
Abstract
Cellular senescence has evolved from an in-vitro model system to study aging in vitro to a multifaceted phenomenon of in-vivo importance as senescent cells in vivo have been identified and their removal delays the onset of age-associated diseases in a mouse model system. From the large emerging class of non-coding RNAs, miRNAs have only recently been functionally implied in the regulatory networks that are modified during the aging process. Here we summarize examples of similarities between the differential expression of miRNAs during senescence and age-associated diseases and suggest that these similarities might emphasize the importance of senescence for the pathogenesis of age-associated diseases. Understanding such a connection on the level of miRNAs might offer valuable opportunities for designing novel diagnostic and therapeutic strategies.
Similar content being viewed by others


Review
Introduction
During aging the incidence of acute and chronic conditions such as neurological disorders, diabetes, degenerative arthritis, and even cancer rises within individuals, so that aging has been termed the substrate on which age-associated diseases grow. Still, the molecular pathways underlying aging are not well understood as large individual heterogeneity of the biological aging process is observed. These interindividual differences are proposed to derive from accumulation of stochastic damage that is counteracted by genetically encoded and environmentally regulated repair systems. At the level of molecules repair works by enzymatic systems while on the cellular level it works by replication and differentiation to maintain tissue homeostasis. However, the replicative potential of somatic and adult stem cells is limited by cellular senescence and recent evidence shows that counteracting senescence or removing senescent cells delays the onset of age-associated pathologies. Here we summarize the current knowledge on how miRNAs might be connecting senescence and age-associated diseases and how such knowledge might be used in the context of biomedical research and medicine.
Cellular senescence
Replicative senescence was discovered almost 50 years ago when Hayflick observed that normal human cells in culture do have a limited replicative potential [1]. The counting mechanism of the amount of replications was found to be telomere shortening due to the end replication problem [2]. After reaching the replicative limit also termed Hayflick limit, cells enter an irreversible growth arrest that is triggered by critically short, unprotected telomeres that induce a DNA damage like signal [3]. This cell cycle arrest is executed by either of the two important cell cycle inhibitors, p21 or p16, and has so far not been reversible by any known combination of growth factors [4].
Triggers of cellular senescence
By now, several other triggers to a replicative senescence-like irreversible growth arrest have been observed (Figure 1), leading to a the broader term ‘cellular senescence’ that includes: (1) replicative senescence; (2) senescence that is induced by various physico-chemical stressors that induce DNA damage and chromatin disruption, such as, for example, oxidative stress leading to the term stress-induced premature senescence (SIPS); as well as (3) hyperoncogenic signaling-induced senescence, for example by constitutively active HRAS [5, 6].

Age-associated functional decline of organs and tissues.
The senescent cell phenotype is characterized by a combination of changes in cell morphology, behavior, structure, and functions. This includes alteration in gene expression [7], protein secretion [8], and inducibility of apoptosis, which increases in senescent fibroblasts [9] and decreases in endothelial cells [10].
Cellular senescence in vivo
By now, the presence and age-related accumulation of senescent cells in vivo has become well accepted [7, 11, 12] in various tissues like skin [13], liver [14], kidney [15–17], vasculature [18, 19], as well as astrocytes in the cortex of the brain [20, 21]. Astrocyte senescence as a component of Alzheimer’s disease.
But is such an accumulation ‘good’ or ‘bad’ for the organism? There seems to no easy answer to this question considering the different faces of senescence [22].
Beneficial functions of senescence include limitation of the extent of fibrosis following liver damage [14]. In addition, senescence has also been well accepted by now as a tumor suppressor mechanism, even in vivo. As senescent cells never re-enter the cell cycle, senescence is considered to prevent malignant transformation of potentially mutated cells.
However, some senescent cells also persist within tissues and are not eliminated by apoptosis or the immune system, such that their altered functional profile might alter tissue microenvironments in ways that can promote both cancer and aging phenotypes [22–24]. Especially in regard to age-associated diseases like atherosclerosis [18, 19, 25] or kidney diseases [26], increasing amounts of senescent cells have been found to at least correlate as will be outlined in more detail below. Causality beyond correlation, however, is supported by the fact that removal of senescent, p16 positive cells in mice delays the onset of at least three prominent age-associated diseases, cataract, sarcopenia, and loss of adipose tissue, even if the model system of BubR1 knock-out mice seems artificial due to its premature aging phenotype [27, 28]. Similarly, inducible onset of telomerase reverses age-related functional decline in a third generation telomerase knock-out mouse [29–31] and a gene therapy using hTERT in old mice delays aging and prolongs the life span [32].
Thus, the detrimentally altered functionality of senescent cells might lead to a vicious circle accelerating senescence and/or loss of cells within tissues, resulting in the age-associated decline of body functions and the rise in age-associated diseases. Such altered functionalities are clearly caused by changes in the gene expression pattern of senescent cells, which includes non-coding RNAs and particularly miRNAs.
MicroRNAs: basics of biogenesis, function, and turnover
MiRNAs comprise a large family of approximately 21-nucleotide-long non-coding RNAs that have emerged as key post-transcriptional regulators of gene expression and have revolutionized our comprehension of the post-transcriptional regulation of gene expression. The first miRNA, lin-4, was discovered by Ambros’s group less than 20 years ago [33]. Since then, the field of small non-coding RNAs has exploded, so that today we are close to developing miRNAs as clinical tools in diagnostics and therapeutic strategies.
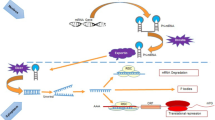
The biogenesis of miRNAs (Figure 2) involves processing from precursor molecules (pri-miRNAs), which are either transcribed by RNA polymerase II as independent genes or can be derived from introns after splicing [34]. The pri-miRNAs are processed by Drosha to pre-miRNAs, exported to the cytoplasm where Dicer cleaves them to the mature approximately 20-bp miRNA 5p/miRNA 3p duplexes. One strand of this duplex is then incorporated into the miRNA-inducing silencing complex (miRISC) [35].

Overview on miRNA biogenesis and translation repression.
Silencing of target mRNAs depends on recognition by base-pair mediated binding. This binding is based on a ‘seed’ region consisting of nucleotides 2 to 8 of the miRNA only. This seed can be supported by 3′ base-pairing after a short bulge of non-complementarity in the ‘canonical’ binding model. In addition, a shorter seed of down to four nucleotides at the 5′ end is still able to silence targets if 3′ compensatory complementary supports miRNA-mRNA binding [36]. Due to this ‘loose’ specificity, one miRNA is able to regulate up to 100 mRNA targets and therefore seems to orchestrate a large variety of cellular processes similar to transcription factors [37, 38]. While most miRNAs have been reported to bind to the 3′ end of their mRNA targets, also 5′ end have been identified as miRNA binding sites [39] and binding within the coding sequence has been found [40].
The variability of miRNA/mRNA targeting, however, also includes a ‘miRNA-escape mechamism’ on the side of mRNAs. Alternative polyadenylation has been shown to generate mRNAs that lack the seed regions and thus can evade miRNA-mediated regulation in stem cells [41, 42], in quiescent versus proliferating T cells [43, 44], but also in cancer cells, where in consequence shorter 30-UTRs arising from alternative cleavage and polyadenylation activate oncogenes [45].
Finally, also miRNA half-life is regulated. It was shown, that miRNAs are subject to degradation by the 5′ → 3′ exoribonuclease XRN-2 both in vitro and in vivo. In vitro, this process involved miRNA release from AGO, followed by degradation by XRN-2, and both release and degradation were prevented when mRNA was present that had binding sites for the miRNA [46]. In vivo, this so-called target mediated miRNA protection (TMMP) acts in opposition to miRNA degradation mediated by XRN-1 and XRN-2 [47].
Summarized, miRNAs are emerging as orchestrators of cell behavior, conferring robustness and balance to biological regulatory loops in many basic biological processes and diseases like cancer. In addition, some functions of miRNAs in controlling aging processes have been uncovered recently as are summarized below: miRNAs regulate lifespan in the nematode Caenorhabditis elegans [48, 49], various miRNAs are regulated during mammalian aging in mouse or human tissues [50, 51], and, especially, miRNAs have been implicated in governing senescence in a variety of human cells [52–55].
MiRNAs and cellular senescence
The identification of miRNAs that contribute to induction and maintenance of senescence might also reveal how cellular functions change to allow or even promote induction of age-associated diseases. The general importance of miRNA biogenesis on senescence has been established by the finding that dicer knock-out induces senescence [56].
During the last few years, several studies have then identified differentially transcribed miRNAs during cellular senescence in various cell types and different senescence inducing conditions including fibroblasts [57–63], keratinocytes [64, 65], endothelial cells [51, 66, 67], renal cells, [51, 68], T-cells [51], human mesenchymal stem cells of different origins [69, 70], UVB-induced senescence of fibroblasts (Greussing et al., in revision), mouse embryonic fibroblasts [71, 72], trabecular meshwork cells [73], and oncogene-induced senescence in human mammary epithelial cells [74]. Most of these miRNAs are still functionally uncharacterized and might be regulated as a consequence of senescence, and thus contribute to the cellular phenotype of senescence. However, some miRNAs are by now clearly involved in the regulation of senescence.
With regard to cell cycle regulation, we outline here only a few examples of miRNAs that are involved in regulating the senescent phenotype, in particular the let-7 family of miRNAs which inhibits KRAS, HMGA2, and c-MYC. In addition, let-7 is involved in aging of the testis stem cells in Drosophila melanogaster [75]. Similarly, miR15a/16-1 cluster and the miR-17-92 cluster are potent regulators of cell cycle progression by targeting CDK6, CARD10, and CDC27 as well as the CDK inhibitor family members p21, p27, and p57 as reviewed recently [76]. Members of the miR-17-92 cluster, the first identified ‘oncomiR’, has also been found as a commonly downregulated microRNA cluster in human replicative [51, 77], and stress-induced senescence [73], as well as organismal aging models. Indeed, inhibition of members of this cluster induces a senescent-like state in human fibroblasts [78], while its upregulation inhibits oncogene-induced senescence [79]. This indicates that this cluster is one additional important player not only in the complex regulatory network of cell cycle and tumorigenesis, but also in aging, emphasizing that these processes are intricately interwoven [52].
With regard to altered functionality of senescent cells, such as, for example, the secretion of cytokines, it is of note that miR-146, which is upregulated in senescent fibroblasts [59] as well as in endothelial cells, is an inhibitor of IL-6 and thus might contribute to the protein secretion alterations observed in senescent cells [59] termed the senescence-associated secretory phenotype (SASP). In addition to such a pro-inflammatory status, members of the miR-200 family that is causally regulating epithelial to mesenchymal transition (EMT), which is an important process in fibrotic as well as metastatic events, has been found as differentially regulated in metformin stress-induced senescence of human fibroblasts [80] as well as in oxidative stress induced senescence of endothelial cells [81]. Finally, miR-24 that is regulated in T cell senescence is also involved in reducing the DNA damage resistance of these cells and thus might contribute to depletion of CD28(−) CD8(+) T cells (Brunner, 2012 #10142).
Replicative senescence, miRNAs, and age-associated diseases
Aging is the substrate on which age-associated diseases are growing
The processes underlying normal aging include accumulation of damage and lack of repair on molecular, cellular, and tissue level ultimately leading to the progressive decline of body functions. Such a decline seems to be an initial event in the pathogenesis of several diseases. Those pathologies that show rapidly increased incidence with higher age and that have advanced age as a single important risk factor are categorized as age-associated diseases. We here rely on a classification of age-associated diseases recently compiled into a comprehensive list by George Martin and colleagues [82] and will here put emphasis on those pathologies that have been connected to cellular senescence (Additional file 1: Table S1). In addition we also summarized all of these most common diseases of the elderly (Table 1). Although many types of cancer can definitely be classified as age-associated diseases, this is not the focus of this review and we kindly recommend some of the very good reviews in the field of cancer and miRNAs [83–85].
Senescence, miRNAs, and cardiovascular diseases
Cardiovascular diseases (CWD) (such as atherosclerosis, diabetes, and hypertension) are the primary cause of death and disability in the Western world. These diseases have long been considered to be age-related in terms of their onset and progression [112]. Vascular aging is associated with endothelial dysfunctions [113–115], arterial stiffening and remodeling [116], impaired angiogenesis [117], defects in vascular repair [118], and with an increasing prevalence of atherosclerosis [114, 119].
A common characteristic of atherosclerosis is neointimal formation, that is alteration of endothelial cell (EC) physiology and hypoplasia of vascular smooth muscle cells (VSMC), which produce a multi-layered compartment internally to the tunica media of the arterial wall, including a gradual narrowing of the vessels lumen which may lead to thrombus formation and vessel occlusion [120].
The reasons for these associations are still unclear, but it is plausible that organismal aging and vascular disease may share common cellular mechanisms. Especially in regard to cellular senescence in vivo, senescent ECs as well as VSMCs have been connected to atherosclerosis [18, 19]. The association between vascular pathology and modification of gene expression gives a reasonable expectation that miRNAs may have a central role in the pathogenesis of vessel diseases (Figure 2).
Endothelial cell senescence and miRNAs
The importance of miRNAs in endothelial physiology (Figure 3) was revealed for the first time through the in-vitro disruption in human ECs of Dicer and Drosha [121–123]. ECs lacking either of these two enzymes showed an impaired ability to form tube structures on matrigel [123]. The generation of an endothelial-specific Dicer knock-out mouse model provided direct evidence that miRNAs are fundamental for the correct vessel development in adulthood in response to angiogenic stimuli [121]. In addition, miRNAs in the serum have been proposed as diagnostic markers for vascular diseases [124–126].

MiRNAs associated with age associated vascular diseases.
In atherosclerosis, an inflammatory response plays a central role in disease progression. In order to maintain the influx of leucocytes to the lesion areas, ECs increase expression of vascular cell adhesion molecules, such as VCAM-1. One of the most abundant miRNAs in endothelial cells, miR-126, directly represses VCAM-1 expression, thus playing an important role in leucocyte recruitment on the endothelial side [88]. Indeed, miR-126 is downregulated in human aortic endothelial cells [67], and circulating levels of VCAM-1 are increased in elderly human subjects [127]in vivo, in stress-induced senescent HUVECs in vitro[128], as well as on the surface of endothelial cells in rats in vivo and in senescent rat ECs in vitro[129]. Therefore, this might contribute to a pro-inflammatory status that allows for disease progression and might explain why upregulated VCAM-1 is suspected to be a causal factor in the pathogenesis of atherosclerosis [130] and is downregulated by a SCM-298, a substance that reduces formation of atherosclerotic plaques in rabbits [131].
Similarly, miR-217 upregulation in human atherosclerotic plaques was observed [66]. In-vitro senescent ECs also show higher levels of miR-217 than early passage cells and functionally, miR-217 was able to induce premature EC senescence with SirT1 as target mRNA [66]. Moreover it was shown, that SirT1 acts in complex with FOXO3, a factor involved in modulating longevity in several model systems also regulates senescence in human cell cultures [93]. Of note, a prominent miRNA highly expressed in senescent cells and inducing cellular senescence, miR-34, also converges on SirT1 as a target. Since high levels of SirT1 have been found protective against atherosclerosis by several different studies as reviewed [132], high levels of SirT1 targeting miRNAs as observed in endothelial senescence might indeed contribute to disease progression.
Vascular smooth muscle cell senescence and miRNAs
Not only endothelial cells, but also vascular smooth muscle cells (VSMCs) play a major role during events of arterial remodeling and atherosclerosis development. Indeed, miR-21 has been found to be deregulated in EC [67] and fibroblast senescence, as a regulator of neointima lesion formation [133]. Downregulation of aberrantly expressed miR-21 decreased neointima formation in rat carotid artery after angioplasty which classifies miR-21 as a potential therapeutic target [133]. Furthermore, miR-143 and miR145 were reported to be downregulated in VSMCs during neointimal formation in rats [133] and that dysregulation of miR-143 and miR-145 genes is causally involved in the aberrant VSMC plasticity encountered during vascular disease [95]. Indeed, miR-143 is also regulated during senescence, although it has been reported only in fibroblasts so far [134].
Diabetes mellitus, type 2
Type 2 diabetes mellitus (T2D) has reached epidemic proportions worldwide [135]. It is estimated that the current 150 million to 220 million people with diabetes will rise up to 300 million in 2025 [136]. T2D is a progressive metabolic disorder characterized by reduced insulin sensitivity, insulin resistance in tissues such as skeletal muscle, liver and adipose tissue, combined with pancreatic β-cell dysfunction, resulting in systemic hyperglycemia [137]. Improper treatment of T2D can lead to severe complications such as heart disease, stroke, kidney failure, blindness, and nerve damage [138].
Cell senescence has recently been postulated as an important cause/consequence of type 2 diabetes and its complications [139].
Circulating miRNAs have also been identified here as potential diagnostic tools [140]. Interestingly, one of the robustly down-regulated miRNAs in this study is miR-126 that is also downregulated in senescent endothelial cells (see above).
Senescence has also been implicated in insulin secretion, since hTERC knock-out mice in the third generation are defective in insulin secretion and glucose metabolism [141]. Remarkably, hTERT gene therapy in old mice restored the age-dependent loss of insulin sensitivity [32]. Similarly, several miRNAs have been implicated in insulin secretion like miR-375, which is one of the most abundant miRNAs in pancreatic islets and beta cells and inhibits insulin secretion via myotrophin (Mtpn) [98]. Mtpn controls release of the neurotransmitter catecholamine [142], that in turn triggers insulin release [98]. In terms of cellular senescence, miR-375 has only been implicated in a chemotherapeutically-induced senescence of the tumor cell line K562.
More recently, miR-130a, miR-200, and miR-410 also were described to be involved in the regulation of insulin secretion [100] and at least members of the miR-200 family are known to contribute to senescence [81].
MiRNAs are not only involved in regulating insulin secretion, but also control insulin signaling in insulin target tissues. In Goto-Kakizaki (GK) rats, which are used as a non-obese model of T2D, members of the miR-29 family are elevated in muscle, fat, and liver, the most important insulin-responsive tissues [143]. This might be causally related to loss of insulin responsiveness, since overexpression of miR-29 in vitro in 3 T3-L1 adipocytes also inhibits insulin and glucose responses. This effect might be due to silencing insulin-induced gene 1 (Insig 1) and caveolin 2 (Cav2) [143], two key insulin-responsive proteins. It is of note that miR-29 is also upregulated during cellular senescence [144].
It can be expected that insulin signaling is also directly regulated by miRNAs. Major players in this pathway are insulin receptor substrate (IRS) proteins. Indeed, miR-145 is established as regulator of IRS1 [145] (Figure 4), however, for IRS2, the central player in the development of T2D and its associated complications, no experimentally confirmed target has been identified so far.

MiRNAs associated with Type 2 Diabetes mellitus.
Kidney diseases
Almost all types of kidney diseases are more common in the elderly having higher risk and incidence of both acute kidney injury (AKI) and end-stage renal disease (ESRD). The precursor state of ESRD, namely generic chronic kidney disease (CKD), is also much more common in the elderly [146]. Furthermore, fibrotic events also diminish kidney functionality. This loss of functionality again is correlated with increase of senescent cells in the kidney [15, 26, 43, 147]. Furthermore, high amounts of senescent cells in kidneys for transplantation are correlated with low transplantation success [16, 17], supporting the idea that senescent cells are ‘bad citizens’ and ‘bad neighbors’ in the kidney of the elderly. MicroRNAs have already been found to be involved in senescence of different kidney cells. In rat mesangial cells, miR-335 as well as miR-34c promote senescence by suppressing antioxidative defense proteins [68]. Loss of miR-335 expression has been found in patients of renal cell carcinoma, which might be in keeping with the idea that miR-335 can act as a tumor suppressor by inducing senescence [148].
In addition, senescent renal proximal tubular epithelial cells have high levels of miRNAs of the miR-200 family including miR-205 [51]. It seems that therefore, senescence of RPTECs and fibrosis might be linked [149]. Indeed, EMT seems to be regulated in renal fibrinogenesis by this family, and miR-200 can ameliorate this condition. It was shown, that five members of the miR-200 family (miR-200a/b/c, miR-141, and miR-429) and miR-205 are specifically downregulated in MDCK cells undergoing EMT [150]. Moreover, miR-200b ameliorates tubulointerstitial fibrosis in obstructed kidneys and thereby might constitute a novel therapeutic targets in kidney disease [151]. Subsequently, the function of the miR-200 family in regulating ZEB1 and ZEB2 and in modulating EMT in a number of different cell types has been confirmed [152–155].
Similarly important for regulating EMT in the kidney are miRNA-192/215 [156], two miRNAs that are specifically high in kidney tissue [91] (Figure 5). miR-192, in particular, also plays a role in diabetic nephropathy [157], as its loss correlates with tubulointerstitial fibrosis and reduction in eGFR in renal biopsies from patients with established diabetic nephropathy [158].

MiRNAs in age associated disease of the kidney.
Finally, miR-29 represses the expression of collagen I and IV at both the mRNA and protein level [110] and is downregulated in senescent RPTECs [51], thus it might contribute to more fibrinogenic material in the aged kidney.
In keeping with the above, in 34 consecutive patients with biopsy-proven hypertensive nephrosclerosis, a progressive disease that results from sclerosis of the small blood vessels in the kidney and is most commonly associated with hypertension or diabetes, intrarenal expression of miR-200a, miR-200b, miR-141, miR-429, miR-205, and miR-192 were increased, and the degree of upregulation correlated with disease severity [159]. Taken together, both cellular senescence as well as miRNAs regulated in cellular senescence have been found to negatively impact on kidney functionality. We therefore suggest that the link between senescence, miRNAs, and kidney disease might not only be correlative, but causal in the aging kidney.
Osteoporosis
The skeleton is continuously remodeled throughout the lifetime of an individual in a dynamic process of bone resorption and bone formation, to replace damaged bone or to respond to metabolic needs [160]. This bone turnover is mediated by the delicate balance of osteoblast and osteoclast numbers and activities. Osteoclasts resorb bone, whereas ostoeblasts synthesize new bone [161]. Dysregulation of either one of these cell types therefore results in an imbalance of bone turnover and pathological consequences, including osteoporosis in case of prevalent bone resorption, resulting in excessive skeletal fragility leading to frailty and a high risk of low-trauma fractures.
Hints for the importance of cellular senescence in the development of osteoporosis come from hTERC knock-out mice [162, 163], hTERT gene therapy that delays the onset of osteoporosis in old mice [32] as well from the fact that removal of senescent p16+ cells also delays the onset of skeletal deformation in the progeroid BubR1 deficient mouse [27]. The cell types mainly studied with regard to senescence are mesenchymal stem cells that are the progenitors for osteoblasts. It has been shown that the replicative potential of MSCs clearly depends on the age of the donor [164], a fact that is not so clear for fibroblast strains [165].
Although, a clear physiological link between osteoporosis and the loss of replicative potential of cells seems to exist, too few studies have yet addressed miRNAs and MSC senescence. Still, we want to point out some candidate miRNAs that have been found to play a role during the differentiation from MSCs to osteoblasts (Figure 6), among them miR-637 [166], miR-133 and miR-135, the miR-29 family [99, 167], and miR-138 [168]. In regard to osteoclasts, only few reports exist and identify miR-233 to reduce formation of osteoclast-like cells in RAW264.7 mouse cells as model system [101, 169].

MiRNAs associated with osteoporosis.
However, so far only miR-2861 has been implicated functionally in osteoporosis, as silencing of it in vivo in mice reduced bone formation and bone mass [170]. In addition, mutations in the pre-miR-2861 in two patients result in lack of mature miR-2861, causing primary osteoporosis [170].
Cataract
Cataracts are a very common eye pathology with advanced age being one of the most prominent risk factors. Most people above the age of 65 years show some changes in lens structure and most will develop a cataract in time [171]. Recently it was shown that cataract formation was significantly accelerated in BubR1H/H mice. However, by removal of senescent cells the onset of cataract formation was significantly delayed, emphasizing the importance of senescence in this regard [27, 28] as well as by hTERT gene therapy [32].
Moreover, it was shown that miRNAs play a role in age-related cataracts [102]. Let-7 miRNA, an important regulator of cellular aging and tissue senescence [102], was demonstrated to be positively associated with patient age and a positive correlation was also observed between cataract and higher expression of let-7b miRNA in patients with age-related cataracts [102]. Moreover it was shown that miR-184 and miR-204 play a role in formation of secondary cataracts, formed mostly after eye surgery, or caused by diabetes or steroid use [105].
Sarcopenia
Sarcopenia can be defined as the age-related loss of muscle mass, strength, and function, and appears to begin in the fourth decade of life and accelerates after the age of approximately 75 years [109, 172]. While many factors contribute to the onset of sarcopenia, one of the main causes is a change in the nature of a small population of muscle stem cells, also called satellite cells. Similar to cataracts, skeletal muscle degeneration was greatly reduced in BubR1H/H muscles after removal of senescent, p16 positive cells [27, 28]. In addition, senescence of muscle cells and satellite cells seems to be implicated in muscle metabolism and disease [173–175].
So far, however, only a few miRNAs were found to be implicated in satellite cell regulation. MiRNA-489 is highly expressed in quiescent satellite cells and is quickly downregulated during satellite-cell activation [107]. Further analysis revealed that miR-489 functions as a regulator of satellite-cell quiescence, as it post-transcriptionally suppresses the oncogene Dek, the protein product of which localizes to the more differentiated daughter cell during asymmetric division of satellite cells and promotes the transient proliferative expansion of myogenic progenitors [107]. Moreover miR-1 and miR-206 can improve human satellite cell differentiation via repressing Pax7, a central player in satellite cell survival, self-renewal, and proliferation [111, 176]. No data are yet available which link these miRNAs to cellular senescence. It will be interesting to what extent such a connection might exist, especially in view to the role of the systemic environment on satellite cell function, since in heterochronic parabiosis of young and old mice the proliferation and regenerative capacity of aged satellite cells was ‘rejuvenated’ [177, 178].
Other age-related diseases
Many more age-associated diseases are known (Additional file 1: Table S1), among them Alzheimer’s disease (AD), Parkinson’s disease, degenerative arthritis, and destructive eye diseases. Except for AD and Parkinson’s disease, to date no reports exist linking miRNAs to these diseases and very recent reviews are available on miRNAs in neurodegenerative diseases [179]. Similarly, we want to refer the reader to recent reviews on miRNAs and cancer [83–85], which is one of the important age-related diseases. In cancer, miRNAs have a potential value as tumor markers and it was shown that deregulation of miRNAs not only results as consequence of cancer progression but also directly promotes tumor initiation and progression in a cause-effect manner.
Conclusion
As cellular senescence is becoming ever more prominent as a mechanism that can drive aging and promote age-related diseases, one of the questions that is only poorly answered remains: how many senescent cells can be found in the elderly in specific tissues and what are the functional changes that tissue specific cells undergo when senescent, as it is clear that cell types as diverse as fibroblasts and epithelial or endothelial cells also will greatly vary when senescent. The comparison of miRNAs involved in cellular senescence to miRNAs involved in age-associated diseases shows that surprisingly many miRNAs are shared in these in-vitro and in-vivo situations. While it is clear that these similarities are merely correlative, a more detailed study on causal links might be a good approach to identify novel diagnostic and therapeutic strategies for age-associated diseases. In addition, since miRNAs are only a small part of the emerging non-coding RNA field, other ncRNAs might emerge equally important for the understanding of the aging process and the pathogenesis of age-associated diseases.
References
Hayflick L: The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965, 37: 614-636. 10.1016/0014-4827(65)90211-9.
Bodnar A, Ooellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright W: Extension of life-span by introduction of telomerase into normal human cells. Science. 1998, 279: 349-352. 10.1126/science.279.5349.349.
Palm W, de Lange T: How shelterin protects mammalian telomeres. Annu Rev Genet. 2008, 42: 301-334. 10.1146/annurev.genet.41.110306.130350.
Ben-Porath I, Weinberg RA: The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005, 37: 961-976. 10.1016/j.biocel.2004.10.013.
Cabrera T, Garrido V, Concha A, MartÃn J, Esquivias J, Oliva MR, Ruiz-Cabello F, Serrano S, Garrido F: Hla molecules in basal cell carcinoma of the skin. Immunobiology. 1992, 185: 440-452. 10.1016/S0171-2985(11)80086-0.
Maruyama J, Naguro I, Takeda K, Ichijo H: Stress-activated map kinase cascades in cellular senescence. Curr Med Chem. 2009, 16: 1229-1235. 10.2174/092986709787846613.
Campisi J, d’Adda di Fagagna F: Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol. 2007, 8: 729-740.
Kuilman T, Peeper DS: Senescence-messaging secretome: Sms-ing cellular stress. Nat Rev Cancer. 2009, 9: 81-94. 10.1038/nrc2560.
Wang E: Regulation of apoptosis resistance and ontogeny of age-dependent diseases. Exp Gerontol. 1997, 32: 471-484. 10.1016/S0531-5565(96)00156-8.
Hampel B, Malisan F, Niederegger H, Testi R, Jansen-Durr P: Differential regulation of apoptotic cell death in senescent human cells. Exp Gerontol. 2004, 39: 1713-1721. 10.1016/j.exger.2004.05.010.
Campisi J, Sedivy J: How does proliferative homeostasis change with age? What causes it and how does it contribute to aging?. J Gerontol A Biol Sci Med Sci. 2009, 64: 164-166.
Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM: Microrna-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. 2009, 36: 181-188. 10.1111/j.1440-1681.2008.05057.x.
Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM: Cellular senescence in aging primates. Science. 2006, 311: 1257-10.1126/science.1122446.
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW: Senescence of activated stellate cells limits liver fibrosis. Cell. 2008, 134: 657-667. 10.1016/j.cell.2008.06.049.
Melk A, Kittikowit W, Sandhu I, Halloran KM, Grimm P, Schmidt BM, Halloran PF: Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003, 63: 2134-2143. 10.1046/j.1523-1755.2003.00032.x.
Melk A, Schmidt BM, Braun H, Vongwiwatana A, Urmson J, Zhu LF, Rayner D, Halloran PF: Effects of donor age and cell senescence on kidney allograft survival. Am J Transplant. 2009, 9: 114-123.
Koppelstaetter C, Schratzberger G, Perco P, Hofer J, Mark W, Ollinger R, Oberbauer R, Schwarz C, Mitterbauer C, Kainz A, Karkoszka H, Wiecek A, Mayer B, Mayer G: Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell. 2008, 7: 491-497. 10.1111/j.1474-9726.2008.00398.x.
Erusalimsky JD: Vascular endothelial senescence: From mechanisms to pathophysiology. J Appl Physiol. 2009, 106: 326-332.
Minamino T, Komuro I: Vascular cell senescence: Contribution to atherosclerosis. Circ Res. 2007, 100: 15-26. 10.1161/01.RES.0000256837.40544.4a.
Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S, Torres C: Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010, 316: 2961-2968. 10.1016/j.yexcr.2010.06.021.
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C: PLoS One. 2012, 7 (9): e45069-10.1371/journal.pone.0045069. Epub 2012 Sep 12
Rodier F, Campisi J: Four faces of cellular senescence. J Cell Biol. 2011, 192: 547-556. 10.1083/jcb.201009094.
Krtolica A, Campisi J: Cancer and aging: A model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002, 34: 1401-1414. 10.1016/S1357-2725(02)00053-5.
Rodier F, Campisi J, Bhaumik D: Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35: 7475-7484. 10.1093/nar/gkm744.
Erusalimsky J, Skene C: Mechanisms of endothelial senescence. Exp Physiol. 2009, 94: 299-304. 10.1113/expphysiol.2008.043133.
Melk A: Senescence of renal cells: Molecular basis and clinical implications. Nephrol Dial Transplant. 2003, 18: 2474-2478. 10.1093/ndt/gfg322.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM: Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011, 479: 232-236. 10.1038/nature10600.
Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, Eberhardt NL, Terzic A, van Deursen JM: Opposing roles for p16ink4a and p19arf in senescence and ageing caused by bubr1 insufficiency. Nat Cell Biol. 2008, 10: 825-836. 10.1038/ncb1744.
Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES, Hu J, Wang W, Xiao Y, Zhang H, Zhang J, Gan B, Perry SR, Jiang S, Li L, Horner JW, Wang YA, Chin L, Depinho RA: Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012, 148: 896-907. 10.1016/j.cell.2012.01.039.
Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadiñanos J, Horner JW, Maratos-Flier E, Depinho RA: Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011, 469: 102-106. 10.1038/nature09603.
Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA: Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011, 470: 359-365. 10.1038/nature09787.
Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA: Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012, 4: 691-704. 10.1002/emmm.201200245.
Lee RC, Feinbaum RL, Ambros V: The c. Elegans heterochronic gene lin-4 encodes small rnas with antisense complementarity to lin-14. Cell. 1993, 75: 843-854. 10.1016/0092-8674(93)90529-Y.
Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC: Mammalian mirtron genes. Mol Cell. 2007, 28: 328-336. 10.1016/j.molcel.2007.09.028.
Krol J, Loedige I, Filipowicz W: The widespread regulation of microrna biogenesis, function and decay. Nat Rev Genet. 2010, 11: 597-610.
Brennecke J, Stark A, Russell RB, Cohen SM: Principles of microrna-target recognition. PLoS Biol. 2005, 3: e85-10.1371/journal.pbio.0030085.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM: Microarray analysis shows that some micrornas downregulate large numbers of target mrnas. Nature. 2005, 433: 769-773. 10.1038/nature03315.
Stefani G, Slack FJ: Small non-coding rnas in animal development. Nat Rev Mol Cell Biol. 2008, 9: 219-230. 10.1038/nrm2347.
Roberts AP, Lewis AP, Jopling CL: Mir-122 activates hepatitis c virus translation by a specialized mechanism requiring particular rna components. Nucleic Acids Res. 2011, 39: 7716-7729. 10.1093/nar/gkr426.
Dai L, Tsai-Morris CH, Sato H, Villar J, Kang JH, Zhang J, Dufau ML: Testis-specific mirna-469 up-regulated in gonadotropin-regulated testicular rna helicase (grth/ddx25)-null mice silences transition protein 2 and protamine 2 messages at sites within coding region: Implications of its role in germ cell development. J Biol Chem. 2011, 286: 44306-44318. 10.1074/jbc.M111.282756.
Di Giammartino DC, Nishida K, Manley JL: Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011, 43: 853-866. 10.1016/j.molcel.2011.08.017.
Tan S, Guo J, Huang Q, Chen X, Li-Ling J, Li Q, Ma F: Retained introns increase putative microrna targets within 3′utrs of human mrna. FEBS Lett. 2007, 581: 1081-1086. 10.1016/j.febslet.2007.02.009.
Melk A, Ramassar V, Helms LM, Moore R, Rayner D, Solez K, Halloran PF: Telomere shortening in kidneys with age. J Am Soc Nephrol. 2000, 11: 444-453.
Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB: Proliferating cells express mrnas with shortened 3′untranslated regions and fewer microrna target sites. Science. 2008, 320: 1643-1647. 10.1126/science.1155390.
Mayr C, Bartel DP: Widespread shortening of 3′utrs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009, 138: 673-684. 10.1016/j.cell.2009.06.016.
Chatterjee S, Grosshans H: Active turnover modulates mature microrna activity in caenorhabditis elegans. Nature. 2009, 461: 546-549. 10.1038/nature08349.
Chatterjee S, Fasler M, Büssing I, Grosshans H: Target-mediated protection of endogenous micrornas in c. Elegans. Dev Cell. 2011, 20: 388-396. 10.1016/j.devcel.2011.02.008.
Pincus Z, Smith-Vikos T, Slack FJ: Microrna predictors of longevity in caenorhabditis elegans. PLoS Genet. 2011, 7: e1002306-10.1371/journal.pgen.1002306.
de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ: Micrornas both promote and antagonize longevity in c. Elegans. Curr Biol. 2010, 20: 2159-2168.
Smith-Vikos T, Slack FJ: Micrornas and their roles in aging. J Cell Sci. 2012, 125: 7-17. 10.1242/jcs.099200.
Hackl M, Brunner S, Fortschegger K, Schreiner C, Micutkova L, Mück C, Laschober GT, Lepperdinger G, Sampson N, Berger P, Herndler-Brandstetter D, Wieser M, Kühnel H, Strasser A, Breitenbach M, Rinnerthaler M, Eckhart L, Mildner M, Tschachler E, Papak C, Trost A, Bauer J, Scheideler M, Trajanoski Z, Grillari-Voglauer R, Grubeck-Loebenstein B, Jansen-Durr P, Grillari J: Mir-17, mir-19b, mir-20a and mir-106a are down-regulated in human aging. Aging Cell. 2010, 9: 291-296. 10.1111/j.1474-9726.2010.00549.x.
Grillari J, Hackl M, Grillari-Voglauer R: Mir-17-92 cluster: Ups and downs in cancer and ageing. Biogerontology. 2010, 11: 501-506. 10.1007/s10522-010-9272-9.
Gorospe M, Abdelmohsen K: Microregulators come of age in senescence. Trends Genet. 2011, 27: 233-241. 10.1016/j.tig.2011.03.005.
Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK: Microrna expression patterns reveal differential expression of target genes with age. PLoS One. 2010, 5: e10724-10.1371/journal.pone.0010724.
Srikantan S, Gorospe M, Abdelmohsen K: Senescence-associated micrornas linked to tumorigenesis. Cell Cycle. 2011, 10: 3211-3212. 10.4161/cc.10.19.17050.
Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, Lawrence JB, Imbalzano AN, Jones SN: Loss of mirna biogenesis induces p19arf-p53 signaling and senescence in primary cells. J Cell Biol. 2008, 181: 1055-1063. 10.1083/jcb.200802105.
Maes OC, Sarojini H, Wang E: Stepwise up-regulation of microrna expression levels from replicating to reversible and irreversible growth arrest states in wi-38 human fibroblasts. J Cell Physiol. 2009, 221: 109-119. 10.1002/jcp.21834.
Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R, Srikantan S, Subrahmanyam R, Martindale JL, Yang X, Ahmed F, Navarro F, Dykxhoorn D, Lieberman J, Gorospe M: P16(ink4a) translation suppressed by mir-24. PLoS One. 2008, 3: e1864-10.1371/journal.pone.0001864.
Bhaumik D, Scott GK, Schokrpur S, Patil CK, Orjalo AV, Rodier F, Lithgow GJ, Campisi J: Micrornas mir-146a/b negatively modulate the senescence-associated inflammatory mediators il-6 and il-8. Aging. 2009, 1: 402-411.
Brosh R, Shalgi R, Liran A, Landan G, Korotayev K, Nguyen GH, Enerly E, Johnsen H, Buganim Y, Solomon H, Goldstein I, Madar S, Goldfinger N, Borresen-Dale AL, Ginsberg D, Harris CC, Pilpel Y, Oren M, Rotter V: P53-repressed mirnas are involved with e2f in a feed-forward loop promoting proliferation. Mol Syst Biol. 2008, 4: 229-
Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K: Microrna profiling in human diploid fibroblasts uncovers mir-519 role in replicative senescence. Aging (Albany NY). 2010, 2: 333-343.
Dhahbi JM, Atamna H, Boffelli D, Magis W, Spindler SR, Martin DI: Deep sequencing reveals novel micrornas and regulation of microrna expression during cell senescence. PLoS One. 2011, 6: e20509-10.1371/journal.pone.0020509.
Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, Bellelli R, Nappi TC, Comegna M, Romano S, Salvatore G, Santoro M, Cimino F: A set of mirnas participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. 2012, 19: 713-721. 10.1038/cdd.2011.143.
Shin KH, Pucar A, Kim RH, Bae SD, Chen W, Kang MK, Park NH: Identification of senescence-inducing micrornas in normal human keratinocytes. Int J Oncol. 2011, 39: 1205-1211.
Rivetti di Val Cervo P, Lena AM, Nicoloso M, Rossi S, Mancini M, Zhou H, Saintigny G, Dellambra E, Odorisio T, Mahe C, Calin GA, Candi E, Melino G: P63-microrna feedback in keratinocyte senescence. Proc Natl Acad Sci USA. 2012, 109: 1133-1138. 10.1073/pnas.1112257109.
Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa-Nicotera M, Ippoliti A, Novelli G, Melino G, Lauro R, Federici M: Microrna 217 modulates endothelial cell senescence via silent information regulator 1. Circulation. 2009, 120: 1524-1532. 10.1161/CIRCULATIONAHA.109.864629.
Rippe C, Blimline M, Magerko KA, Lawson BR, LaRocca TJ, Donato AJ, Seals DR: Microrna changes in human arterial endothelial cells with senescence: Relation to apoptosis, enos and inflammation. Exp Gerontol. 2012, 47: 45-51. 10.1016/j.exger.2011.10.004.
Bai XY, Ma Y, Ding R, Fu B, Shi S, Chen XM: Mir-335 and mir-34a promote renal senescence by suppressing mitochondrial antioxidative enzymes. J Am Soc Nephrol. 2011, 22: 1252-1261. 10.1681/ASN.2010040367.
Wagner W, Horn P, Castoldi M, Diehlmann A, Bork S, Saffrich R, Benes V, Blake J, Pfister S, Eckstein V, Ho AD: Replicative senescence of mesenchymal stem cells: A continuous and organized process. PLoS One. 2008, 3: e2213-10.1371/journal.pone.0002213.
Kim YJ, Hwang SH, Lee SY, Shin KK, Cho HH, Bae YC, Jung JS: Mir-486-5p induces replicative senescence of human adipose tissue-derived mesenchymal stem cells and its expression is controlled by high glucose. Stem Cells Dev. 2012, 21: 1749-1760. 10.1089/scd.2011.0429.
Poliseno L, Pitto L, Simili M, Mariani L, Riccardi L, Ciucci A, Rizzo M, Evangelista M, Mercatanti A, Pandolfi PP, Rainaldi G: The proto-oncogene lrf is under post-transcriptional control of mir-20a: Implications for senescence. PLoS One. 2008, 3: e2542-10.1371/journal.pone.0002542.
Pitto L, Rizzo M, Simili M, Colligiani D, Evangelista M, Mercatanti A, Mariani L, Cremisi F, Rainaldi G: Mir-290 acts as a physiological effector of senescence in mouse embryo fibroblasts. Physiol Genomics. 2009, 39: 210-218. 10.1152/physiolgenomics.00085.2009.
Li G, Luna C, Qiu J, Epstein DL, Gonzalez P: Alterations in microrna expression in stress-induced cellular senescence. Mech Ageing Dev. 2009, 130: 731-741. 10.1016/j.mad.2009.09.002.
Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, Beach DH: Multiple micrornas rescue from ras-induced senescence by inhibiting p21(waf1/cip1). Oncogene. 2010, 29: 2262-2271. 10.1038/onc.2009.497.
Toledano H, D’Alterio C, Czech B, Levine E, Jones DL: The let-7-imp axis regulates ageing of the drosophila testis stem-cell niche. Nature. 2012, 485: 605-610. 10.1038/nature11061.
Chivukula RR, Mendell JT: Circular reasoning: Micrornas and cell-cycle control. Trends Biochem Sci. 2008, 33: 474-481. 10.1016/j.tibs.2008.06.008.
de Zhuo X, Niu XH, Chen YC, Xin DQ, Guo YL, Mao ZB: Vitamin D3 up-regulated protein 1(VDUP1) is regulated by FOXO3A and miR-17-5p at the transcriptional and post-transcriptional levels, respectively, in senescent fibroblasts. J Biol Chem. 2010, 285: 31491-31501. 10.1074/jbc.M109.068387.
Wang M, Cheng Z, Tian T, Chen J, Dou F, Guo M, Cong YS: Differential expression of oncogenic mirnas in proliferating and senescent human fibroblasts. Mol Cell Biochem. 2011, 352: 271-279. 10.1007/s11010-011-0763-z.
Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, Xiao C, Hu WY, Han J, Sun P: The mir-17-92 cluster of micrornas confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 2010, 70: 8547-8557. 10.1158/0008-5472.CAN-10-1938.
Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Quirantes R, Segura-Carretero A, Micol V, Joven J, Bosch-Barrera J, Del Barco S, Martin-Castillo B, Vellon L, Menendez JA: Metformin lowers the threshold for stress-induced senescence: A role for the microrna-200 family and mir-205. Cell Cycle. 2012, 11: 1235-1246. 10.4161/cc.11.6.19665.
Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, Antonini A, Martelli F, Capogrossi MC: Mir-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via zeb1 inhibition. Cell Death Differ. 2011, 18: 1628-1639. 10.1038/cdd.2011.42.
Martin GM, Bergman A, Barzilai N: Genetic determinants of human health span and life span: Progress and new opportunities. PLoS Genet. 2007, 3: e125-10.1371/journal.pgen.0030125.
Lovat F, Valeri N, Croce CM: Micrornas in the pathogenesis of cancer. Semin Oncol. 2011, 38: 724-733. 10.1053/j.seminoncol.2011.08.006.
Wang Y, Liang Y, Lu Q: Microrna epigenetic alterations: Predicting biomarkers and therapeutic targets in human diseases. Clin Genet. 2008, 74: 307-315. 10.1111/j.1399-0004.2008.01075.x.
Kato M, Slack FJ: Micrornas: Small molecules with big roles - c. Elegans to human cancer. Biol Cell. 2008, 100: 71-81.
Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, Jandeleit-Dahm K, Burns WC, Thomas MC, Cooper ME, Kantharidis P: miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes. 2011, 60: 280-287. 10.2337/db10-0892.
Raitoharju E, Lyytikäinen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kähönen M, Karhunen PJ, Laaksonen R, Lehtimäki T: Mir-21, mir-210, mir-34a, and mir-146a/b are up-regulated in human atherosclerotic plaques in the tampere vascular study. Atherosclerosis. 2011, 219: 211-217. 10.1016/j.atherosclerosis.2011.07.020.
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ: Microrna-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A. 2008, 105: 1516-1521. 10.1073/pnas.0707493105.
Chen CZ, Li L, Lodish HF, Bartel DP: Micrornas modulate hematopoietic lineage differentiation. Science. 2004, 303: 83-86. 10.1126/science.1091903.
Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K: Lymphoproliferative disease and autoimmunity in mice with increased mir-17-92 expression in lymphocytes. Nat Immunol. 2008, 9: 405-414. 10.1038/ni1575.
Sun Y, Koo S, White N, Peralta E, Esau C, Dean NM, Perera RJ: Development of a micro-array to detect human and mouse micrornas and characterization of expression in human organs. Nucleic Acids Res. 2004, 32: e188-10.1093/nar/gnh186.
Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF: Mir-150, a microrna expressed in mature b and t cells, blocks early b cell development when expressed prematurely. Proc Natl Acad Sci U S A. 2007, 104: 7080-7085. 10.1073/pnas.0702409104.
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME: Stress-dependent regulation of foxo transcription factors by the sirt1 deacetylase. Science. 2004, 303: 2011-2015. 10.1126/science.1094637.
Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R: Microrna-143 regulates adipocyte differentiation. J Biol Chem. 2004, 279: 52361-52365. 10.1074/jbc.C400438200.
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G: The knockout of mir-143 and −145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16: 1590-1598. 10.1038/cdd.2009.153.
Li Z, Hassan MQ, Volinia S, van Wijnen AJ, Stein JL, Croce CM, Lian JB, Stein GS: A microrna signature for a bmp2-induced osteoblast lineage commitment program. Proc Natl Acad Sci U S A. 2008, 105: 13906-13911. 10.1073/pnas.0804438105.
Goettsch C, Rauner M, Pacyna N, Hempel U, Bornstein SR, Hofbauer LC: Mir-125b regulates calcification of vascular smooth muscle cells. Am J Pathol. 2011, 179: 1594-1600. 10.1016/j.ajpath.2011.06.016.
Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M: A pancreatic islet-specific microrna regulates insulin secretion. Nature. 2004, 432: 226-230. 10.1038/nature03076.
Kapinas K, Kessler CB, Delany AM: Mir-29 suppression of osteonectin in osteoblasts: Regulation during differentiation and by canonical wnt signaling. J Cell Biochem. 2009, 108: 216-224. 10.1002/jcb.22243.
Hennessy E, Clynes M, Jeppesen PB, O’Driscoll L: Identification of micrornas with a role in glucose stimulated insulin secretion by expression profiling of min6 cells. Biochem Biophys Res Commun. 2010, 396: 457-462. 10.1016/j.bbrc.2010.04.116.
Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, Kawamura A, Nakamura K, Takeuchi T, Tanabe M: An evolutionarily conserved mechanism for microrna-223 expression revealed by microrna gene profiling. Cell. 2007, 129: 617-631. 10.1016/j.cell.2007.02.048.
Peng CH, Liu JH, Woung LC, Lin TJ, Chiou SH, Tseng PC, Du WY, Cheng CK, Hu CC, Chien KH, Chen SJ: Micrornas and cataracts: Correlation among let-7 expression, age and the severity of lens opacity. Br J Ophthalmol. 2012, 96: 747-751. 10.1136/bjophthalmol-2011-300585.
Krek A, Grün D, Poy M, Wolf R, Rosenberg L, Epstein E, MacMenamin P, da Piedade I, Gunsalus K, Stoffel M, Rajewsky N: Combinatorial microrna target predictions. Nat Genet. 2005, 37: 495-500. 10.1038/ng1536.
Hughes AE, Bradley DT, Campbell M, Lechner J, Dash DP, Simpson DA, Willoughby CE: Mutation altering the mir-184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011, 89: 628-633. 10.1016/j.ajhg.2011.09.014.
Hoffmann A, Huang Y, Suetsugu-Maki R, Ringelberg CS, Tomlinson CR, Rio-Tsonis KD, Tsonis PA: Implication of the mir-184 and mir-204 competitive rna network in control of mouse secondary cataract. Mol Med. 2012, 18: 528-538.
Najafi-Shoushtari SH: Micrornas in cardiometabolic disease. Curr Atheroscler Rep. 2011, 13: 202-207. 10.1007/s11883-011-0179-y.
Cheung TH, Quach NL, Charville GW, Liu L, Park L, Edalati A, Yoo B, Hoang P, Rando TA: Maintenance of muscle stem-cell quiescence by microrna-489. Nature. 2012, 482: 524-528. 10.1038/nature10834.
Sun H, Li QW, Lv XY, Ai JZ, Yang QT, Duan JJ, Bian GH, Xiao Y, Wang YD, Zhang Z, Liu YH, Tan RZ, Yang Y, Wei YQ, Zhou Q: Microrna-17 post-transcriptionally regulates polycystic kidney disease-2 gene and promotes cell proliferation. Mol Biol Rep. 2010, 37: 2951-2958. 10.1007/s11033-009-9861-3.
Vandervoot AA, Symons TB: Functional and metabolic consequences of sarcopenia. Can J Appl Physiol. 2001, 26: 90-101. 10.1139/h01-007.
Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, Koh P, Thomas M, Jandeleit-Dahm K, Gregorevic P, Cooper ME, Kantharidis P: Suppression of microrna-29 expression by tgf-β1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012, 23: 252-265. 10.1681/ASN.2011010055.
Koning M, Werker PM, van der Schaft DW, Bank RA, Harmsen MC: Microrna-1 and microrna-206 improve differentiation potential of human satellite cells: A novel approach for tissue engineering of skeletal muscle. Tissue Eng Part A. 2011, 18: 889-898.
RR K: Heart and cardiovascular system. Handbook of the Biology of Aging. Edited by: Finch CE, Hayflick L. 1977, New York: Van Nostrand Reinhold, 281-317.
van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF: Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000, 192: 1731-1744. 10.1084/jem.192.12.1731.
Weingand KW, Clarkson TB, Adams MR, Bostrom AD: Effects of age and/or puberty on coronary artery atherosclerosis in cynomolgus monkeys. Atherosclerosis. 1986, 62: 137-144. 10.1016/0021-9150(86)90059-6.
Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE: Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. 1994, 24: 471-476. 10.1016/0735-1097(94)90305-0.
Lakatta EG, Levy D: Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part i: Aging arteries: A “Set up” For vascular disease. Circulation. 2003, 107: 139-146. 10.1161/01.CIR.0000048892.83521.58.
Rivard A, Fabre JE, Silver M, Chen D, Murohara T, Kearney M, Magner M, Asahara T, Isner JM: Age-dependent impairment of angiogenesis. Circulation. 1999, 99: 111-120. 10.1161/01.CIR.99.1.111.
Weinsaft JW, Edelberg JM: Aging-associated changes in vascular activity: A potential link to geriatric cardiovascular disease. Am J Geriatr Cardiol. 2001, 10: 348-354. 10.1111/j.1076-7460.2001.00833.x.
Eggen DA, Solberg LA: Variation of atherosclerosis with age. Lab Invest. 1968, 18: 571-579.
Quintavalle M, Condorelli G, Elia L: Arterial remodeling and atherosclerosis: Mirnas involvement. Vascul Pharmacol. 2011, 55: 106-110. 10.1016/j.vph.2011.08.216.
Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC: Dicer dependent micrornas regulate gene expression and functions in human endothelial cells. Circ Res. 2007, 100: 1164-1173. 10.1161/01.RES.0000265065.26744.17.
Cheng WH, Muftic D, Muftuoglu M, Dawut L, Morris C, Helleday T, Shiloh Y, Bohr VA: Wrn is required for atm activation and the s-phase checkpoint in response to interstrand crosslink-induced dna double strand breaks. Mol Biol Cell. 2008, 19: 3923-3933. 10.1091/mbc.E07-07-0698.
Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S: Role of dicer and drosha for endothelial microrna expression and angiogenesis. Circ Res. 2007, 101: 59-68. 10.1161/CIRCRESAHA.107.153916.
Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S: Circulating micrornas in patients with coronary artery disease. Circ Res. 2010, 107: 677-684. 10.1161/CIRCRESAHA.109.215566.
Li T, Cao H, Zhuang J, Wan J, Guan M, Yu B, Li X, Zhang W: Identification of mir-130a, mir-27b and mir-210 as serum biomarkers for atherosclerosis obliterans. Clin Chim Acta. 2011, 412: 66-70. 10.1016/j.cca.2010.09.029.
Zampetaki A, Willeit P, Tilling L, Drozdov I, Prokopi M, Renard JM, Mayr A, Weger S, Schett G, Shah A, Boulanger CM, Willeit J, Chowienczyk PJ, Kiechl S, Mayr M: Prospective study on circulating micrornas and risk of myocardial infarction. J Am Coll Cardiol. 2012, 60: 290-299. 10.1016/j.jacc.2012.03.056.
Richter V, Rassoul F, Purschwitz K, Hentschel B, Reuter W, Kuntze T: Circulating vascular cell adhesion molecules vcam-1, icam-1, and e-selectin in dependence on aging. Gerontology. 2003, 49: 293-300. 10.1159/000071710.
Sermsathanasawadi N, Ishii H, Igarashi K, Miura M, Yoshida M, Inoue Y, Iwai T: Enhanced adhesion of early endothelial progenitor cells to radiation-induced senescence-like vascular endothelial cells in vitro. J Radiat Res. 2009, 50: 469-475. 10.1269/jrr.09036.
Zou Y, Yoon S, Jung KJ, Kim CH, Son TG, Kim MS, Kim YJ, Lee J, Yu BP, Chung HY: Upregulation of aortic adhesion molecules during aging. J Gerontol A Biol Sci Med Sci. 2006, 61: 232-244. 10.1093/gerona/61.3.232.
Malik I, Danesh J, Whincup P, Bhatia V, Papacosta O, Walker M, Lennon L, Thomson A, Haskard D: Soluble adhesion molecules and prediction of coronary heart disease: A prospective study and meta-analysis. Lancet. 2001, 358: 971-976. 10.1016/S0140-6736(01)06104-9.
Zhang Y, Guo W, Wen Y, Xiong Q, Liu H, Wu J, Zou Y, Zhu Y: Scm-198 attenuates early atherosclerotic lesions in hypercholesterolemic rabbits via modulation of the inflammatory and oxidative stress pathways. Atherosclerosis. 2012, 224: 43-50. 10.1016/j.atherosclerosis.2012.06.066.
Stein S, Matter CM: Protective roles of sirt1 in atherosclerosis. Cell Cycle. 2011, 10: 640-647. 10.4161/cc.10.4.14863.
Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C: Microrna expression signature and antisense-mediated depletion reveal an essential role of microrna in vascular neointimal lesion formation. Circ Res. 2007, 100: 1579-1588. 10.1161/CIRCRESAHA.106.141986.
Bonifacio LN, Jarstfer MB: Mirna profile associated with replicative senescence, extended cell culture, and ectopic telomerase expression in human foreskin fibroblasts. PLoS One. 2010, 5: e12519-10.1371/journal.pone.0012519.
Ferland-McCollough D, Ozanne SE, Siddle K, Willis AE, Bushell M: The involvement of micrornas in type 2 diabetes. Biochem Soc Trans. 2010, 38: 1565-1570. 10.1042/BST0381565.
Zimmet P, Alberti KG, Shaw J: Global and societal implications of the diabetes epidemic. Nature. 2001, 414: 782-787. 10.1038/414782a.
Ribbing J, Hamrén B, Svensson MK, Karlsson MO: A model for glucose, insulin, and beta-cell dynamics in subjects with insulin resistance and patients with type 2 diabetes. J Clin Pharmacol. 2010, 50: 861-872. 10.1177/0091270009349711.
Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA: The hormone resistin links obesity to diabetes. Nature. 2001, 409: 307-312. 10.1038/35053000.
Testa R, Ceriello A: Pathogenetic loop between diabetes and cell senescence. Diabetes Care. 2007, 30: 2974-2975. 10.2337/dc07-1534.
Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, Mayr A, Weger S, Oberhollenzer F, Bonora E, Shah A, Willeit J, Mayr M: Plasma microrna profiling reveals loss of endothelial mir-126 and other micrornas in type 2 diabetes. Circ Res. 2010, 107: 810-817. 10.1161/CIRCRESAHA.110.226357.
Kuhlow D, Florian S, von Figura G, Weimer S, Schulz N, Petzke KJ, Zarse K, Pfeiffer AF, Rudolph KL, Ristow M: Telomerase deficiency impairs glucose metabolism and insulin secretion. Aging (Albany NY). 2010, 2: 650-658.
Hiwatashi Y, Kurahashi Y, Hatada R, Ueno S, Honma T, Yanagihara N, Yanase H, Iwanaga T, Ohizumi Y, Yamakuni T: Glucocorticoid inhibits expression of v-1, a catecholamine biosynthesis regulatory protein, in cultured adrenal medullary cells. FEBS Lett. 2002, 528: 166-170. 10.1016/S0014-5793(02)03293-3.
He A, Zhu L, Gupta N, Chang Y, Fang F: Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3 t3-l1 adipocytes. Mol Endocrinol. 2007, 21: 2785-2794. 10.1210/me.2007-0167.
Martinez I, Cazalla D, Almstead LL, Steitz JA, DiMaio D: Mir-29 and mir-30 regulate b-myb expression during cellular senescence. Proc Natl Acad Sci U S A. 2011, 108: 522-527. 10.1073/pnas.1017346108.
Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, DeAngelis T, Baserga R: Micro rna 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem. 2007, 282: 32582-32590. 10.1074/jbc.M702806200.
Winearls CG, Glassock RJ: Classification of chronic kidney disease in the elderly: Pitfalls and errors. Nephron Clin Pract. 2011, c2-c4. Suppl 1
Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF: Expression of p16ink4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65: 510-520. 10.1111/j.1523-1755.2004.00438.x.
White NM, Bao TT, Grigull J, Youssef YM, Girgis A, Diamandis M, Fatoohi E, Metias M, Honey RJ, Stewart R, Pace KT, Bjarnason GA, Yousef GM: Mirna profiling for clear cell renal cell carcinoma: Biomarker discovery and identification of potential controls and consequences of mirna dysregulation. J Urol. 2011, 186: 1077-1083. 10.1016/j.juro.2011.04.110.
Monteforte R, Wagner S, Koenigsrainer A, Grillari J, Grillari-Voglauer R, Wieser M: Senescent renal proximal tubular epithelial cells undergo partial epithelial-to-mesenchymal transition. in review
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ: The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting zeb1 and sip1. Nat Cell Biol. 2008, 10: 593-601. 10.1038/ncb1722.
Oba S, Kumano S, Suzuki E, Nishimatsu H, Takahashi M, Takamori H, Kasuya M, Ogawa Y, Sato K, Kimura K, Homma Y, Hirata Y, Fujita T: Mir-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS One. 2010, 5: e13614-10.1371/journal.pone.0013614.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T: A reciprocal repression between zeb1 and members of the mir-200 family promotes emt and invasion in cancer cells. EMBO Rep. 2008, 9: 582-589. 10.1038/embor.2008.74.
Korpal M, Lee ES, Hu G, Kang Y: The mir-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of e-cadherin transcriptional repressors zeb1 and zeb2. J Biol Chem. 2008, 283: 14910-14914. 10.1074/jbc.C800074200.
Park SM, Gaur AB, Lengyel E, Peter ME: The mir-200 family determines the epithelial phenotype of cancer cells by targeting the e-cadherin repressors zeb1 and zeb2. Genes Dev. 2008, 22: 894-907. 10.1101/gad.1640608.
Xiong M, Jiang L, Zhou Y, Qiu W, Fang L, Tan R, Wen P, Yang J: The mir-200 family regulates tgf-β1-induced renal tubular epithelial to mesenchymal transition through smad pathway by targeting zeb1 and zeb2 expression. Am J Physiol Renal Physiol. 2012, 302: F369-F379. 10.1152/ajprenal.00268.2011.
Wang B, Herman-Edelstein M, Koh P, Burns W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM, Cooper ME, Kantharidis P: E-cadherin expression is regulated by mir-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes. 2010, 59: 1794-1802. 10.2337/db09-1736.
Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, Natarajan R: Microrna-192 in diabetic kidney glomeruli and its function in tgf-beta-induced collagen expression via inhibition of e-box repressors. Proc Natl Acad Sci U S A. 2007, 104: 3432-3437. 10.1073/pnas.0611192104.
Li JY, Yong TY, Michael MZ, Gleadle JM: Review: The role of micrornas in kidney disease. Nephrology (Carlton). 2010, 15: 599-608. 10.1111/j.1440-1797.2010.01363.x.
Wang G, Kwan BC, Lai FM, Choi PC, Chow KM, Li PK, Szeto CC: Intrarenal expression of mirnas in patients with hypertensive nephrosclerosis. Am J Hypertens. 2010, 23: 78-84. 10.1038/ajh.2009.208.
Raisz LG: Physiology and pathophysiology of bone remodeling. Clin Chem. 1999, 45: 1353-1358.
Kapinas K, Delany AM: Microrna biogenesis and regulation of bone remodeling. Arthritis Res Ther. 2011, 13: 220-10.1186/ar3325.
Saeed H, Abdallah BM, Ditzel N, Catala-Lehnen P, Qiu W, Amling M, Kassem M: Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J Bone Miner Res. 2011, 26: 1494-1505. 10.1002/jbmr.349.
Kassem M, Marie PJ: Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell. 2011, 10: 191-197. 10.1111/j.1474-9726.2011.00669.x.
Laschober GT, Brunauer R, Jamnig A, Singh S, Hafen U, Fehrer C, Kloss F, Gassner R, Lepperdinger G: Age-specific changes of mesenchymal stem cells are paralleled by upregulation of cd106 expression as a response to an inflammatory environment. Rejuvenation Res. 2011, 14: 119-131. 10.1089/rej.2010.1077.
Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck J: Relationship between donor age and the replicative lifespan of human cells in culture: A reevaluation. Proc Natl Acad Sci USA. 1998, 95: 10614-10619. 10.1073/pnas.95.18.10614.
Zhang JF, Fu WM, He ML, Wang H, Wang WM, Yu SC, Bian XW, Zhou J, Lin MC, Lu G, Poon WS, Kung HF: Mir-637 maintains the balance between adipocytes and osteoblasts by directly targeting osterix. Mol Biol Cell. 22: 3955-3961.
Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P: Microrna 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mrnas encoding extracellular matrix proteins. Proc Natl Acad Sci U S A. 2008, 105: 5874-5878. 10.1073/pnas.0801130105.
Eskildsen T, Taipaleenmaki H, Stenvang J, Abdallah BM, Ditzel N, Nossent AY, Bak M, Kauppinen S, Kassem M: Microrna-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc Natl Acad Sci U S A. 108: 6139-6144.
Sugatani T, Hruska KA: Impaired micro-rna pathways diminish osteoclast differentiation and function. J Biol Chem. 2009, 284: 4667-4678.
Li H, Xie H, Liu W, Hu R, Huang B, Tan YF, Xu K, Sheng ZF, Zhou HD, Wu XP, Luo XH: A novel microrna targeting hdac5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J Clin Invest. 2009, 119: 3666-3677. 10.1172/JCI39832.
Stefek M, Karasu C: Eye lens in aging and diabetes: Effect of quercetin. Rejuvenation Res. 2011, 14: 525-534. 10.1089/rej.2011.1170.
Waters DL, Baumgartner RN, Garry PJ: Sarcopenia: Current perspectives. J Nutr Health Aging. 2000, 4: 133-139.
Mouly V, Aamiri A, Bigot A, Cooper RN, Di Donna S, Furling D, Gidaro T, Jacquemin V, Mamchaoui K, Negroni E, Perie S, Renault V, Silva-Barbosa SD, Butler-Browne GS: The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiol Scand. 2005, 184: 3-15. 10.1111/j.1365-201X.2005.01417.x.
Bigot A, Jacquemin V, Debacq-Chainiaux F, Butler-Browne GS, Toussaint O, Furling D, Mouly V: Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biol Cell. 2008, 100: 189-199. 10.1042/BC20070085.
Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, Mouly V, Furling D: Large ctg repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol. 2009, 174: 1435-1442. 10.2353/ajpath.2009.080560.
Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, Wang DZ: Microrna-1 and microrna-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing pax7. J Cell Biol. 2010, 190: 867-879. 10.1083/jcb.200911036.
Conboy I, Rando T: Aging, stem cells and tissue regeneration: Lessons from muscle. Cell Cycle. 2005, 4: 407-410. 10.4161/cc.4.3.1518.
Conboy I, Conboy M, Wagers A, Girma E, Weissman I, Rando T: Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005, 433: 760-764. 10.1038/nature03260.
Junn E, Mouradian MM: Micrornas in neurodegenerative diseases and their therapeutic potential. Pharmacol Ther. 2012, 133: 142-150. 10.1016/j.pharmthera.2011.10.002.
Acknowledgements
This work has been supported by the GEN-AU Project 820982 ‘Non-coding RNAs’, FWF project P 24498-B20 of the Austrian Science Fund, as well as grants by the Herzfelder’sche Familienstiftung and C.E.R.I.E.S.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JG is co-founder and CSO of Evercyte Gmbh.
Authors’ contributions
ES planned and wrote the manuscript. JG planned, designed and wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Schraml, E., Grillari, J. From cellular senescence to age-associated diseases: the miRNA connection. Longev Healthspan 1, 10 (2012). https://doi.org/10.1186/2046-2395-1-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2046-2395-1-10