
Abstract
Background
Haynaldia villosa (L.) Schur (syn. Dasypyrum villosum L. Candargy, 2n = 14, genome VV) is the tertiary gene pool of wheat, and thus a potential resource of genes for wheat improvement. Among other, wheat yellow mosaic (WYM) resistance gene Wss1 and a take-all resistance gene were identified on the short arm of chromosome 4 V (4VS) of H. villosa. We had obtained introgressions on 4VS chromosome arm, with the objective of utilizing the target genes. However, monitoring these introgressions has been a daunting task because of inadequate knowledge as to H.villosa genome, as reflected by the lack of specific markers.
Results
This study aims to develop 4VS-specific markers by combination of chromosome sorting and next-generation sequencing. The short arm of chromosome 4VS of H.villosa was flow-sorted using a FACSVantage SE flow cytometer and sorter, and then sequenced by Illumina sequencing. The sequence of H. villosa 4VS was assembled by the software Hecate, and then was compared with the sequence assemblies of wheat chromosome arms 4AL, 4BS and 4DS and Ae. tauschii 4DS, with the objectives of identifying exon-exon junctions and localizing introns on chromosome 4VS of H. villosa. The intron length polymorphisms suitable for designing H. villosa primers were evaluated with criteria. Consequently, we designed a total of 359 intron targeting (IT) markers, among which 232 (64.62%) markers were specific for tracing the 4VS chromatin in the wheat background.
Conclusion
The combination of chromosome sorting and next-generation sequencing to develop specific IT markers for 4VS of H. villosa has high success rate and specificity, thus being applicable for the development of chromosome-specific markers for alien chromatin in wheat breeding.
Similar content being viewed by others

Background
Wild relatives of wheat are the tertiary gene pool of bread wheat and contain many favorable genes for wheat improvement, such as disease resistance, drought tolerance, salt tolerance, winter hardiness and adaptability to poor soil [1–3]. For the utilization of these elite genes, chromosome engineering has been used to produce small alien introgressions. Irrespective of the enormous genetic variation in wild germplasm and sophisticated techniques available for alien gene transfer, it is still difficult to efficiently identify introgressed chromatin when alien introgression was used in wheat improvement.
Cytological approaches including chromosome banding, genomic in situ hybridization (GISH) and fluorescence in situ hybridization (FISH), have been extensively applied to identify and characterize introgression lines in wheat, but with limitation of low throughput [4]. Molecular markers are high efficient for identifying alien chromatin, while the numbers of molecular markers are very low in particular for the tertiary gene pool species of wheat. Hence, there is urgent need to exploit a system for high-throughput method for developing molecular markers to identify alien introgression and translocation lines.
Introns are an attractive source of polymorphism for marker development, because insertions/deletions and base substitutions are more common within intron than within exon sequences [5]. Intron length polymorphism has been considered as a convenient and reliable source of informative markers with high interspecies transferability [6]. Based on the orthologous gene conservation between rice and wheat [7], a set of PCR-based landmark unique gene (PLUG) primers were developed, and subsequently the PLUG primers were shown as being suitable to detect polymorphisms among wheat A, B and D genomes [8]. The PLUG markers not only can identify homology between wheat and alien chromosomes, thus being useful in marker assisted selection (MAS), comparative genomics, alien chromosome tracing, taxonomic studies and genotyping [9–12].
Genome mapping and sequencing in large and polyploid genomes especially wheat remain daunting tasks. However, the recent technological advances in flow cytometric sorting makes possible the dissecting of large genomes into individual chromosome, reducing sample complexity and enabling analysis at the subgenomic level [13]. Flow cytometric chromosome sorting has been successfully applied in bread and durum wheat [13]. Recently, flow-sorting of individual chromosomes has been performed in wild relatives of wheat, such as Ae. geniculata, Ae. umbellulata, Ae. comosa, Ae. markgrafii, Ae. triuncialis, Ae.cylindrica, T. urartu, Ae. speltoides, Ae. tauschii and Ae. biuncialis [14–18]. In combination with the flow sorting and the next-generation sequencing to develop DNA sequence-based markers of wild relatives of wheat have already been used to trace alien segments in wheat breeding. For example, the single-nucleotide polymorphism (SNP) markers specific for the short arm of chromosome 5MgS of Aegilops geniculata were successfully developed by chromosome sorting and next generation sequencing platform and the SNPs identified could be employed to accurately detect 5MgS introgressions in common wheat [14].
Haynaldia villosa (L.) Schur (syn. Dasypyrum villosum L. Candargy, 2n = 14, genome VV) is the tertiary gene pool of wheat, carrying many important genes for wheat improvement, such as resistant to several wheat diseases, including powdery mildew, eyespot, take-all, wheat spindle streak mosaic virus (WSSMV), and tolerant to drought and cold stresses, good tiller ability, and high grain protein content [19]. Previously, we identified wheat yellow mosaic resistance gene Wss1 [20] located on the short arm of chromosome 4 V (4VS). A set of small fragment translocation lines involving 4VS were obtained using ph1b induction system, by which the Wss1 was mapped to the terminal region of 4VS [21]. Here, we report an efficient method for developing chromosome-specific IT markers for the alien 4VS by chromosome sorting and next-generation sequencing, in order to utilize the beneficial genes of wild relatives for wheat breeding.
Methods
Plant materials
T. durum-H.villosa amphiploid (AABBVV), T. aestivum-H. villosa substitution line DS4V(4D), 4VS ditelosomic addition line, T. aestivum-H. villosa translocation lines T4DL · 4VS, T4VL · 4DS, NAU421, NAU428, NAU429, NAU433 and NAU435 [21]were developed at the Cytogenetics Institute, Nanjing Agricultural University (CINAU, hereafter). H. villosa (VV, accession No. 91C43) was obtained from Cambridge Botanical Garden, UK. Common wheat cv. Chinese Spring (AABBDD) maintained at CINAU was used as control. Chromosome composition of these materials is shown in Additional file 1: Figure S1.
Chromosome sorting and DNA sequencing
Aqueous suspensions of chromosome 4VS of H. villosa were prepared from synchronized meristem root tip cells following Vrana et al. [22] and Kubaláková et al. [23, 24]. The chromosomes in suspension were stained with 2 μg/ml DAPI (4′, 6-diamidino-2-phenylindole) and the 4VS telosomes were sorted using a FACSVantage SE flow cytometer and sorter (Becton Dickinson, San Jose, USA). Purity in the sorted fractions was determined after FISH with probes for GAA microsatellite and pSc119.2 repeat on chromosomes sorted onto microscope slides. DNA of the sorted chromosome arms was purified and amplified by multiple displacement amplification (MDA) as described by Šimková et al. [25]. Three independent amplification products were combined to reduce amplification bias. The amplified DNA was purified by ethanol precipitation before sequencing.
About 10 μg of MDA-amplified DNA was used to create the two shotgun DNA-seq libraries of 500-700 bp and 700-1300 bp inserted-size. The libraries were sequenced in a single lane of Illumina HiSeq 2000 platform. The sequence read data were deposited in the (NCBI) Sequence Read Archive (SRA) and is available under accession number SRR3741672. De novo assembly of the Illumina paired-end reads was performed using the software Hecate (unpublished, http://bgi-international.com/us/) using different k-mer sizes (41, 45, 49 and 63). The result of the 45-mer run provided the assembly with the best sequence coverage and N50 size, and therefore was determined to generate 4VS scaffolds.
Sequence resources for primer design
DNA sequences used in this work included H. villosa 4VS assembly, common wheat cv. Chinese Spring chromosome arms 4AL, 4BS and 4DS assemblies and annotated genes on 4DS [26] and annotated genes in the draft genome sequence assembly of Ae. tauschii [27].
Homology and alignment analysis
The flowchart of designing 4VS specific markers is shown in Fig. 1. In the first step, we choose a set of genes to calculate exon-exon junction sizes in genomic sequences of homoeolog arms 4AL, 4BS, 4DS and 4DS. The set contained genes from the annotated Ae. tauschii chromosome 4D mapped within 60 Mb of pseudomolecule and all annotated genes on Chinese Spring 4DS. We removed repeats in the two gene sets by Blastn analysis using the cut-off parameters e-value > 1e-5, coverage >80%. In the second step, all genes were compared with genomic sequences of Chinese Spring 4AL, 4BS, 4DS and H. villosa 4VS using a local Blastn program. All genes matching 4AL, 4BS, 4DS and 4VS assemblies and possessing at least one predicted exon-exon junction were selected. Intron sizes of corresponding genes were then calculated and compared against each other. Genes whose intron size in 4VS differed at least 10% from that of 4AL, 4BS and 4DS simultaneously in the same homologues allele were chosen for designing the markers. The primers were designed in the exons flanking the targeted introns.

Schematic representation of the development system of PCR-based IT markers specific for the chromosome 4VS of Haynaldia villosa and 8% non-denaturing poly-acrylamide gels electrophoresis of PCR product of IT marker CINAU687. a Schematic representation of the development system of PCR-based IT markers specific for the chromosome 4VS of Haynaldia villosa. b 8% non-denaturing poly-acrylamide gels electrophoresis of PCR product of IT marker CINAU687. Grey boxes represented the exons and grey lines represented the introns. Red line, light blue line, deep blue line and light green line showed the intron 7 region of gene fragment AEGTA27279 corresponding to the intron regions of the scaffold of IWGSC_CSS_4AL_scaff_7149580, IWGSC_CSS_4BS_scaff_4952162, IWGSC_CSS_4DS_scaff_2323695 and Hecate_SCF226, respectively). The base number of intron 7 in the gene fragment of subgenome AA, BB, DD and VV was 658 bp, 367 bp, 415 bp and 468 bp, respectively
Primer design
IT primers were designed in exon regions flanking a targeted intron using the online software Primer 3 (Version 4.0, http:// frodo.wi.mit.edu/primer3/) according to the H. villosa sequences, assuming that exon regions and exon-intron structures of orthologous genes are highly conserved among grass species. The primers designed were expected to amplify 4AL, 4BS, 4DS and 4VS genomic DNA, simultaneously. The following criteria were used to design PCR primers for these markers: melting temperature 55–65 °C (optimum: 60 °C), primer length ranging between 18–25 bases (optimum: 20), desired size of amplified fragments estimated was approximately 50 bp more than targeted intron. All primers were synthesized by Shanghai Invitrogen Biotechnology Company Ltd. (Shanghai, China).
DNA extraction and PCR
Genomic DNA was isolated from 2 g fresh leaves of plants at three-leaf stage with SDS-phenol-chloroform method according to Sharp et al. [28] and Devos et al. [29] and purified to eliminate RNA, amylase and other unwanted compounds. The purity and concentration of DNA was assessed with microplate reader (M200, TECan, Switzerland). The DNA was finally diluted to concentration of 50 ng/μl and stored at −20 °C until use.
PCR amplification was carried out in a 10 μl reaction volume containing 40 ng genomic DNA, 2 μM each of the primer pairs, 2.5 mM each dNTPs, 2.5 mM MgCl2, 1 × PCR buffer (10 mM Tris–HCl, pH 8.5, 50 mM KCl), and 0.5 U Taq DNA polymerase with a PTC-200 thermal cycler (Bio-Rad, Hercules, CA, USA). The amplification was conducted an initial denaturation at 94 °C for 3 min, followed by 35 cycles at 94 °C for 30 s, annealing of different primers at 50, 55, or 60 °C for 50 s at a ramp rate of 0.5 °C/s, 72 °C for 1 min 10 s, and a final extension at 72 °C for 10 min. PCR products were resolved in 8% non-denaturing poly-acrylamide gels (Acr : Bis = 19 : 1 or 39 : 1) and the band patterns were visualized by silver staining [30].
Results
Shotguns sequencing of H. villosa chromosome 4VS and its assembly
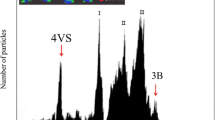
The analysis of DAPI-stained, chromosome suspensions prepared from a wheat-H. villosa 4VS addition line resulted in histograms with five peaks of fluorescence intensity (flow karyotypes). The leftmost peak represented telocentric chromosome 4VS, which was well resolved from the chromosome composite peaks I, II, III and chromosome 3B peak of the bread wheat (Additional file 2: Figure S2). The flow-sorted telocentric 4VS has more than 89% purity. DNA amplified from flow-sorted t4VS was sequenced by the Illumina technology. In total, we generated a high-quality of 33.5 Gb paired-end reads. A total length of 170.6 Mb assembled sequences was obtained, comprising 201,193 scaffolds.
Development of PCR-based IT markers
A gene set is collected first which might have homologous alleles on 4AL, 4BS, 4DS and 4VS, simultaneously. Genes from 4VS are the first choice but poorly annotated. Genes on 4DS either from Ae. tauschii or common wheat, are the second choice as they are presumably most related to 4VS. Ae. tauschii 4DS was defined as follow. All annotated genes along the pseudomolecule of Ae. tauschii 4D were aligned by Blastn search against the 4VS assembly. Genes within about 60 Mb(From 0–60 Mb) had the highest frequency of Blastn hits (Data not shown). This 60-Mb interval is presumed to be Ae. tauschii 4DS which contained 1203 genes. A total of 1821 genes were annotated on 4DS of Chinses Spring. If one gene in Chinese Spring 4DS showed high homologous to that in the Ae. tauschii 4DS by Blastn analysis, they are acknowledged as the same (Additional file 3: Figure S3). All genes from Ae. tauschii were reserved, while redundant genes from Chinese Spring were removed, considering that the average length of genes in Ae. tauschii is longer than that in Chinese Spring release. Finally, we identified 1157 genes on Chinese Spring 4DS were retained. Finally, a gene set totaling 2360 genes from 4DS were established and aligned to genomic sequences of 4AL, 4BS, 4DS and 4VS using Blastn. A total of 2075 genes have blastn hits on all of four chromosome arms. Out of them, 626 genes contained at least one intron and a total of 1774 introns in the sequences of 4AL, 4BS, 4DS and 4VS with an average of 2.83 introns per gene. A total of 595 introns in 4VS differed by at least 10% as compared to those in wheat 4AL, 4BS and 4DS. They are termed “targeted intron” which meet the criteria for primer design. These 595 introns were assigned to 367 genes of which some have two or more introns. To abide “one genome marker” rule, only one such intron for each gene were selected to develop IT marker. Except for 8 genes that failed to design primers, a total of 359 primer pairs were designed which spanned the targeted intron, using the online software Primer 3 V0.4.0 (http://frodo.wi.mit.edu/primer 3/).
Take one gene for designing primers at the targeted intron for an example. The principle to develop specific IT markers of H. villosa 4VS was displayed in Fig. 1 . A gene (AEGTA27279) fragment from Ae. tauschii was aligned to DNA sequences of chromosome arms 4AL, 4BS, and 4DS of Chinese Spring and H. villosa 4VS. The gene has blastn hits on scaffolds IWGSC_CSS_4AL_scaff_7149580, IWGSC_CSS_4BS_scaff_495216, and IWGSC_CSS_4DS_scaff_2323695 on 4AL, 4BS and 4DS, and 4VS scaffold Hecate_SCF226, respectively. We compared the size of every intron of homologues alleles and found that the length of intron 7 differed among A, B, D of wheat and V genome of H. villosa, that they are 685 bp, 367 bp, 415 bp and 468 bp, respectively. IT marker was developed by designing primers on exons 7 and 8 flanking the intron 7 locus.
Validation and the efficiency of the IT markers development by PCR
To verify the performance of the IT marker system, a total of 359 IT markers were designed using the online software Primer 3 V0.4.0 (http:// frodo.wi.mit.edu/primer3/). PCR was performed with the designed 359 primer sets using genomic DNA from the wheat cv. Chinese Spring, T. durum-H. villosa amphiploid (AABBVV), H. villosa and T. aestivum-H. villosa translocation line T4DL · 4VS, T. aestivum-H. villosa substitution line DS4V(4D), 4VS ditelosomic addition line and T. aestivum-H. villosa translocation line T4VL · 4DS as template. Only 14 out of the 359 primer pairs produced no amplicons, while PCR with 345 primer pairs resulted in amplicons in all four templates.
In order to assign the 345 IT markers to chromosome arm 4VS of H. villosa, PCR products obtained in wheat cv. Chinese Spring, T. durum-H. villosa amphiploid (AABBVV), H. villosa and T. aestivum-H. villosa translocation line T4DL · 4VS were separated on non-denaturing polyacrylamide gel. If the amplification of a primer pair generated a distinct PCR product shown as polymorphic band in H. villosa, T. durum-H. villosa amphiploid and T. aestivum-H. villosa translocation line T4DL · 4VS, but not in cv. Chinese Spring, the primer pair can be used as chromosome arm 4VS-specific marker. In total, 232 markers were found specific for 4VS (Additional file 4: Table S1). Consequently, the success rate of developing 4VS chromosome arm-specific molecular markers was as high as 64.62%.
Polyacrylamide gel electrophoresis separated PCR products into two, three or four bands. Four bands were obtained with 21 primer sets (Type I), three bands with the 55 primer sets (Type II 29; Type III 5; and Type VI 21), two bands with the 156 primer sets (Type V 17 and Type VII 139) (Additional file 5: Table S2). Examples of amplification products obtained using six primer pairs are given in Fig. 2. The representative 6 types IT markers corresponding to the sequence information in subgenome AA, BB, DD and VV and the sequences of forward and reverse primers and the size of intron of the representative 6 types IT markers are given in Tables 1 and 2, respectively. The size of the products amplified by these six primer pairs ranged from 250 to 1800 bp, and each product was larger than the size predicted from the sequences of wheat and H. villosa intron, suggesting that all PCR products contained introns and parts of exons.

8% non-denaturing poly-acrylamide gels electrophoresis of PCR products. Arrows show 4VS-specific fragments
Considering the size of PCR products, the four bands amplified by the Type I markers originated from chromosomes 4A, 4B, 4D and 4VS, respectively. These 4VS-specific molecular markers developed are co-dominant and are useful to simultaneously trace the alien 4VS chromosome arm and its wheat homoeologous group.
For the Type II, Type III and Type IV markers, according to the size of introns in the subgenomes A, B, D and V, the three bands were amplified from chromosome 4D, 4A and 4 V; 4D, 4B and 4 V; 4A, 4B and 4 V, respectively. For the Type V and VII markers, the two bands detected on polyacrylamide gels consisted of the products derived from 4 VS chromosome arm, and a Chinese Spring chromosome (possibly one of 4A or 4B or 4D;or two of 4A and 4D,or 4D and 4B or 4A and 4D; or three chromosomes 4A, 4D and 4B).
Based on PCR product size polymorphisms, a total of 93 (40.09%) IT markers could be assigned to subgenomes A, B and D, 139 IT markers could not be assigned to subgenomes A, B and D. We compared the intron sizes of subgenomes A, B and D and found that the difference in the intron size between homoelogs was small.
Discussion
Wild relatives of wheat contain a large number of favorable genes for resistance to biotic and abiotic stresses and can be used for wheat improvement. But now, monitoring alien introgressions in wheat background is difficult because of the shortage of genetic and molecular mapping information on the wild relatives. DNA markers can detect small amounts of alien chromatin that cannot be recognized cytogenetically, thus having high efficiency in the identification of the introgressed fragments. Simple sequence repeat (SSR) was used as the molecular markers to screen the individual chromosome of wild relatives of wheat. However, they were low in transferability to wild relatives and were lack in locus specificity, and therefore had limitation in application to wheat breeding [31]. Taken the H. villosa as an example, only 9 of the 276 wheat microsatellites had high polymorphisms suitable for molecular markers [32]. Random amplified polymorphic DNAs (RAPD), although easy to develop, still suffer from poor reproducibility [33]. Bin-mapped expressed sequence tags (ESTs) were also explored as a source of markers, but only a few of them were polymorphic [34]. Zhao et al. [35] designed 607 primer pairs from wheat EST sequences and found that only 58 (9.23%) of the primers amplified specific bands from chromosome 4 V. Up to now, there is still fewer molecular markers specific for individual chromosome of wild relatives that can be used, this is partially due to the lack of genome information of the relatives, as reflected by the fact that most of the markers were designed on the basis of genome of wheat, rice, or other species.
Flow cytometric chromosome sorting is an efficient method for dramatically simplifying genome analysis by reducing DNA sequence complexity [36]. And it combination with next-generation sequencing technology could obtain the sequence composition of single chromosome of wild relatives, thus being highly productive in development and validation of molecular markers [36]. As next-generation sequencing technologies become more economical, making possible the high throughput mining of molecular markers specific for chromosome. Moreover, all wheat chromosomes have been sequenced using flow sorting and next-generation sequencing, which should be useful for the developing molecular markers specific for alien chromosome [26]. Tiwari et al. [14] developed a total of 2178 unique, 5MgS-specific SNPs of the Ae. geniculata by the combination of chromosome sorting and the next generation sequencing platform, and showed that this approach has high-throughput for the discovery of markers specific for wild relative. In the present study, we first flow-sorted the chromosome arm 4VS of H. villosa, thereby drastically reducing the amount of work on DNA sequencing of this wild relatives. Based on the genome sequence of 4VS, we designed IT markers, which are based on the sequence conservation of orthologous genes and therefore have higher transferability between Triticeae species. A total of 359 IT markers were designed and 232 (64.62%) markers proved to be specific for H. villosa 4VS. Compared to RAPD, SSR, and EST-PCR markers developed by conventional methods [32, 33, 35], the IT markers designing by chromosome sorting and next-generation sequencing is more efficient with high success rate and specificity.
In our lab, Zhao et al. used wheat ph1b mutant to induce translocations of 4VS chromosome fragments to further physically map the Wss1 to specific chromosome region [21]. However, due to the lower density of markers used in determination of the translocated fragments, a limited resolution of physical maps consisting of 13 bins were obtained. The IT markers developed in this study will dramatically increase the density of 4VS physical map or cytological map using these structural aberrants involving 4VS chromosome. The development of a large number of IT markers will be invaluable to trace alien chromatin in a wheat background, for comparative genome mapping, chromosomal evolutionary analysis, and gene introgression during wheat improvement using H. villosa as gene donor. Twenty-one 4VS-specific molecular markers developed in this work are co-dominant and are useful to simultaneously trace the alien 4VS chromosome arm and its wheat homoeologues.
In the present research, we randomly used 100 specific IT markers for the physical mapping analysis and tracing alien chromatin in wheat background. Using template DNA from five wheat-H. villosa 4VS translocation lines (NAU421, NAU428, NAU429, NAU433 and NAU435) and wheat-H. villosa T4DL · 4VS translocation line, we examined the presence or absence of the 100 specific IT markers. Thus, the 18, 55, 69, 24 and 44 IT markers were assigned to the region of wheat-H. villosa 4VS translocation lines (NAU421, NAU428, NAU429, NAU433 and NAU435), respectively(Additional file 6: Table S3).
Wheat spindle streak mosaic virus (WSSMV) and wheat yellow mosaic virus (WYMV) resistance gene Wss1 were previously mapped to the 4VS arm of H. villosa, using 4 V (4D) substitution and T4DL · 4VS translocation lines [20]. For a more accurate mapping of Wss1 gene, a ph1b mutant of cv. Chinese Spring was used to induce new translocations with smaller 4VS chromosome fragments. Based on the resistance evaluation, GISH and molecular marker analysis of the available translocations, the gene(s) conferring the WYMV resistance on 4VS were mapped to the distal region of 4VS in the bin of FL 0.78–1.00 [21]. If the resistance genes are fully explored and used, they would greatly enrich the available resistance germplasm resources for wheat. Wheat-H. villosa 4VS translocation lines (NAU421, NAU428, NAU429) contained wheat yellow mosaic virus (WYMV) resistance gene, Wss1. So these IT markers located in the terminal region will be helpful for marker-assisted introgression of the genes of interest, such as Wss1 gene, into elite cultivars of the common wheat.
Miftahudin et al. [37] demonstrated that chromosome 4A have undergone two reciprocal translocations and two inversions events that placed most of the ancestral short arm on the modern long arm (4AL). In our lab, we also used ChromoWiz to define the 4VS syntenic regions in wheat chromosomes and found that they were enriched on wheat group 4 chromosomes 4AL. To develop 4VS-specific IT marker, we used DNA sequences obtained from flow-sorted chromosome arm 4VS of H. villosa was compared with the sequence assemblies of wheat homoeologous group 4 chromosomes (4AL, 4BS and 4DS) and Ae. tauschii 4DS to identify exon-exon junctions and localize introns on chromosome 4VS of H. villosa.
Conclusions
In this study, we flow-sorted a ditelosomic addition wheat-H. villosa line to isolate the short arm of the H.villosa 4VS chromosome with ~89% purity identified by FISH using a FACSVantage SE flow cytometer and sorter. This approach reduced DNA sample complexity and permitted the development of markers specific for the short arm of 4VS. The sequence of H. villosa 4VS was assembled by the software Hecate, and then was compared with the sequence assemblies of wheat homoeologous group 4 chromosomes and Ae. tauschii 4DS to identify exon-exon junctions and localize introns on chromosome 4VS of H. villosa. The intron length polymorphisms suitable for designing H. villosa primers were evaluated with criteria, whose intron size of genes in 4VS differed at least 10% from that of 4AL, 4BS and 4DS simultaneously in the same homologues allele were chosen for designing the markers. Lastly, we designed a total of 359 IT markers, among which 232 (64.62%) markers were found specific for 4VS, with the success rate being as high as 64.62%. Collectively, this approach of combination of chromosome sorting and genome sequencing can be applicable for development of species/genome-specific markers to trace alien chromatin in wheat background, or for comparative genome mapping, chromosomal evolutionary analysis.
Abbreviations
- ESTs:
-
Expressed sequence tags
- FISH:
-
Fluorescence in situ hybridization
- GISH:
-
Genomic in situ hybridization
- IT:
-
Intron targeting
- MAS:
-
Marker assisted selection
- PLUG:
-
PCR-based landmark unique gene
- RAPD:
-
Random amplified polymorphic DNAs
- SNP:
-
Single-nucleotide polymorphism
- SSR:
-
Simple sequence repeat
- WSSMV:
-
Wheat spindle streak mosaic virus
- WYMV:
-
Wheat yellow mosaic virus.
References
Colmer TD, Flowers TJ, Munns R. Use of wild relatives to improve salt tolerance in wheat. J Exp Bot. 2006;57(5):1059–78.
Friebe B, Jiang J, Raupp WJ, McIntosh RA, Gill BS. Characterization of wheat-alien translocations conferring resistance to diseases and pests: Current status. Euphytica. 1996;91(1):59–87.3.
Triebe B, Mukai Y, Dhaliwal HS, Martin TJ, Gill BS. Identification of alien chromatin specifying resistance to wheat streak mosaic and greenbug in wheat germ plasm by C-banding and insitu hybridization. Theor Appl Genet. 1991;81(3):381–89.
Lukaszewski AJ, Lapinski B, Rybka K. Limitations of in situ hybridization with total genomic DNA in routine screening for alien introgressions in wheat. Cytogenet Genome Res. 2005;109(1–3):373–77.
Kimura M. Rare variant alleles in the light of the neutral theory. Mol Biol Evol. 1983;1(1):84–93.
Poczai P, Varga I, Laos M, Cseh A, Bell N, Valkonen JP, et al. Advances in plant gene-targeted and functional markers: a review. Plant Methods. 2013;9(1):6.7.
Ishikawa G, Yonemaru J, Saito M, Nakamura T. PCR-based landmark unique gene (PLUG) markers effectively assign homoeologous wheat genes to A, B and D genomes. BMC genomics. 2007;8:135.
Ishikawa G, Nakamura T, Ashida T, Saito M, Nasuda S, Endo TR, et al. Localization of anchor loci representing five hundred annotated rice genes to wheat chromosomes using PLUG markers. Theor Appl Genet. 2009;118(3):499–514.
Poczai P, Cernak I, Gorji AM, Nagy S, Taller J, Polgar Z. Development of intron targeting (IT) markers for potato and cross-species amplification in Solanum nigrum (Solanaceae). Am J Bot. 2010;97(12):e142–5.
Li J, Endo TR, Saito M, Ishikawa G, Nakamura T, Nasuda S. Homoeologous relationship of rye chromosome arms as detected with wheat PLUG markers. Chromosoma. 2013;122(6):555–64.
Hu LJ, Liu C, Zeng ZX, Li GR, Song XJ, Yang ZJ. Genomic rearrangement between wheat and Thinopyrum elongatum revealed by mapped functional molecular markers. Genes Genom. 2012;34(1):67–75.
Zhan HX, Li GR, Chang ZJ, Zhang XJ, Li X, Yang ZJ. Molecular identification of a new wheat-Thinopyrum intermedium cryptic translocation line for resistance to powdery mildew. IJBBB. 2013;4(3):376–8.
Dolezel J, Vrana J, Safar J, Bartos J, Kubalakova M, Simkova H. Chromosomes in the flow to simplify genome analysis. Funct Integr Genomic. 2012;12(3):397–416.
Tiwari VK, Wang SC, Sehgal S, Vrana J, Friebe B, Kubalakova M, et al. SNP Discovery for mapping alien introgressions in wheat. BMC Genomics. 2014;15.
Molnar I, Kubalakova M, Simkova H, Cseh A, Molnar-Lang M, Dolezel J. Chromosome isolation by flow sorting in Aegilops umbellulata and Ae. comosa and their allotetraploid hybrids Ae. biuncialis and Ae. geniculata. Plos One. 2011;6(11):e27708.
Molnar I, Vrana J, Farkas A, Kubalakova M, Cseh A, Molnar-Lang M, et al. Flow sorting of C-genome chromosomes from wild relatives of wheat Aegilops markgrafii, Ae. triuncialis and Ae. cylindrica, and their molecular organization. Ann Bot. 2015;116(2):189–200.
Molnar I, Kubalakova M, Simkova H, Farkas A, Cseh A, Megyeri M, et al. Flow cytometric chromosome sorting from diploid progenitors of bread wheat, T. urartu, Ae. speltoides and Ae. tauschii. Theor Appl Genet. 2014;127(5):1091–104.
Molnar I, Simkova H, Leverington-Waite M, Goram R, Cseh A, Vrana J, et al. Syntenic relationships between the U and M genomes of Aegilops, wheat and the model species Brachypodium and rice as revealed by COS markers. Plos One. 2013;8(8):e70844.
Gradzielewska A. The genus Dasypyrum - Part 2. Dasypyrum villosum - a wild species used in wheat improvement. Euphytica. 2006;152(3):441–54.
Zhang QP, Li Q, Wang X, Wang HY, Lang SP, Wang YN, et al. Development and characterization of a Triticum aestivum-Haynaldia villosa translocation line T4VS · 4DL conferring resistance to wheat spindle streak mosaic virus. Euphytica. 2005;145(3):317–20.
Zhao RH, Wang HY, Xiao J, Bie T, Cheng SH, Jia Q, et al. Induction of 4VS chromosome recombinants using the CS ph1b mutant and mapping of the wheat yellow mosaic virus resistance gene from Haynaldia villosa. Theor Appl Genet. 2013;126(12):2921–30.
Vrana J, Kubalakova M, Simkova H, Cihalikova J, Lysak MA, Dolezel J. Flow sorting of mitotic chromosomes in common wheat (Triticum aestivum L.). Genetics. 2000;156(4):2033–41.
Kubalakova M, Valarik M, Bartos J, Vrana J, Cihalikova J, Molnar-Lang M, et al. Analysis and sorting of rye (Secale cereale L.) chromosomes using flow cytometry. Genome. 2003;46(5):893–905.
Kubalakova M, Vrana J, Cihalikova J, Simkova H, Dolezel J. Flow karyotyping and chromosome sorting in bread wheat (Triticum aestivum L.). Theor Appl Genet. 2002;104(8):1362–72.
Simkova H, Svensson JT, Condamine P, Hribova E, Suchankova P, Bhat PR, et al. Coupling amplified DNA from flow-sorted chromosomes to high-density SNP mapping in barley. BMC Genomics. 2008;9.
Mayer KFX, Rogers J, Dolezel J, Pozniak C, Eversole K, Feuillet C, et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014; 345(6194):1251788.
Jia JZ, Zhao SC, Kong XY, Li YR, Zhao GY, He WM, et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature. 2013;496(7443):91–5.
Sharp PJ, Chao S, Desai S, Gale MD. The isolation, characterization and application in the Triticeae of a set of wheat RFLP probes identifying each homoeologous chromosome arm. Theor Appl Genet. 1989;78(3):342–8.
Devos KM, Atkinson MD, Chinoy CN, Liu CJ, Gale MD. RFLP-based genetic map of the homoeologous group 3 chromosomes of wheat and rye. Theor Appl Genet. 1992;83(8):931–9.
Bassam BJ, Gresshoff PM. Silver staining DNA in polyacrylamide gels. Nat Protoc. 2007;2(11):2649–54.
Mullan DJ, Platteter A, Teakle NL, Appels R, Colmer TD, Anderson JM, et al. EST-derived SSR markers from defined regions of the wheat genome to identify Lophopyrum elongatum specific loci. Genome. 2005;48(5):811–22.
Zhang W, Gao AL, Zhou B, Chen PD. Screening and applying wheat microsatellite markers to trace individual Haynaldia villosa chromosomes. Acta Genet Sin. 2006;33(3):236–43.
Liu SB, Tang ZH, You MS, Li BY, Song JM, Liu GT. Development and application of a genome specific PCR marker for Haynaldia villosa. Acta Genet Sin. 2003;30(4):350–6.
Qi LL, Friebe B, Zhang P, Gill BS. Homoeologous recombination, chromosome engineering and crop improvement. Chromosome Res. 2007;15(1):3–19.
Zhao RH, Wang HY, Jia Q, Xiao J, Yuan CX, Zhang YJ, et al. Development of EST-PCR markers for the chromosome 4 V of Haynaldia villosa and their application in Identification of 4 V chromosome structural aberrants. J Integr Agr. 2014;13(2):282–9.
Dolezel J, Vrana J, Capal P, Kubalakova M, Buresova V, Simkova H. Advances in plant chromosome genomics. Biotechnol Adv. 2014;32(1):122–36.
Miftahudin RK, Ma XF, Mahmoud AA, Layton J, Milla MAR, et al. Analysis of expressed sequence tag loci on wheat chromosome group 4. Genetics. 2004;168(2):651–63.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Key Research and Development Program (2016YFD0102001) to XEW, the National Natural Science Foundation of China (Grant No. 31571653, 31201204, 31501305) to HYW and JX, the National High Technology Research Program (‘863’ Program) of China (Grant No. 2011AA10010201) and ‘948’ Project of Ministry of Agriculture (2015-Z41) to XEW, the Fundamental Research Funds for the Central Universities (KYZ201403; KJ2013003) to HYW and JX, the Program of Introducing Talents of Discipline to Universities (No.B08025) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Six Talent Peaks project in Jiangsu Province to XEW. JD has been supported by the Ministry of Education, Youth and Sports of the Czech Republic (grant LO1204 from the National Program of Sustainability I).
Availability of data and materials
Scaffold sequences for Common wheat cv. Chinese Spring chromosome arms 4A, 4B, 4D and Aegilops tauschii 4D as well as the annotated genes on 4DS of CS and Aegil-ops tauschii 4D were downloaded from the http://plants.ensembl.org/index.html. The sequence read data of 4VS chromosome were deposited in the (NCBI) Sequence Read Archive (SRA) and is available under accession number SRR3741672.
Authors’ contributions
Author Contribution Statement: WXE, WHY and XJ designed experimental plan. DKL, YCX and WHY performed all experiments. XJ, WYF and CAZ carried out sequence analysis and designed IT markers. ZRH and CPD contributed to the development of the germplasm used in the experiments. ZSZ managed the materials in the field. WHY, WXE and JD wrote the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
The chromosome composition of these materials used in the experiment. (PNG 138 kb)
Additional file 2: Figure S2.
Histogram of relative DAPI fluorescence (flow karyotype). Histogram obtained after flow cytometric analysis of mitotic metaphase chromosomes of T. aestivum-H. villosa ditelosomic additional line Dt4VS. Peak corresponding to telosomes 4VS (red arrow pointed) is well discriminated, which facilitated their flow sorting. Sorted chromosome arms were identified after FISH with probes for GAA (green) and pSc119.2 (red) repeat, which results in characteristic banding pattern (inset). X-axis: relative DAPI fluorescence intensity; Y-axis: number of particles. (PNG 232 kb)
Additional file 3: Figure S3.
The numbers of genes annotated in Chinese Spring 4DS chromosome and 60-Mb of Ae. tauschii 4D. The Venn diagrams displayed the numbers of genes differently annotated in Chinese Spring 4DS chromosome and 60-Mb of Ae. tauschii 4D (outer cycle), and the number of shared conserved genes among the two chromosomes (inner cycle). (PNG 133 kb)
Additional file 4: Table S1.
The IT markers corresponding to the sequence information in subgenome AA, BB, DD and VV (DOCX 38 kb)
Additional file 5: Table S2.
The sequences of forward and reverse primers and the size of intron of the IT markers (DOCX 48 kb)
Additional file 6: Table S3.
PCR products of 100 IT markers. (DOCX 101 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Wang, H., Dai, K., Xiao, J. et al. Development of intron targeting (IT) markers specific for chromosome arm 4VS of Haynaldia villosa by chromosome sorting and next-generation sequencing. BMC Genomics 18, 167 (2017). https://doi.org/10.1186/s12864-017-3567-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3567-z