenosine to promote the release of sleep regulatory substances. Adenosine itself is a sleep regulatory substance as stated in reviews [43, 74], although causal roles for the molecule in sleep homeostasis are debated [74].
Genes within the SPDY family constitute a new group of candidate biomarkers for the effects of TSD. In differential expression analysis of both Treatment and PVT lapses, Transcript Clusters for SPDY genes were up-regulated (Figs. 4 and 5), and WGCNA grouped several members of the SPDY family in a co-expression module (White) correlated with PVT lapses (Fig. 3, Additional file 6: Table S5). The SPDY members can activate cyclin-dependent kinases independent of cyclin activity, and they function in cell cycle progression, meiotic maturation, and the DNA damage response [75, 76]. To the authors’ knowledge the SPDY gene family has not previously been associated with sleep deprivation, although Cyclin A has been linked to sleep-wake transitions and the sleep homeostat in Drosophila [77]. The best known member of this family, SpdyA (also known as Spy1), was shown to be expressed in the lumbar spinal cord of adult rats and may function in nerve regeneration [78, 79]. Meanwhile, a growing body of research points to effects of sleep deprivation on adult neurogenesis, albeit results seem to vary among studies and may depend on the extent of sleep deprivation [80]. Combining these findings one can hypothesize that sleep deprivation induces the SPDY family, thereby altering cell cycle progression and neurogenesis. However, this gene family has high sequence homology making unambiguous identification of the relevant family members difficult.
Future directions
The present investigation was aimed at biomarker discovery, and preliminary analysis of biomarker function and associated molecular networks. As with any such project, further work is required for biomarker validation [16]. This should include additional comparisons with published datasets, and collection of new data from more subjects. Collecting nighttime blood samples from control individuals without disturbing their sleep would be helpful for refining the association of biomarkers with neurobehavioral impairment across the circadian cycle. Also, many of the genes associated with neurobehavioral impairment from TSD in this study are connected to several different regulatory pathways, suggesting the potential for pleiotropic roles. For example, Networks A, B, D, and P for PVT all contain six of the 28 differentially expressed PVT genes, Aquaporin 9 (AQP9), Arrestin Domain Containing 3 (ARRDC3), CPEB4, ELOVL Fatty Acid Elongase 5 (ELOVL5), HIF1A, and Lymphocyte Cytosolic Protein 1 (LCP1) (Fig. 6, Additional File 2: Supplementary text – Figs. S6–S8). Moreover, these four networks all contain two ligand-dependent transcription factors, Nuclear Receptor Subfamily 3, Group C, Member 1 (NR3C1), and Peroxisome Proliferator Activated Receptor Alpha (PPARA). While all four networks clearly predict down-regulation of PPARA, for the glucocorticoid receptor NR3C1 evidence of down-regulation is stronger in networks A and P. The PPARA protein is important to coordinating rhythmic gene expression, and it interacts with the period gene PER2 [81]. Ultimately, confirmation of predicted pathway networks and regulatory molecules will require targeted laboratory studies, and a different approach will be needed to test for causation and verify mechanistic insights (e.g., gene knockout or silencing assays).
In this study (Additional file 3: Table S2, Additional file 5: Table S4), as in prior research on sleep and gene expression [22], fold change values typically were low. Validation of biomarker panels will require additional data collection from more subjects to increase power, and to adequately represent the continuum of sleep deprivation responses. However, identification of similar themes in multiple different analyses increases confidence in the results presented here. For example, the WGCNA co-expression module most strongly correlated with PVT lapses contains several SPDY genes, congruent with identification of SPDY members in the PVT differential expression list. Of course findings here are specific for measurements in blood. Gene expression may vary across fluid and tissue types, although concordance as high as 80% has been reported between the transcriptome of blood and major tissues [82, 83].
In the future, separate analyses of fatigue resistant individuals to determine any gene expression patterns unique to them could advance understanding of the ability to maintain neurobehavioral functioning during sleep deprivation. Because the current study only included three fatigue resistant subjects, this is left to future studies with a larger sample size.
Conclusion
In this study, gene expression was associated with not only total sleep deprivation, but also PVT lapses of attention following sleep loss. The 13 genes unique to the PVT analysis may be particularly promising candidates for exploring the relationship between TSD and the capacity for sustained attention at a molecular level. This has added relevance in light of findings that sleep deprivation affects multiple distinct aspects of cognition differentially [7, 33, 84]. Although the PVT is perhaps the most widely used neurobehavioral assay in sleep deprivation research, tests are available for other components of cognition, and gene expression patterns associated with other processes such as decision making should be examined. These comparisons may yield new biomarker panels or identify genes that overlap with the PVT list here, providing novel insights regarding the molecular changes associated with the response of diverse aspects of neurobehavioral performance to sleep loss. Other fields such as proteomics and metabolomics may yield further biomarker candidates and capture additional phenomena, such as post-translational effects.
References
Franzen PL, Siegle GJ, Buysse DJ. Relationships between affect, vigilance, and sleepiness following sleep deprivation. J Sleep Res. 2008;17:34–41. https://doi.org/10.1111/j.1365-2869.2008.00635.x.
Van Dongen HPA, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–26.
Killgore WDS. Effects of sleep deprivation on cognition. In: Gerard AK, Hans PAD, editors. Human sleep and cognition, part I: basic research. Progress in brain research, vol. 185. Amsterdam: Elsevier; 2010. p. 105–29.
Laposky AD, Van Cauter E, Diez-Roux AV. Reducing health disparities: the role of sleep deficiency and sleep disorders. Sleep Med. 2016;18:3–6. doi: https://doi.org/10.1016/j.sleep.2015.01.007.
Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–9. doi: https://doi.org/10.1016/S0140-6736(99)01376-8.
Lim J, Dinges DF. A meta-analysis of the impact of short-term sleep deprivation on cognitive variables. Psychol Bull. 2010;136:375–89. https://doi.org/10.1037/a0018883.
Whitney P, Hinson JM, Jackson ML, Van Dongen HPA. Feedback blunting: total sleep deprivation impairs decision making that requires updating based on feedback. Sleep. 2015;38:745–54. https://doi.org/10.5665/sleep.4668.
Lim J, Dinges DF. Sleep deprivation and vigilant attention. Ann N Y Acad Sci. 2008;1129:305–22. https://doi.org/10.1196/annals.1417.002.
Dinges DF, Powell JW. Microcomputer analyses of performance on a portable, simple visual RT task during sustained operations. Behav Res Methods Instrum Comput. 1985;17:652–5. https://doi.org/10.3758/bf03200977.
Doran SM, Van Dongen HPA, Dinges DF. Sustained attention performance during sleep deprivation: evidence of state instability. Arch Ital Biol. 2001;139:253–67.
Davies SK, Ang JE, Revell VL, Holmes B, Mann A, Robertson FP, et al. Effect of sleep deprivation on the human metabolome. Proc Natl Acad Sci U S A. 2014;111:10761–6. https://doi.org/10.1073/pnas.1402663111.
Goel N. “Omics” approaches for sleep and circadian rhythm research: biomarkers for identifying differential vulnerability to sleep loss. Curr Sleep Med Rep. 2015;1:38–46. https://doi.org/10.1007/s40675-014-0003-7.
Weljie AM, Meerlo P, Goel N, Sengupta A, Kayser MS, Abel T, et al. Oxalic acid and diacylglycerol 36:3 are cross-species markers of sleep debt. Proc Natl Acad Sci U S A. 2015;112:2569–74. https://doi.org/10.1073/pnas.1417432112.
Archer SN, Oster H. How sleep and wakefulness influence circadian rhythmicity: effects of insufficient and mistimed sleep on the animal and human transcriptome. J Sleep Res. 2015;24:476–93. https://doi.org/10.1111/jsr.12307.
Pellegrino R, Sunaga DY, Guindalini C, Martins RCS, Mazzotti DR, Wei Z, et al. Whole blood genome-wide gene expression profile in males after prolonged wakefulness and sleep recovery. Physiol Genomics. 2012;44:1003–12. https://doi.org/10.1152/physiolgenomics.00058.2012.
Mullington JM, Abbott SM, Carroll JE, Davis CJ, Dijk D-J, Dinges DF, et al. Developing biomarker arrays predicting sleep and circadian-coupled risks to health. Sleep. 2016;39:727–36. https://doi.org/10.5665/sleep.5616.
Möller-Levet CS, Archer SN, Bucca G, Laing EE, Slak A, Kabiljo R, et al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc Natl Acad Sci U S A. 2013;110:E1132–41. https://doi.org/10.1073/pnas.1217154110.
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95.
Van Dongen HPA, Baynard MD, Maislin G, Dinges DF. Systematic interindividual differences in neurobehavioral impairment from sleep loss: evidence of trait-like differential vulnerability. Sleep. 2004;27:423–33.
Patanaik A, Kwoh CK, Chua ECP, Gooley JJ, Chee MWL. Classifying vulnerability to sleep deprivation using baseline measures of psychomotor vigilance. Sleep. 2015;38:723–34. https://doi.org/10.5665/sleep.4664.
Van Dongen HPA, Bender AM, Dinges DF. Systematic individual differences in sleep homeostatic and circadian rhythm contributions to neurobehavioral impairment during sleep deprivation. Accid Anal Prev. 2012;45:11–6. https://doi.org/10.1016/j.aap.2011.09.018.
Arnardottir ES, Nikonova EV, Shockley KR, Podtelezhnikov AA, Anafi RC, Tanis KQ, et al. Blood-gene expression reveals reduced circadian rhythmicity in individuals resistant to sleep deprivation. Sleep. 2014;37:1589–600. https://doi.org/10.5665/sleep.4064.
Rupp TL, Wesensten NJ, Newman R, Balkin TJ. PER3 and ADORA2A polymorphisms impact neurobehavioral performance during sleep restriction. J Sleep Res. 2013;22:160–5. https://doi.org/10.1111/j.1365-2869.2012.01062.x.
Satterfield BC, Wisor JP, Field SA, Schmidt MA, Van Dongen HPA. TNFα G308A polymorphism is associated with resilience to sleep deprivation-induced psychomotor vigilance performance impairment in healthy young adults. Brain Behav Immun. 2015;47:66–74. doi: https://doi.org/10.1016/j.bbi.2014.12.009.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. https://doi.org/10.1093/nar/gkv007.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:1–13. https://doi.org/10.1186/1471-2105-9-559.
Kumar L, Futschik ME. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2139991/
Reverter A, Hudson NJ, Nagaraj SH, Pérez-Enciso M, Dalrymple BP. Regulatory impact factors: unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics. 2010;26:896–904. https://doi.org/10.1093/bioinformatics/btq051.
Uyhelji HA, Cheng C, Besansky NJ. Transcriptomic differences between euryhaline and stenohaline malaria vector sibling species in response to salinity stress. Mol Ecol. 2016;25:2210–25. https://doi.org/10.1111/mec.13609.
Liu G, Loraine AE, Shigeta R, Cline M, Cheng J, Valmeekam V, et al. NetAffx: Affymetrix probesets and annotations. Nucleic Acids Res. 2003;31:82–6. https://doi.org/10.1093/nar/gkg121.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. https://www.nature.com/articles/nprot.2008.211.
Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 2014;30:523–30. https://doi.org/10.1093/bioinformatics/btt703.
Jackson ML, Gunzelmann G, Whitney P, Hinson JM, Belenky G, Rabat A, et al. Deconstructing and reconstructing cognitive performance in sleep deprivation. Sleep Med Rev. 2013;17:215–25. http://dx.doi.org/10.1016/j.smrv.2012.06.007
Mackiewicz M, Zimmerman JE, Shockley KR, Churchill GA, Pack AI. What are microarrays teaching us about sleep? Trends Mol Med. 2009;15:79–87. doi: https://doi.org/10.1016/j.molmed.2008.12.002.
Charlesworth A, Meijer HA, de Moor CH. Specificity factors in cytoplasmic polyadenylation. Wiley Interdiscip Rev RNA. 2013;4:437–61. https://doi.org/10.1002/wrna.1171.
Richter JD. CPEB: a life in translation. Trends in Biochem Sci. 2007;32:279–85. https://doi.org/10.1016/j.tibs.2007.04.004.
Grønli J, Dagestad G, Milde AM, Murison R, Bramham CR. Post-transcriptional effects and interactions between chronic mild stress and acute sleep deprivation: regulation of translation factor and cytoplasmic polyadenylation element-binding protein phosphorylation. Behav Brain Res. 2012;235:251–62. doi: https://doi.org/10.1016/j.bbr.2012.08.008.
Gingras A-C, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–26. https://doi.org/10.1101/gad.887201.
da Costa SA, Ribeiro S. Sleep deprivation and gene expression. In: Meerlo P, Benca RM, Abel T, editors. Sleep, neuronal plasticity, and brain function. Current topics in behavioral neurosciences, vol. 25. New York: Springer; 2015. p. 65–90.
Hall JM. Bradykinin receptors. General Pharmacol: Vasc S 1997;28:1–6. https://doi.org/10.1016/S0306-3623(96)00174-7.
Tang X, Metzger D, Leeman S, Amar S. LPS-induced TNF-α factor (LITAF)-deficient mice express reduced LPS-induced cytokine: evidence for LITAF-dependent LPS signaling pathways. Proc Natl Acad Sci U S A. 2006;103:13777–82. https://doi.org/10.1073/pnas.0605988103.
Krueger JM. The role of cytokines in sleep regulation. Curr Pharm Des. 2008;14:3408–16. doi: https://doi.org/10.2174/138161208786549281.
Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM. Biochemical regulation of sleep and sleep biomarkers. J Clin Sleep Med. 2011;7:S38–42. https://doi.org/10.5664/JCSM.1360.
Mullington JM, Simpson NS, Meier-Ewert HK, Haack M. Sleep loss and inflammation. Best Pract Res Clin Endocrinol Metab. 2010;24:775–84. https://doi.org/10.1016/j.beem.2010.08.014.
Aho V, Ollila HM, Rantanen V, Kronholm E, Surakka I, van Leeuwen WMA, et al. Partial sleep restriction activates immune response-related gene expression pathways: experimental and epidemiological studies in humans. PLoS One. 2013;8:e77184. https://doi.org/10.1371/journal.pone.0077184.
Terao A, Steininger TL, Hyder K, Apte-Deshpande A, Ding J, Rishipathak D, et al. Differential increase in the expression of heat shock protein family members during sleep deprivation and during sleep. Neuroscience. 2003;116:187–200. doi: https://doi.org/10.1016/S0306-4522(02)00695-4.
Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, et al. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics. 2007;31:441–57. https://doi.org/10.1152/physiolgenomics.00275.2006.
Jaspers T, Morrell M, Simonds A, Adcock I, Durham A. The role of hypoxia and the circadian rhythm in sleep apnoea. Eur Respir J. 2015;46:OA298. https://doi.org/10.1183/13993003.congress-2015.OA298.
Ray S, Reddy AB. Cross-talk between circadian clocks, sleep-wake cycles, and metabolic networks: dispelling the darkness. BioEssays. 2016;38:394–405. https://doi.org/10.1002/bies.201500056.
Deboer T, Vansteensel MJ, Détár L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat Neurosci. 2003;6:1086–90. https://doi.org/10.1038/nn1122
Nakashima A, Kawamoto T, Honda KK, Ueshima T, Noshiro M, Iwata T, et al. DEC1 modulates the circadian phase of clock gene expression. Mol Cell Biol. 2008;28:4080–92. https://doi.org/10.1128/mcb.02168-07.
Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature 2002;419:841–4. https://doi.org/10.1038/nature01123.
Pellegrino R, Kavakli IH, Goel N, Cardinale CJ, Dinges DF, Kuna ST, et al. A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep. 2014;37:1327–36. https://doi.org/10.5665/sleep.3924.
Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. https://doi.org/10.1146/annurev-biochem-060308-103103.
Kay DB, Davis CJ. Sleep and microRNAs (miRNAs) in neurodegenerative diseases. In: Sahu SC, editor. microRNAs in toxicology and medicine. West Sussex: Wiley; 2013. p. 201–13. https://doi.org/10.1002/9781118695999.ch13.
Zhang W, Wang P, Chen S, Zhang Z, Liang T, Liu C. Rhythmic expression of miR-27b-3p targets the clock gene Bmal1 at the posttranscriptional level in the mouse liver. The FASEB J. 2016;30:2151–60. https://doi.org/10.1096/fj.201500120.
Figueredo DS, Barbosa MR, Gitaí DLG, de Andrade TG. Predicted microRNAs for mammalian circadian rhythms. J Biol Rhythm. 2013;28:107–16. https://doi.org/10.1177/0748730413476827.
Chen R, D’Alessandro M, Lee C. miRNAs are required for generating a time delay critical for the circadian oscillator. Curr Biol. 2013;23:1959–68. doi: https://doi.org/10.1016/j.cub.2013.08.005.
Heegaard NHH, Carlsen AL, Lilje B, Ng KL, Rønne ME, Jørgensen HL, et al. Diurnal variations of human circulating cell-free micro-RNA. PLoS One. 2016;11:e0160577. https://doi.org/10.1371/journal.pone.0160577.
Shende VR, Goldrick MM, Ramani S, Earnest DJ. Expression and rhythmic modulation of circulating microRNAs targeting the clock gene Bmal1 in mice. PLoS One. 2011;6:e22586. https://doi.org/10.1371/journal.pone.0022586.
Shimomura K, Kumar V, Koike N, Kim T-K, Chong J, Buhr ED, et al. Usf1, a suppressor of the circadian Clock mutant, reveals the nature of the DNA-binding of the CLOCK:BMAL1 complex in mice. elife. 2013;2:e00426. https://doi.org/10.7554/eLife.00426.
Landgraf D, Shostak A, Oster H. Clock genes and sleep. Pflügers Arch. 2012;463:3–14. https://doi.org/10.1007/s00424-011-1003-9.
Siepka SM, Yoo S-H, Park J, Lee C, Takahashi JS. Genetics and neurobiology of circadian clocks in mammals. Cold Spring Harb Symp Quant Biol. 2007;72:251–9. https://doi.org/10.1101/sqb.2007.72.052.
Reiff RE, Ali BR, Baron B, Yu TW, Ben-Salem S, Coulter ME, et al. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum Mol Genet. 2014;23:3456–66. https://doi.org/10.1093/hmg/ddu054.
Gumz ML, Rabinowitz L. Role of circadian rhythms in potassium homeostasis. Semin in Nephrol. 2013;33:229–36. doi: https://doi.org/10.1016/j.semnephrol.2013.04.003.
Flourakis M, Kula-Eversole E, Hutchison Alan L, Han Tae H, Aranda K, Moose Devon L, et al. A conserved bicycle model for circadian clock control of membrane excitability. Cell. 2015;162:836–48. https://doi.org/10.1016/j.cell.2015.07.036.
Ding F, O’Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science. 2016;352:550–5. https://doi.org/10.1126/science.aad4821.
Tatsuki F, Sunagawa Genshiro A, Shi S, Susaki Etsuo A, Yukinaga H, Perrin D, et al. Involvement of Ca2+-dependent hyperpolarization in sleep duration in mammals. Neuron. 2016;90:70–85. https://doi.org/10.1016/j.neuron.2016.02.032.
Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–84. https://doi.org/10.1124/mi.2.3.168.
Graves L, Pack A, Abel T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001;24:237–43. doi: https://doi.org/10.1016/S0166-2236(00)01744-6.
Havekes R, Bruinenberg VM, Tudor JC, Ferri SL, Baumann A, Meerlo P, et al. Transiently increasing cAMP levels selectively in hippocampal excitatory neurons during sleep deprivation prevents memory deficits caused by sleep loss. J Neurosci. 2014;34:15715–21. https://doi.org/10.1523/jneurosci.2403-14.2014.
Narasimamurthy R, Hatori M, Nayak SK, Liu F, Panda S, Verma IM. Circadian clock protein cryptochrome regulates the expression of proinflammatory cytokines. Proc Natl Acad Sci U S A. 2012;109:12662–7. https://doi.org/10.1073/pnas.1209965109.
Van Dongen HPA, Belenky G, Krueger JM. A local, bottom-up perspective on sleep deprivation and neurobehavioral performance. Curr Top Med Chem. 2011;11:2414–22. doi: https://doi.org/10.2174/156802611797470286.
Holst SC, Landolt H-P. Sleep Homeostasis, Metabolism, and Adenosine. Curr Sleep Med Rep. 2015;1:27–37. https://doi.org/10.1007/s40675-014-0007-3.
Chauhan S, Zheng X, Tan YY, Tay B-H, Lim S, Venkatesh B, et al. Evolution of the Cdk-activator speedy/RINGO in vertebrates. Cell Mol Life Sci. 2012;69:3835–50. https://doi.org/10.1007/s00018-012-1050-1.
Gastwirt RF, McAndrew CW, Donoghue DJ. Speedy/RINGO regulation of CDKs in cell cycle, checkpoint activation and apoptosis. Cell Cycle. 2007;6:1188–93. https://doi.org/10.4161/cc.6.10.4252.
Rogulja D, Young MW. Control of sleep by cyclin a and its regulator. Science. 2012;335:1617–21. https://doi.org/10.1126/science.1212476.
Cao J, Yang J, Wang Y, Xu J, Zhou Z, Cheng C, et al. Temporal-spatial expressions of Spy1 in rat sciatic nerve after crush. Cell Mol Neurobiol. 2013;33:213–21. https://doi.org/10.1007/s10571-012-9887-2.
Huang Y, Liu Y, Chen Y, Yu X, Yang J, Lu M, et al. Peripheral nerve lesion induces an up-regulation of Spy1 in rat spinal cord. Cell Mol Neurobiol. 2009;29:403–11. https://doi.org/10.1007/s10571-008-9332-8.
Fernandes C, Rocha NBF, Rocha S, Herrera-Solís A, Salas-Pacheco J, García-García F, et al. Detrimental role of prolonged sleep deprivation on adult neurogenesis. Front Cell Neurosci. 2015;9:Article 140. https://doi.org/10.3389/fncel.2015.00140.
Schmutz I, Ripperger JA, Baeriswyl-Aebischer S, Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev. 2010;24:345–57. https://doi.org/10.1101/gad.564110.
Sunde RA. mRNA transcripts as molecular biomarkers in medicine and nutrition. J Nutr Biochem. 2010;21:665–70. doi: https://doi.org/10.1016/j.jnutbio.2009.11.012.
Liew C-C, Ma J, Tang H-C, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–32. doi: https://doi.org/10.1016/j.lab.2005.10.005.
Tucker AM, Whitney P, Belenky G, Hinson JM, Van Dongen HPA. Effects of sleep deprivation on dissociated components of executive functioning. Sleep. 2010;33:47–57. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2802247/.
Acknowledgements
We thank Qi Yanminn for assistance with microarray samples and Daniel Williams for aid with qPCR. We also thank the staff of the Sleep and Performance Research Center, particularly Lindsey A. Tompkins, Brieann C. Satterfield, Amy M. Bender, and Devon A. Grant, for aid with subject recruitment and screening, laboratory experiments, and blood collections. Additionally, we are grateful to reviewers whose comments improved this report.
Funding
We acknowledge the FAA Office of Aerospace Medicine and Office of Aviation Accident Investigation and Prevention for sponsoring this research. Research was supported by FAA grant DTFAAC-11-A-00003 and NIH grant R01HL105768.
Availability of data and materials
Genomics data is archived at the NCBI GEO online repository, accession GSE98582 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98582. Other datasets supporting the conclusions of this article are included within the article (and its additional files).
Author information
Authors and Affiliations
Contributions
DMB, HVD, and MLJ designed and oversaw research; VLW acquired blood samples and assisted with initial processing and quality interpretation; DMK performed microarrays and qPCR; and HAU analyzed data and interpreted findings. HAU drafted this manuscript with input from all coauthors including critical review of writing and data portrayal. All authors have read and approved this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All individuals provided written informed consent, and Institutional Review Board approval was obtained at both Washington State University (WSU), and the Civil Aerospace Medical Institute (CAMI) of the Federal Aviation Administration.
Consent for publication
Not applicable; publication does not contain personally identifiable information and all subjects provided written informed consent. Analysis was performed using de-identified data.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Description of the samples. Values of N/A for the RNA integrity number correspond to three samples where this could not be calculated. No RNA was collected for five timepoints with unsuccessful blood draws. (XLSX 20 kb)
Additional file 2:
Supplementary text and figures. Detailed study methods, results, and supplementary Figures S1-S9. (PDF 1546 kb)
Additional file 3:
Table S2. List of Treatment effect Transcript Clusters with fold change, P-value, and FDR for the Treatment effect at the Experimental phase, Mfuzz Treatment group, and annotations from the NetAffx™ batch annotation tool. Notes on gene family, comments, and mRNA assignment are from review of the Affymetrix annotation package (HuGene-1_0-st-v1.na35.hg19.probeset.csv) for Transcript Cluster IDs without gene level (e.g., title) annotation or with mixed hybridization targets. Mixed hybridization targets suggest the potential for the Transcript Cluster to be associated with more than one gene. Three slashes designate multiple annotations associated with the same Transcript Cluster. Column human blood designates whether or not the gene was found in an IPA® biomarker screen for genes detectable in human blood. (XLSX 88 kb)
Additional file 4:
Table S3. Comparison of Treatment and PVT lists, based on the first gene symbol listed for each Transcript Cluster, with datasets from literature. Worksheet tab “Summary” presents the numbers of genes found in common between the Treatment and PVT lists with published datasets. Subsequent worksheet tabs provide detailed comparisons for individual published datasets. Identification of a match required finding the exact same gene symbol in published datasets as the first listed gene symbol in the Treatment and PVT list. Thus, overlap may be underestimated if different annotations were given to the same gene. (XLSX 96 kb)
Additional file 5:
Table S4. List of Psychomotor Vigilance Test (PVT) effect Transcript Clusters with P-value and FDR for the PVT effect, Mfuzz PVT group, and annotations from the NetAffx™ batch annotation tool. Notes on gene family, comments, and mRNA assignment are from review of Affymetrix annotation package (HuGene-1_0-st-v1.na35.hg19.probeset.csv) for Transcript Cluster IDs without gene level (e.g., title) annotation or with mixed hybridization targets. Mixed hybridization targets suggest the potential for the Transcript Cluster to be associated with more than one gene. Three slashes designate multiple annotations associated with the same Transcript Cluster. Genes also found in the Treatment list are noted. Column human blood designates whether or not the gene was found in an IPA® biomarker screen for genes detectable in human blood. (XLSX 24 kb)
Additional file 6:
Table S5. WGCNA module membership for all Transcript Clusters passing the low-expression threshold. Correlation coefficients (r) and P-values are given for the Pearson correlation between each Transcript Cluster and each of three variables: Treatment, PVT lapses, and Time of Day. Annotations are taken from the Affymetrix batch NetAffx™ tool, with /// separating multiple annotations given to the same Transcript Cluster. (XLSX 2732 kb)
Additional File 7:
Table S6. Results of Ingenuity Pathway Analysis® Causal Networks as run on the Treatment list. (XLSX 33 kb)
Additional File 8:
Table S7. Results of Ingenuity Pathway Analysis® Causal Networks as run on the PVT list. (XLSX 20 kb)
Additional file 9:
Table S8. Results of linear mixed-effects models on microRNA qPCR data, including tests for a relation to Treatment, Psychomotor Vigilance Test (PVT), and Time of Day. (XLSX 12 kb)
Rights and permissions
Open Access This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
About this article

Cite this article
Uyhelji, H.A., Kupfer, D.M., White, V.L. et al. Exploring gene expression biomarker candidates for neurobehavioral impairment from total sleep deprivation. BMC Genomics 19, 341 (2018). https://doi.org/10.1186/s12864-018-4664-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4664-3