
Abstract
The tissue protective functions of the hematopoietic growth factor erythropoietin (EPO) are independent of its action on erythropoiesis. EPO and its receptors (EPOR) are expressed in multiple brain cells during brain development and upregulated in the adult brain after injury. Peripherally administered EPO crosses the blood-brain barrier and activates in the brain anti-apoptotic, anti-oxidant and anti-inflammatory signaling in neurons, glial and cerebrovascular endothelial cells and stimulates angiogenesis and neurogenesis. These mechanisms underlie its potent tissue protective effects in experimental models of stroke, cerebral hemorrhage, traumatic brain injury, neuroinflammatory and neurodegenerative disease. The preclinical data in support of the use of EPO in brain disease have already been translated to first clinical pilot studies with encouraging results with the use of EPO as a neuroprotective agent.
Similar content being viewed by others

The cytokine erythropoetin (EPO)
The cytokine erythropoietin (EPO) is a 34 kD glycoprotein which was originally described to stimulate erythropoiesis. EPO supports the proliferation and differentiation of erythroid progenitor cells and is critical for their survival [1]. The main site of EPO production is fetal liver and adult kidney [1]. Mice deficient for EPO or EPO receptor (EPOR) genes die at embryonic day 13 (E13) because of severe anemia caused by deficiency in definitive erythropoiesis [2–4]. Over the last decade it has become clear that EPO acts as growth and survival factors for multiple tissues expressing the EPO receptor [1]. The number of described targets of EPO action continues to grow.
EPO Receptor (EPOR)
EPO acts by binding to its specific transmembrane receptor (EPOR). EPOR belongs to the single-chain cytokine type I receptor family [5]. These receptors are characterized by an extracellular N-terminal domain with conserved cysteines and a WSXWS-motif, a single hydrophobic transmembrane segment and a cytosolic domain with no intrinsic kinase activity [5]. Two transmembrane EPOR molecules form a homodimer that binds one EPO molecule leading to a conformational change and tight bonding of the two EPOR monomers which in turn activate two Janus family tyrosine kinase 2 (JAK2) molecules which associate with cytoplasmic domain of the EPOR. Once activated, JAK2 phosphorylates distal parts of receptors which subsequently serve as docking sites for downstream signaling molecules. Multiple signal transduction pathways are activated downstream of EPOR/JAK2 [1, 5]. In neurons these include signal transducers and activators of transcription (Stat), phosphatidylinositol 3-kinase (PI3K)/Akt, Ras/extracellular signal regulated kinase (ERK1/2), nuclear factor-kappa-B (NF-κB), and calcium [6–8]. Best investigated from these are PI3K/Akt and Ras-MAPK pathways, both of which are important for the antiapoptotic and trophic effects of EPO [8–18]. NFκB pathway plays a role in antiapoptotic activity of EPO in neurons [7, 19, 20] as well as in EPO-mediated propagation of neural stem cells [21]. In addition EPO by activating phospholipase Cγ modulates intracellular calcium concentration, electrical activity and neurotransmitter release in PC12 cells [22–24] as well as in hippocampal neurons and brain slices [25, 26]. The role of the Stat transcriptional factors in regard to EPO effects in the neural cells remains unclear; EPO induces phosphorylation of Stat5 in neurons [8, 19, 27], neural stem cells [21] and neuroblastoma cells [17, 28]. Using primary hippocampal neurons from Stat5a/b knock-out mice we have recently shown that the presence of functional Stat5 is required for the trophic but not for the protective effect of EPO against glutamate-induced toxicity [9].
EPO signaling is terminated by activation of phosphatases which dephosphorylate JAK2. The ligand-receptor complex is then internalized and degraded by the proteasome [1, 5]. In hematopoietic cell lines 60% of the internalized EPO is re-secreted [29].
The brain EPO/EPOR system
mRNA and protein of EPO and EPOR are detected in brain (hippocampus, internal capsule, cortex, midbrain), as well as in vitro in neurons, astrocytes, oligodendrocytes, microglia and cerebral endothelial cells [24, 30–44] suggesting that this factor can function in the brain in a paracrine and/or autocrine manner. In the developing mouse brain expression of EPO and EPOR peaks during midgestation and decreases to adult levels in late gestation [43]. Brain specific ablation of EPOR leads to deficits in neural cell proliferation and neuronal survival in the embryonic brain and in post-stroke neurogenesis in the adult brain [45, 46]. Expression of EPO and EPOR in the adult brain is stress-responsive and is regulated by oxygen supply: Both receptor and ligand expression is upregulated after hypoxia or ischemia [36, 42, 43, 47–50]. Other stimuli such as hypoglycemia, insulin release, reactive oxygen species and insulin-like growth factor activate hypoxia-inducible factor and lead to increased expression of EPO [51, 52]. Proinflammatory cytokines downregulate expression of EPO mRNA but increase that of EPOR in astrocytes [34]
Based on the loss of some of the tissue protective effects of EPO and its non-hematopoietic derivative, the carbamylated EPO (CEPO) in mice lacking the common β chain shared by the members of the IL-3 receptor family, Brines and Cerami have proposed that the cytoprotective effects of EPO and CEPO are mediated by a heteromeric receptor complex comprised of one EPOR subunit and a dimer of the common β chain [6, 53]. Immunoreactivity of the common β chain has been detected in brain tissue with a pattern reminiscent to that of the classical EPOR [53]. Furthermore, the common β chain can be coimmunoprecipitated with EPOR antibodies from the P19 embryonal carcinoma cells [53], but the existence of the proposed heteromeric receptor structure in primary cells or tissues has yet to be directly proven. In a recent study no expression of the common β subunit was detected in neuronal PC-12 cells even if EPO rescued these cells from staurosporine-induced apoptosis [28]. Interestingly, the classical EPOR is required for EPO-stimulated neuronal differentiation and survival but not for neurogenesis induced by CEPO suggesting that differential receptor interactions mediate the effects of EPO and CEPO in brain cells [28, 54].
Neuroprotection by EPO is independent of hematopoesis
The neuroprotective actions of EPO can be separated from its hematopoietic actions, a fact that is of value for therapeutic applications where the increase in red cell mass is not desired. EPO and EPO derivatives are directly neuroprotective in cell culture models (see below) and after direct application into the brain [36, 55, 56]. Moreover, CEPO and other EPO derivatives which do not bind to EPOR in myeloid cells and thus lack hematopoietic activity display potent tissue protective activities [57–59]. Expression of EPO and the classical EPOR in brain cells is induced by hypoxic-ischemic stress and contributes to ischemic tolerance [14, 24, 32, 37, 41, 49, 50, 60–66] while neutralization of the brain endogenous EPO augments ischemic damage [56]. Brain-specific genetic ablation of the classical EPOR impairs post-stroke neurogenesis and neuronal survival [45, 46] whereas transgenic brain specific overexpression of human EPO is associated with reductions in postischemic infarct volume, brain swelling and functional deficits in a transient stroke model [16].
Multimodal neuroprotective profile
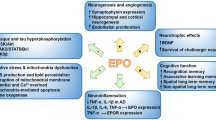
EPO has been reported to induce a broad range of cellular responses in the brain directed to protect and repair tissue damage (Figure 1). EPO is neuroprotective in a variety of hypoxic, hypoglycemic, and excitotoxic in vitro models [7, 8, 10, 14, 17, 18, 20, 21, 42, 45, 50, 57, 67–72]. A fundamental mechanism of EPO-induced neuroprotection in cultured neurons is its ability to inhibit apoptosis reducing both DNA damage and cell membrane asymmetry [7, 8, 10, 14, 17, 18, 20, 21, 42, 45, 50, 57, 67–72]. Necrotic cell death (for example, after glutamate exposure) is also be attenuated by EPO [9, 69, 73]. Why astroglial cultures are protected by EPO from nitric oxide- and staurosporine-induced death but not from arsenic trioxide- or glutamate-induced toxicity is not fully understood [73, 74].

Multimodal neuroprotective profile of erythropoietin (EPO). BBB - blood brain barrier; EC - endothelial cells.
Another tissue-protective mechanism of EPO is its ability to protect cells against oxidative damage [75, 76]. EPO inhibits lipid peroxidation by increasing the activities of cytosolic antioxidant enzymes such as superoxide dismutase and glutathione peroxidase [77–79].
EPO attenuates inflammation by reducing reactive astrocytosis and microglia activation and by inhibiting immune cells recruitment into the injured area [47, 58, 59, 70, 80–85]. In cerebrovascular endothelial cell cultures EPO down-regulates TNF-α-induced gene expression of interleukin-6 (IL-6), IL-1beta, CXCR4, and IL-1alpha [86]. It also directly counteracts interferon-γ- and lipopolyssaccharide-induced cytotoxicity in oligodendrocytes, preserves white matter [87] and reduces TNF-α release and its effects in cultured Schwann cells [88].
EPO protects vascular integrity and stimulates angiogenesis [89–92]. It preserves blood-brain barrier integrity during injury by restoring expression of tight junction proteins [90, 93, 94], by reducing vascular inflammation [95] and reactive free radical expression [90, 93, 94, 96]. In vasculogenesis EPO stimulates proliferation of endothelial precursor cells, production of matrix metalloproteinase-2, migration of endothelial cells into vascular sites and formation of capillary tubes [90–92, 97, 98]. EPO displays direct antiapoptotic activity in cerebral endothelial cells during oxidative stress and ischemic injury as well [91]. Stimulation of endothelial nitric oxide synthase (eNOS) activity has been shown to contribute to the improvements by EPO after experimental cerebral hemorrhage [99–101]. Interestingly, plasma and tissue levels of NO are markedly increased in transgenic rats overexpressing EPO [102] whereas the vascular protection by EPO is abolished in eNOS-deficient mice [103].
EPO promotes differentiation towards neurons in several neuroblastoma cell lines, in neural stem cell cultures derived from both embryonic and adult neuronal germinal zones, as well as in embryonic neural progenitor-cell cultures [21, 45, 46, 54, 89, 104–108]. Neuronal differentiation of adult neural progenitor cell by EPO seem to require activation of the sonic hedgehog signaling and up-regulation of suppressor of cytokine signaling-2 (SOCS2) [54, 105]. EPO increases proliferation of oligodendrocyte progenitors and promotes differentiation of oligodendrocytes in culture [33, 34]. EPOR-/- fetuses exhibit increased apoptosis in the brain and a reduction in the number of neural progenitor cells, as well as increased sensitivity to hypoxia prior to significant anaemia or heart defects in the embryo proper [42, 45, 46]. Moreover, adult mice that lack EPOR in the brain have significantly reduced neurogenesis in the subventricular zone and demonstrate impaired migration of precursors into infracted cortex [46]. Nevertheless, expression of EPO or EPOR on neural cells is not indispensable for brain development [42, 45, 46].
The reported neurotrophic effects of EPO include the ability to stimulate axonal regrowth, neurite formation, dendritic sprouting, electrical activity and modulate intracellular calcium and neurotransmitter synthesis and release [9, 13, 22, 23, 25, 26, 46, 109–113]. A recent study demonstrated a calcium sensitive activation of cAMP response element binding protein (CREB) and induction of brain-derived neurotrophic factor (BDNF) gene expression by EPO in primary hippocampal neurons [25]. In rat hippocampal slices, EPO improved synaptic transmission during and following oxygen and glucose deprivation [13]. However, it has not been directly shown that EPO induces formation of new synapses.
Animal Studies
The preclinical data in support of the use of EPO in human brain disease have explosively increased since the first discovery of its neuroprotective action. In particular, the preclinical evidence in support for testing EPO in human acute ischemic stroke fulfills most of the STAIR criteria [114] such as testing by several laboratories using both temporary and permanent stroke models, post-treatment at several doses and exploration of therapeutic window, characterization of pharmacokinetic profile in respect to blood-brain-barrier penetration after peripheral administration, measurement of histological and functional outcome with prolonged survival.
Cerebral ischemia
The in vivo potential of EPO to protect neurons against ischemic neuronal damage was first provided by the Sasaki group. Their landmark finding was that application of recombinant human (rh)EPO directly into the cerebral ventricles of gerbils prevented ischemia-induced learning disabilities and protected hippocampal pyramidal CA1 neurons from lethal ischemia while neutralization of the endogenous brain EPO by infusions of soluble EPOR before a nonlethal mild ischemia induced neuronal death [56]. Since the circulating EPO, as a large, highly glycosylated negatively charged molecule, was thought not to cross the blood-brain-barrier [91, 115, 116], the early studies used direct intracerebroventricular route of administration of EPO to demonstrate its potent tissue protective activity in focal and global models of cerebral ischemia [36, 55, 56]. The first evidence for a neuroprotective effect of EPO by peripheral route of administration was provided by Brines et al. (2000) who demonstrated in a focal stroke model reduction of infarct volumes by intraperitoneally applied high dose rhEPO (5000 U/kg) up to 6 h after reperfusion. Immunohistochemical detection of biotinylated rhEPO 5 hours after its intraperitoneal injection at the therapeutically effective dose (5000 U/kg) further provided evidence that EPO can cross the blood-brain barrier [117]. Studies in several species including man have confirmed the ability for high dose EPO to cross the blood-brain barrier in therapeutic effective concentrations [118–122]. To date EPO and its derivatives have shown to reduce histological damage and improve functional outcome when given as intraperitoneal or even intranasal post-treatment after experimental stroke [57–59, 70, 85, 89, 123], global cerebral ischemia [124], neonatal stroke and hypoxia-ischemia [107, 125–128]. For example, a comprehensive dosing study using post-treatment with EPO and CEPO starting at 6 h after an embolic middle cerebral artery occlusion in rats demonstrated reduction of functional deficits and infarct volume up to 28 days models of middle cerebral artery occlusion [59]. Induction of EPO and its intracellular signaling intermediates represents a significant component of tolerance induced by ischemic or hypoxic preconditioning [60–62, 65]. Here activation of the antiapoptotic and anti-inflammatory signaling seems to play a major role in the EPO-induced neuroprotection [14, 19, 129].
Cerebral hemorrhage
Post-treatment with EPO starting at 2 h after induction of intracerebral hemorrhage (ICH) by intraparenchymal injections of collagenase or autologous blood dose-dependently reduced volume of hemorrhage, brain edema, perihematomal apoptosis and inflammation in a rat model [99]. Functional recovery was faster and more efficient in the EPO-treated group and was associated with reduction in hemispheric brain atrophy 5 weeks after the induction of ICH [99]. Cerebral vasospasm and ischemic brain damage after subarachnoid hemorrhage (SAH) by autologous blood injections into the cisterna magna in rabbits are reduced by EPO administered either by intraperitoneal injections of rhEPO or by delivery of adenoviral vectors encoding the human EPO into cisterna magna immediately after induction of SAH [130–133]. Mortality and functional deficits 3 days after induction of SAH were reduced in EPO treated rabbits [130–132]. In a rat model of SAH, the impaired autoregulatory response of cerebral blood flow to intravenous noradrenaline was restored by a single subcutaneous bolus of EPO [134].
Traumatic brain and spinal cord injury
Administration of EPO and EPO-analogs in experimental models of traumatic brain and spinal cord injury leads to morphological, functional and cognitive recovery that can be attributed to a multitude of cytoprotective mechanisms including inhibition of apoptosis, anti-inflammatory and anti-oxidant actions, restoration of blood-brain barrier integrity, stimulation of neurogenesis and angiogenesis [67, 95, 96, 104, 117, 135–146]. Induction of EPO and its protective down stream signaling via Akt seems also to account for the protective effect of heat acclimation stress in a closed head injury model [147, 148]. Brain edema after experimental brain injury can effectively be attenuated by post-treatment with EPO [95, 138, 140]. A reduction of cytotoxic and vasogenic edema may be anticipated based on the direct actions of EPO on glutamate release [112] and on the endothelial barrier function (see above). It is not clear to date which from the panoply of neurorestorative effects of EPO are responsible for the long-term prevention of trauma-induced brain atrophy, cognitive and neurobehavioral dysfunction [104, 135–137, 146]. In this context it is interesting to note that chronic peripheral administration of EPO has been reported to improve spatial memory function and cognitive functioning in the context of an aversion task also in healthy mice [119, 149]. Improved hippocampal functioning after a single intravenous bolus of EPO was recently shown in a study using functional magnetic resonance imaging in healthy human volunteers [150].
Degeneration & neuroinflammation
EPO and its analogs offer protection also in models of neurodegenerative and neuroinflammatory disease. In experimental autoimmunencephalitis (EAE), an animal model for multiple sclerosis (MS), treatment with EPO and EPO analogs can improve functional recovery, reduce tissue damage, inflammatory responses and blood-brain barrier leakage [47, 80–84, 117]. Beneficial effects of EPO have also been reported in models of peripheral axonal nerve injury, injury-induced Wallerian degeneration and HIV-associated sensory neuropathy [88, 151, 152]. Here, the anti-cytokine, anti-apoptotic, anti-oxidative and trophic effects on both neurons and oligodendrocyte progenitor cells by EPO seem to play an important role in reducing inflammation and preserving myelination and neuronal function [35, 47, 80–84, 86–88, 151].
Chronic neurodegeneration might also be a target for EPO therapy as EPO and its analogs can counteract degenerative processes in experimental models of Parkinson disease and amyotrophic lateral sclerosis (ALS) by inducing anti-oxidant enzymes, inhibiting apoptosis and stimulating axonal regeneration [78, 153–155]. EPO improved graft survival of embryonic ventral mesencephalic dopamine neurons when transplanted into the striatum of 6-hydroxy-dopamine lesioned rats [156]. However, not all degenerative diseases seem to respond to EPO therapy [157].
Translation to human brain disease
EPO and its receptor are abundantly expressed in the developing human brain decreasing to a weak constitutive expression in the adult [30, 39, 41, 66, 119, 158]. Hypoxia rapidly induces expression of brain EPO as evidenced by the increased expression of EPO in cerebrospinal fluid (CSF) or postmortem brain tissue in humans with traumatic brain injury, SAH, stroke and hypoxia [31, 41, 66, 121, 159, 160]. Expression of EPOR has also been detected on myelin sheath of radicular nerves and in the epineurial blood vessels of sural nerves in the human peripheral nervous system [161, 162]. Measurements of endogenous levels of EPO in CSF of patients with neurodegenerative diseases has revealed EPO in CSF of patients with ALS to be lower than in controls whereas patients with Alzheimer disease (AD) or vascular dementia had EPO levels comparable to control persons [163, 164]. Astrocytic EPOR immunoreactivity in postmortem hippocampus and temporal cortex from subjects with AD or chronic schizophrenia has been reported to be increased as compared to age-matched control brains [119, 158]. In AD, however, no association of EPOR-positive astrocytes was found with summary measures of global cognition or AD pathology [158].
Millions of patients have already been receiving EPO as a highly efficacious and safe treatment for anemia [1]. Indeed, the first proof-of-concept study on use of EPO in human acute ischemic stroke has already demonstrated that treatment of stroke patients with intravenous high dose EPO is not only well tolerated but is associated with improvement in clinical outcome at 30 days [120]. Encouraging results with the use of EPO as a neuroprotective agent have been recently reported in clinical pilot studies after out-of-hospital cardiac arrest [165], ureamia-associated peripheral neuropathy [166], chronic schizophrenia [167] and MS [145]. A small pilot study to probe the safety and efficacy of EPO in SAH was recently preliminarily terminated with no conclusive results because of low recruitment rate [168]. To date a randomized multicenter double blinded placebo-controlled clinical trial in acute ischemic stroke is running (for details see http://www.epo-study.de) reflecting the beginning of exploration of EPO and its analogs for clinical neuroprotection in large phase II/III setting.
References
Jelkmann W: Erythropoietin after a century of research: younger than ever. Eur J Haematol 2007, 78: 183–205. 10.1111/j.1600-0609.2007.00818.x
Lin CS, Lim SK, D'Agati V, Costantini F: Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes Dev 1996, 10: 154–164. 10.1101/gad.10.2.154
Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML: Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 1999, 126: 3597–3605.
Wu H, Liu X, Jaenisch R, Lodish HF: Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995, 83: 59–67. 10.1016/0092-8674(95)90234-1
Youssoufian H, Longmore G, Neumann D, Yoshimura A, Lodish HF: Structure, function, and activation of the erythropoietin receptor. Blood 1993, 81: 2223–2236.
Brines M, Cerami A: Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci 2005, 6: 484–494. 10.1038/nrn1687
Digicaylioglu M, Lipton SA: Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 2001, 412: 641–647. 10.1038/35088074
Sirén AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P: Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA 2001, 98: 4044–4049. 10.1073/pnas.051606598
Byts N, Samoylenko A, Fasshauer T, Ivanisevic M, Hennighausen L, Ehrenreich H, Sirén AL: Essential role for Stat5 in the neurotrophic but not in the neuroprotective effect of erythropoietin. Cell Death and Differentiation 2008, 15: 783–792. 10.1038/cdd.2008.1
Kilic U, Kilic E, Soliz J, Bassetti CI, Gassmann M, Hermann DM: Erythropoietin protects from axotomy-induced degeneration of retinal ganglion cells by activating ERK-1/-2. Faseb J 2005, 19: 249–251.
Digicaylioglu M, Garden G, Timberlake S, Fletcher L, Lipton SA: Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc Natl Acad Sci USA 2004, 101: 9855–9860. 10.1073/pnas.0403172101
Chong ZZ, Kang JQ, Maiese K: Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br J Pharmacol 2003, 138: 1107–1118. 10.1038/sj.bjp.0705161
Weber A, Maier RF, Hoffmann U, Grips M, Hoppenz M, Aktas AG, Heinemann U, Obladen M, Schuchmann S: Erythropoietin improves synaptic transmission during and following ischemia in rat hippocampal slice cultures. Brain Res 2002, 958: 305–311. 10.1016/S0006-8993(02)03604-1
Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A: Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci 2002, 22: 10291–10301.
Chong ZZ, Lin SH, Kang JQ, Maiese K: Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res 2003, 71: 659–669. 10.1002/jnr.10528
Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM: Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. Faseb J 2005, 19: 2026–2028.
Um M, Lodish HF: Antiapoptotic effects of erythropoietin in differentiated neuroblastoma SH-SY5Y cells require activation of both the STAT5 and AKT signaling pathways. J Biol Chem 2006, 281: 5648–5656. 10.1074/jbc.M510943200
Wu Y, Shang Y, Sun S, Liang H, Liu R: Erythropoietin prevents PC12 cells from 1-methyl-4-phenylpyridinium ion-induced apoptosis via the Akt/GSK-3beta/caspase-3 mediated signaling pathway. Apoptosis 2007, 12: 1365–1375. 10.1007/s10495-007-0065-9
Liu J, Narasimhan P, Yu F, Chan PH: Neuroprotection by Hypoxic Preconditioning Involves Oxidative Stress-Mediated Expression of Hypoxia-Inducible Factor and Erythropoietin. Stroke 2005, 36: 1264–1269. 10.1161/01.STR.0000166180.91042.02
Chong ZZ, Li F, Maiese K: Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr Neurovasc Res 2005, 2: 387–399. 10.2174/156720205774962683
Shingo T, Sorokan ST, Shimazaki T, Weiss S: Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci 2001, 21: 9733–9743.
Kawakami M, Iwasaki S, Sato K, Takahashi M: Erythropoietin inhibits calcium-induced neurotransmitter release from clonal neuronal cells. Biochem Biophys Res Commun 2000, 279: 293–297. 10.1006/bbrc.2000.3926
Koshimura K, Murakami Y, Sohmiya M, Tanaka J, Kato Y: Effects of erythropoietin on neuronal activity. J Neurochem 1999, 72: 2565–2572. 10.1046/j.1471-4159.1999.0722565.x
Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F Jr, Tabira T, Sasaki R: Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem 1993, 268: 11208–11216.
Viviani B, Bartesaghi S, Corsini E, Villa P, Ghezzi P, Garau A, Galli CL, Marinovich M: Erythropoietin protects primary hippocampal neurons increasing the expression of brain-derived neurotrophic factor. J Neurochem 2005, 93: 412–421. 10.1111/j.1471-4159.2005.03033.x
Yamamoto M, Koshimura K, Kawaguchi M, Sohmiya M, Murakami Y, Kato Y: Stimulating effect of erythropoietin on the release of dopamine and acetylcholine from the rat brain slice. Neurosci Lett 2000, 292: 131–133. 10.1016/S0304-3940(00)01441-5
Zhang F, Wang S, Cao G, Gao Y, Chen J: Signal transducers and activators of transcription 5 contributes to erythropoietin-mediated neuroprotection against hippocampal neuronal death after transient global cerebral ischemia. Neurobiol Dis 2007, 25: 45–53. 10.1016/j.nbd.2006.08.007
Um M, Gross AW, Lodish HF: A "classical" homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal 2007, 19: 634–645. 10.1016/j.cellsig.2006.08.014
Gross AW, Lodish HF: Cellular trafficking and degradation of erythropoietin and novel erythropoiesis stimulating protein (NESP). J Biol Chem 2006, 281: 2024–2032. 10.1074/jbc.M510493200
Juul SE, Anderson DK, Li Y, Christensen RD: Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatr Res 1998, 43: 40–49. 10.1203/00006450-199801000-00007
Marti HH, Gassmann M, Wenger RH, Kvietikova I, Morganti-Kossmann MC, Kossmann T, Trentz O, Bauer C: Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney Int 1997, 51: 416–418. 10.1038/ki.1997.55
Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, Gassmann M: Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci USA 1995, 92: 3717–3720. 10.1073/pnas.92.9.3717
Sugawa M, Sakurai Y, Ishikawa-Ieda Y, Suzuki H, Asou H: Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res 2002, 44: 391–403. 10.1016/S0168-0102(02)00161-X
Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, Kim SU: Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol 2001, 60: 386–392.
Li X, Gonias SL, Campana WM: Schwann cells express erythropoietin receptor and represent a major target for Epo in peripheral nerve injury. Glia 2005, 51: 254–265. 10.1002/glia.20202
Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E: A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab 1999, 19: 643–651. 10.1097/00004647-199906000-00007
Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R: A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. J Biol Chem 1994, 269: 19488–19493.
Chin K, Yu X, Beleslin-Cokic B, Liu C, Shen K, Mohrenweiser HW, Noguchi CT: Production and processing of erythropoietin receptor transcripts in brain. Brain Res Mol Brain Res 2000, 81: 29–42. 10.1016/S0169-328X(00)00157-1
Dame C, Bartmann P, Wolber E, Fahnenstich H, Hofmann D, Fandrey J: Erythropoietin gene expression in different areas of the developing human central nervous system. Brain Res Dev Brain Res 2000, 125: 69–74. 10.1016/S0165-3806(00)00118-8
Knabe W, Knerlich F, Washausen S, Kietzmann T, Sirén AL, Brunnett G, Kuhn HJ, Ehrenreich H: Expression patterns of erythropoietin and its receptor in the developing midbrain. Anat Embryol (Berl) 2004, 207: 503–512. 10.1007/s00429-003-0365-y
Sirén AL, Knerlich F, Poser W, Gleiter CH, Brück W, Ehrenreich H: Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol 2001, 101: 271–276.
Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, Lin CS, Nikodem VM, Hempstead B, Flanders KC, Costantini F, Noguchi CT: Erythropoietin receptor signalling is required for normal brain development. Development 2002, 129: 505–516.
Liu ZY, Chin K, Noguchi CT: Tissue specific expression of human erythropoietin receptor in transgenic mice. Dev Biol 1994, 166: 159–169. 10.1006/dbio.1994.1304
Yamaji R, Okada T, Moriya M, Naito M, Tsuruo T, Miyatake K, Nakano Y: Brain capillary endothelial cells express two forms of erythropoietin receptor mRNA. Eur J Biochem 1996, 239: 494–500. 10.1111/j.1432-1033.1996.0494u.x
Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT: Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem 2007, 282: 25875–25883. 10.1074/jbc.M701988200
Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, Liu X, Wu H, Carmichael ST: A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci 2006, 26: 1269–1274. 10.1523/JNEUROSCI.4480-05.2006
Diem R, Sattler MB, Merkler D, Demmer I, Maier K, Stadelmann C, Ehrenreich H, Bahr M: Combined therapy with methylprednisolone and erythropoietin in a model of multiple sclerosis. Brain 2005, 128: 375–385. 10.1093/brain/awh365
Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I, Mackenzie ET, Petit E: Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia 2000, 30: 271–278. 10.1002/(SICI)1098-1136(200005)30:3<271::AID-GLIA6>3.0.CO;2-H
Fandrey J: Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol 2004, 286: R977–988.
Lewczuk P, Hasselblatt M, Kamrowski-Kruck H, Heyer A, Unzicker C, Sirén AL, Ehrenreich H: Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport 2000, 11: 3485–3488. 10.1097/00001756-200011090-00017
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT: Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 1998, 95: 11715–11720. 10.1073/pnas.95.20.11715
Masuda S, Chikuma M, Sasaki R: Insulin-like growth factors and insulin stimulate erythropoietin production in primary cultured astrocytes. Brain Res 1997, 746: 63–70. 10.1016/S0006-8993(96)01186-9
Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su-Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A: Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA 2004, 101: 14907–14912. 10.1073/pnas.0406491101
Wang L, Zhang ZG, Gregg SR, Zhang RL, Jiao Z, LeTourneau Y, Liu X, Feng Y, Gerwien J, Torup L, Leist M, Noguchi CT, Chen ZY, Chopp M: The Sonic hedgehog pathway mediates carbamylated erythropoietin-enhanced proliferation and differentiation of adult neural progenitor cells. J Biol Chem 2007, 282: 32462–32470. 10.1074/jbc.M706880200
Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S, Masuda S, Sasaki R: Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem Biophys Res Commun 1998, 253: 26–32. 10.1006/bbrc.1998.9748
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R: In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA 1998, 95: 4635–4640. 10.1073/pnas.95.8.4635
Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, Kallunki P, Larsen AK, Helboe L, Christensen S, Pedersen LO, Nielsen M, Torup L, Sager T, Sfacteria A, Erbayraktar S, Erbayraktar Z, Gokmen N, Yilmaz O, Cerami-Hand C, Xie QW, Coleman T, Cerami A, Brines M: Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 2004, 305: 239–242. 10.1126/science.1098313
Villa P, van Beek J, Larsen AK, Gerwien J, Christensen S, Cerami A, Brines M, Leist M, Ghezzi P, Torup L: Reduced functional deficits, neuroinflammation, and secondary tissue damage after treatment of stroke by nonerythropoietic erythropoietin derivatives. J Cereb Blood Flow Metab 2007, 27: 552–563. 10.1038/sj.jcbfm.9600370
Wang Y, Zhang ZG, Rhodes K, Renzi M, Zhang RL, Kapke A, Lu M, Pool C, Heavner G, Chopp M: Post-ischemic treatment with erythropoietin or carbamylated erythropoietin reduces infarction and improves neurological outcome in a rat model of focal cerebral ischemia. Br J Pharmacol 2007, 151: 1377–1384. 10.1038/sj.bjp.0707285
Bergeron M, Gidday JM, Yu AY, Semenza GL, Ferriero DM, Sharp FR: Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann Neurol 2000, 48: 285–296. 10.1002/1531-8249(200009)48:3<285::AID-ANA2>3.0.CO;2-8
Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P: Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab 2002, 22: 393–403. 10.1097/00004647-200204000-00003
Dirnagl U, Simon RP, Hallenbeck JM: Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003, 26: 248–254. 10.1016/S0166-2236(03)00071-7
Marti HH: Erythropoietin and the hypoxic brain. J Exp Biol 2004, 207: 3233–3242. 10.1242/jeb.01049
Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Henn V, Yonekawa Y, Bauer C, Gassmann M: Erythropoietin gene expression in human, monkey and murine brain. Eur J Neurosci 1996, 8: 666–676. 10.1111/j.1460-9568.1996.tb01252.x
Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A: Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34: 1981–1986. 10.1161/01.STR.0000080381.76409.B2
Sairanen T, Karjalainen-Lindsberg ML, Paetau A, Ijas P, Lindsberg PJ: Apoptosis dominant in the periinfarct area of human ischaemic stroke--a possible target of antiapoptotic treatments. Brain 2006, 129: 189–199. 10.1093/brain/awh645
Celik M, Gokmen N, Erbayraktar S, Akhisaroglu M, Konakc S, Ulukus C, Genc S, Genc K, Sagiroglu E, Cerami A, Brines M: Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci USA 2002, 99: 2258–2263. 10.1073/pnas.042693799
Ehrenreich H, Hasselblatt M, Knerlich F, von Ahsen N, Jacob S, Sperling S, Woldt H, Vehmeyer K, Nave KA, Sirén AL: A hematopoietic growth factor, thrombopoietin, has a proapoptotic role in the brain. Proc Natl Acad Sci USA 2005, 102: 862–867. 10.1073/pnas.0406008102
Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R: Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1997, 76: 105–116. 10.1016/S0306-4522(96)00306-5
Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P: Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med 2003, 198: 971–975. 10.1084/jem.20021067
Wen TC, Sadamoto Y, Tanaka J, Zhu PX, Nakata K, Ma YJ, Hata R, Sakanaka M: Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J Neurosci Res 2002, 67: 795–803. 10.1002/jnr.10166
Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR: Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci 1999, 19: 9821–9830.
Sinor AD, Greenberg DA: Erythropoietin protects cultured cortical neurons, but not astroglia, from hypoxia and AMPA toxicity. Neurosci Lett 2000, 290: 213–215. 10.1016/S0304-3940(00)01361-6
Diaz Z, Assaraf MI, Miller WH Jr, Schipper HM: Astroglial cytoprotection by erythropoietin pre-conditioning: implications for ischemic and degenerative CNS disorders. J Neurochem 2005, 93: 392–402. 10.1111/j.1471-4159.2005.03038.x
Wu Y, Shang Y, Sun S, Liu R: Antioxidant effect of erythropoietin on 1-methyl-4-phenylpyridinium-induced neurotoxicity in PC12 cells. Eur J Pharmacol 2007, 564: 47–56. 10.1016/j.ejphar.2007.02.020
Solaroglu I, Solaroglu A, Kaptanoglu E, Dede S, Haberal A, Beskonakli E, Kilinc K: Erythropoietin prevents ischemia-reperfusion from inducing oxidative damage in fetal rat brain. Childs Nerv Syst 2003, 19: 19–22.
Chattopadhyay A, Choudhury TD, Bandyopadhyay D, Datta AG: Protective effect of erythropoietin on the oxidative damage of erythrocyte membrane by hydroxyl radical. Biochem Pharmacol 2000, 59: 419–425. 10.1016/S0006-2952(99)00277-4
Genc S, Akhisaroglu M, Kuralay F, Genc K: Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci Lett 2002, 321: 73–76. 10.1016/S0304-3940(02)00041-1
Kumral A, Gonenc S, Acikgoz O, Sonmez A, Genc K, Yilmaz O, Gokmen N, Duman N, Ozkan H: Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxic-ischemic brain injury in neonatal rats. Biol Neonate 2005, 87: 15–18. 10.1159/000080490
Agnello D, Bigini P, Villa P, Mennini T, Cerami A, Brines ML, Ghezzi P: Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res 2002, 952: 128–134. 10.1016/S0006-8993(02)03239-0
Li W, Maeda Y, Yuan RR, Elkabes S, Cook S, Dowling P: Beneficial effect of erythropoietin on experimental allergic encephalomyelitis. Ann Neurol 2004, 56: 767–777. 10.1002/ana.20274
Sättler MB, Merkler D, Maier K, Stadelmann C, Ehrenreich H, Bahr M, Diem R: Neuroprotective effects and intracellular signaling pathways of erythropoietin in a rat model of multiple sclerosis. Cell Death Differ 2004, 11(Suppl 2):S181–192. 10.1038/sj.cdd.4401504
Zhang J, Li Y, Cui Y, Chen J, Lu M, Elias SB, Chopp M: Erythropoietin treatment improves neurological functional recovery in EAE mice. Brain Res 2005, 1034: 34–39. 10.1016/j.brainres.2004.11.036
Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, Bigini P, Barbera S, Fumagalli E, Mennini T, Vezzani A, Rizzi M, Coleman T, Cerami A, Brines M, Ghezzi P, Bianchi R: Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol 2006, 172: 27–37. 10.1016/j.jneuroim.2005.10.016
Yu YP, Xu QQ, Zhang Q, Zhang WP, Zhang LH, Wei EQ: Intranasal recombinant human erythropoietin protects rats against focal cerebral ischemia. Neurosci Lett 2005, 387: 5–10. 10.1016/j.neulet.2005.07.008
Avasarala JR, Konduru SS: Recombinant Erythropoietin Down-Regulates IL-6 and CXCR4 Genes in TNF-alpha-Treated Primary Cultures of Human Microvascular Endothelial Cells: Implications for Multiple Sclerosis. J Mol Neurosci 2005, 25: 183–190. 10.1385/JMN:25:2:183
Genc K, Genc S, Baskin H, Semin I: Erythropoietin decreases cytotoxicity and nitric oxide formation induced by inflammatory stimuli in rat oligodendrocytes. Physiol Res 2006, 55: 33–38.
Campana WM, Li X, Shubayev VI, Angert M, Cai K, Myers RR: Erythropoietin reduces Schwann cell TNF-alpha, Wallerian degeneration and pain-related behaviors after peripheral nerve injury. Eur J Neurosci 2006, 23: 617–626. 10.1111/j.1460-9568.2006.04606.x
Wang L, Zhang Z, Wang Y, Zhang R, Chopp M: Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2004, 35: 1732–1737. 10.1161/01.STR.0000132196.49028.a4
Ribatti D, Vacca A, Roccaro AM, Crivellato E, Presta M: Erythropoietin as an angiogenic factor. Eur J Clin Invest 2003, 33: 891–896. 10.1046/j.1365-2362.2003.01245.x
Marti HH, Bernaudin M, Petit E, Bauer C: Neuroprotection and Angiogenesis: Dual Role of Erythropoietin in Brain Ischemia. News Physiol Sci 2000, 15: 225–229.
Jaquet K, Krause K, Tawakol-Khodai M, Geidel S, Kuck KH: Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc Res 2002, 64: 326–333. 10.1006/mvre.2002.2426
Martinez-Estrada OM, Rodriguez-Millan E, Gonzalez-De Vicente E, Reina M, Vilaro S, Fabre M: Erythropoietin protects the in vitro blood-brain barrier against VEGF-induced permeability. Eur J Neurosci 2003, 18: 2538–2544. 10.1046/j.1460-9568.2003.02987.x
Li Y, Lu ZY, Ogle M, Wei L: Erythropoietin prevents blood brain barrier damage induced by focal cerebral ischemia in mice. Neurochem Res 2007, 32: 2132–2141. 10.1007/s11064-007-9387-9
Chen G, Shi JX, Hang CH, Xie W, Liu J, Liu X: Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: a potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO). Neurosci Lett 2007, 425: 177–182. 10.1016/j.neulet.2007.08.022
Ozturk E, Demirbilek S, Kadir But A, Saricicek V, Gulec M, Akyol O, Ozcan Ersoy M: Antioxidant properties of propofol and erythropoietin after closed head injury in rats. Prog Neuropsychopharmacol Biol Psychiatry 2005, 29: 922–927. 10.1016/j.pnpbp.2005.04.028
Muller-Ehmsen J, Schmidt A, Krausgrill B, Schwinger RH, Bloch W: Role of erythropoietin for angiogenesis and vasculogenesis: from embryonic development through adulthood. Am J Physiol Heart Circ Physiol 2006, 290: H331–340. 10.1152/ajpheart.01269.2004
Wang L, Zhang ZG, Zhang RL, Gregg SR, Hozeska-Solgot A, LeTourneau Y, Wang Y, Chopp M: Matrix metalloproteinase 2 (MMP2) and MMP9 secreted by erythropoietin-activated endothelial cells promote neural progenitor cell migration. J Neurosci 2006, 26: 5996–6003. 10.1523/JNEUROSCI.5380-05.2006
Lee ST, Chu K, Sinn DI, Jung KH, Kim EH, Kim SJ, Kim JM, Ko SY, Kim M, Roh JK: Erythropoietin reduces perihematomal inflammation and cell death with eNOS and STAT3 activations in experimental intracerebral hemorrhage. J Neurochem 2006, 96: 1728–1739. 10.1111/j.1471-4159.2006.03697.x
Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT: Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104: 2073–2080. 10.1182/blood-2004-02-0744
Santhanam AV, Smith LA, Nath KA, Katusic ZS: In vivo stimulatory effect of erythropoietin on endothelial nitric oxide synthase in cerebral arteries. Am J Physiol Heart Circ Physiol 2006, 291: H781–786. 10.1152/ajpheart.00045.2006
Ruschitzka FT, Wenger RH, Stallmach T, Quaschning T, de Wit C, Wagner K, Labugger R, Kelm M, Noll G, Rulicke T, Shaw S, Lindberg RL, Rodenwaldt B, Lutz H, Bauer C, Luscher TF, Gassmann M: Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc Natl Acad Sci USA 2000, 97: 11609–11613. 10.1073/pnas.97.21.11609
d'Uscio LV, Smith LA, Santhanam AV, Richardson D, Nath KA, Katusic ZS: Essential role of endothelial nitric oxide synthase in vascular effects of erythropoietin. Hypertension 2007, 49: 1142–1148. 10.1161/HYPERTENSIONAHA.106.085704
Lu D, Mahmood A, Qu C, Goussev A, Schallert T, Chopp M: Erythropoietin enhances neurogenesis and restores spatial memory in rats after traumatic brain injury. J Neurotrauma 2005, 22: 1011–1017. 10.1089/neu.2005.22.1011
Wang L, Zhang Z, Zhang R, Hafner MS, Wong HK, Jiao Z, Chopp M: Erythropoietin up-regulates SOCS2 in neuronal progenitor cells derived from SVZ of adult rat. Neuroreport 2004, 15: 1225–1229.
Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R: Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci 2000, 20: 7377–7383.
Gonzalez FF, McQuillen P, Mu D, Chang Y, Wendland M, Vexler Z, Ferriero DM: Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev Neurosci 2007, 29: 321–330. 10.1159/000105473
Ling ZD, Potter ED, Lipton JW, Carvey PM: Differentiation of mesencephalic progenitor cells into dopaminergic neurons by cytokines. Exp Neurol 1998, 149: 411–423. 10.1006/exnr.1998.6715
Konishi Y, Chui DH, Hirose H, Kunishita T, Tabira T: Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res 1993, 609: 29–35. 10.1016/0006-8993(93)90850-M
Tabira T, Konishi Y, Gallyas F Jr: Neurotrophic effect of hematopoietic cytokines on cholinergic and other neurons in vitro. Int J Dev Neurosci 1995, 13: 241–252. 10.1016/0736-5748(94)00020-4
Campana WM, Misasi R, O'Brien JS: Identification of a neurotrophic sequence in erythropoietin. Int J Mol Med 1998, 1: 235–241.
Kawakami M, Sekiguchi M, Sato K, Kozaki S, Takahashi M: Erythropoietin receptor-mediated inhibition of exocytotic glutamate release confers neuroprotection during chemical ischemia. J Biol Chem 2001, 276: 39469–39475. 10.1074/jbc.M105832200
Lipton SA: Erythropoietin for neurologic protection and diabetic neuropathy. N Engl J Med 2004, 350: 2516–2517. 10.1056/NEJMcibr041121
Recommendations for standards regarding preclinical neuroprotective and restorative drug development Stroke 1999, 30: 2752–2758.
Cerami A: Beyond erythropoiesis: novel applications for recombinant human erythropoietin. Semin Hematol 2001, 38: 33–39. 10.1016/S0037-1963(01)90128-3
Sasaki R, Masuda S, Nagao M: Erythropoietin: multiple physiological functions and regulation of biosynthesis. Biosci Biotechnol Biochem 2000, 64: 1775–1793. 10.1271/bbb.64.1775
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A: Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci USA 2000, 97: 10526–10531. 10.1073/pnas.97.19.10526
Banks WA, Jumbe NL, Farrell CL, Niehoff ML, Heatherington AC: Passage of erythropoietic agents across the blood-brain barrier: a comparison of human and murine erythropoietin and the analog darbepoetin alfa. Eur J Pharmacol 2004, 505: 93–101. 10.1016/j.ejphar.2004.10.035
Ehrenreich H, Degner D, Meller J, Brines M, Behe M, Hasselblatt M, Woldt H, Falkai P, Knerlich F, Jacob S, von Ahsen N, Maier W, Brück W, Ruther E, Cerami A, Becker W, Sirén AL: Erythropoietin: a candidate compound for neuroprotection in schizophrenia. Mol Psychiatry 2004, 9: 42–54.
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Ruther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Sirén AL: Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 2002, 8: 495–505.
Juul SE, Stallings SA, Christensen RD: Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatr Res 1999, 46: 543–547. 10.1203/00006450-199911000-00009
Statler PA, McPherson RJ, Bauer LA, Kellert BA, Juul SE: Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr Res 2007, 61: 671–675.
Belayev L, Khoutorova L, Zhao W, Vigdorchik A, Belayev A, Busto R, Magal E, Ginsberg MD: Neuroprotective effect of darbepoetin alfa, a novel recombinant erythropoietic protein, in focal cerebral ischemia in rats. Stroke 2005, 36: 1071–1076. 10.1161/01.STR.0000163083.59201.34
Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, Frisina N, Caputi AP, Buemi M: Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol 2000, 401: 349–356. 10.1016/S0014-2999(00)00466-0
Spandou E, Papadopoulou Z, Soubasi V, Karkavelas G, Simeonidou C, Pazaiti A, Guiba-Tziampiri O: Erythropoietin prevents long-term sensorimotor deficits and brain injury following neonatal hypoxia-ischemia in rats. Brain Res 2005, 1045: 22–30.
Chang YS, Mu D, Wendland M, Sheldon RA, Vexler ZS, McQuillen PS, Ferriero DM: Erythropoietin Improves Functional and Histological Outcome in Neonatal Stroke. Pediatr Res 2005, 58: 106–111. 10.1203/01.PDR.0000163616.89767.69
Kumral A, Uysal N, Tugyan K, Sonmez A, Yilmaz O, Gokmen N, Kiray M, Genc S, Duman N, Koroglu TF, Ozkan H, Genc K: Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav Brain Res 2004, 153: 77–86. 10.1016/j.bbr.2003.11.002
Aydin A, Genc K, Akhisaroglu M, Yorukoglu K, Gokmen N, Gonullu E: Erythropoietin exerts neuroprotective effect in neonatal rat model of hypoxic-ischemic brain injury. Brain Dev 2003, 25: 494–498. 10.1016/S0387-7604(03)00039-1
Malhotra S, Savitz SI, Ocava L, Rosenbaum DM: Ischemic preconditioning is mediated by erythropoietin through PI-3 kinase signaling in an animal model of transient ischemic attack. J Neurosci Res 2006, 83: 19–27. 10.1002/jnr.20705
Alafaci C, Salpietro F, Grasso G, Sfacteria A, Passalacqua M, Morabito A, Tripodo E, Calapai G, Buemi M, Tomasello F: Effect of recombinant human erythropoietin on cerebral ischemia following experimental subarachnoid hemorrhage. Eur J Pharmacol 2000, 406: 219–225. 10.1016/S0014-2999(00)00691-9
Buemi M, Grasso G, Corica F, Calapai G, Salpietro FM, Casuscelli T, Sfacteria A, Aloisi C, Alafaci C, Sturiale A, Frisina N, Tomasello F: In vivo evidence that erythropoietin has a neuroprotective effect during subarachnoid hemorrhage. Eur J Pharmacol 2000, 392: 31–34. 10.1016/S0014-2999(00)00081-9
Grasso G, Buemi M, Alafaci C, Sfacteria A, Passalacqua M, Sturiale A, Calapai G, De Vico G, Piedimonte G, Salpietro FM, Tomasello F: Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc Natl Acad Sci USA 2002, 99: 5627–5631. 10.1073/pnas.082097299
Santhanam AV, Smith LA, Akiyama M, Rosales AG, Bailey KR, Katusic ZS: Role of endothelial NO synthase phosphorylation in cerebrovascular protective effect of recombinant erythropoietin during subarachnoid hemorrhage-induced cerebral vasospasm. Stroke 2005, 36: 2731–2737. 10.1161/01.STR.0000190021.85035.5b
Springborg JB, Ma X, Rochat P, Knudsen GM, Amtorp O, Paulson OB, Juhler M, Olsen NV: A single subcutaneous bolus of erythropoietin normalizes cerebral blood flow autoregulation after subarachnoid haemorrhage in rats. Br J Pharmacol 2002, 135: 823–829. 10.1038/sj.bjp.0704521
Sirén AL, Radyushkin K, Boretius S, Kammer D, Riechers CC, Natt O, Sargin D, Watanabe T, Sperling S, Michaelis T, Price J, Meyer B, Frahm J, Ehrenreich H: Global brain atrophy after unilateral parietal lesion and its prevention by erythropoietin. Brain 2006, 129: 480–489. 10.1093/brain/awh703
Xiong Y, Mahmood A, Lu D, Qu C, Goussev A, Schallert T, Chopp M: Role of gender in outcome after traumatic brain injury and therapeutic effect of erythropoietin in mice. Brain Res 2007, 1185: 301–312. 10.1016/j.brainres.2007.09.052
Mahmood A, Lu D, Qu C, Goussev A, Zhang ZG, Lu C, Chopp M: Treatment of traumatic brain injury in rats with erythropoietin and carbamylated erythropoietin. J Neurosurg 2007, 107: 392–397. 10.3171/JNS-07/08/0392
Grasso G, Sfacteria A, Meli F, Fodale V, Buemi M, Iacopino DG: Neuroprotection by erythropoietin administration after experimental traumatic brain injury. Brain Res 2007, 1182: 99–105. 10.1016/j.brainres.2007.08.078
Liao ZB, Zhi XG, Shi QH, He ZH: Recombinant human erythropoietin administration protects cortical neurons from traumatic brain injury in rats. Eur J Neurol 2008, 15: 140–149.
Verdonck O, Lahrech H, Francony G, Carle O, Farion R, Looij Y, Remy C, Segebarth C, Payen JF: Erythropoietin protects from post-traumatic edema in the rat brain. J Cereb Blood Flow Metab 2007, 27: 1369–1376. 10.1038/sj.jcbfm.9600443
Mala H, Rodriguez Castro M, Dall Jorgensen K, Mogensen J: Effects of erythropoietin on posttraumatic place learning in fimbria-fornix transected rats after a 30-day postoperative pause. J Neurotrauma 2007, 24: 1647–1657. 10.1089/neu.2007.0292
Cherian L, Goodman JC, Robertson C: Neuroprotection with erythropoietin administration following controlled cortical impact injury in rats. J Pharmacol Exp Ther 2007, 322: 789–794. 10.1124/jpet.107.119628
Gorio A, Gokmen N, Erbayraktar S, Yilmaz O, Madaschi L, Cichetti C, Di Giulio AM, Vardar E, Cerami A, Brines M: Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci USA 2002, 99: 9450–9455. 10.1073/pnas.142287899
Grasso G, Sfacteria A, Erbayraktar S, Passalacqua M, Meli F, Gokmen N, Yilmaz O, La Torre D, Buemi M, Iacopino DG, Coleman T, Cerami A, Brines M, Tomasello F: Amelioration of spinal cord compressive injury by pharmacological preconditioning with erythropoietin and a nonerythropoietic erythropoietin derivative. J Neurosurg Spine 2006, 4: 310–318. 10.3171/spi.2006.4.4.310
Ehrenreich H, Fischer B, Norra C, Schellenberger F, Stender N, Stiefel M, Sirén AL, Paulus W, Nave KA, Gold R, Bartels C: Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 2007, 130: 2577–2588. 10.1093/brain/awm203
Yatsiv I, Grigoriadis N, Simeonidou C, Stahel PF, Schmidt OI, Alexandrovitch AG, Tsenter J, Shohami E: Erythropoietin is neuroprotective, improves functional recovery, and reduces neuronal apoptosis and inflammation in a rodent model of experimental closed head injury. Faseb J 2005, 19: 1701–1703.
Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E: Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. J Cereb Blood Flow Metab 2005, 25: 1456–1465. 10.1038/sj.jcbfm.9600142
Shein NA, Tsenter J, Alexandrovich AG, Horowitz M, Shohami E: Akt phosphorylation is required for heat acclimation-induced neuroprotection. J Neurochem 2007, 103: 1523–1529. 10.1111/j.1471-4159.2007.04862.x
Hengemihle JM, Abugo O, Rifkind J, Spangler E, Danon D, Ingram DK: Chronic treatment with human recombinant erythropoietin increases hematocrit and improves water maze performance in mice. Physiol Behav 1996, 59: 153–156. 10.1016/0031-9384(95)02046-2
Miskowiak K, O'Sullivan U, Harmer CJ: Erythropoietin enhances hippocampal response during memory retrieval in humans. J Neurosci 2007, 27: 2788–2792. 10.1523/JNEUROSCI.5013-06.2007
Keswani SC, Buldanlioglu U, Fischer A, Reed N, Polley M, Liang H, Zhou C, Jack C, Leitz GJ, Hoke A: A novel endogenous erythropoietin mediated pathway prevents axonal degeneration. Ann Neurol 2004, 56: 815–826. 10.1002/ana.20285
Keswani SC, Leitz GJ, Hoke A: Erythropoietin is neuroprotective in models of HIV sensory neuropathy. Neurosci Lett 2004, 371: 102–105. 10.1016/j.neulet.2004.08.080
Genc S, Kuralay F, Genc K, Akhisaroglu M, Fadiloglu S, Yorukoglu K, Fadiloglu M, Gure A: Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci Lett 2001, 298: 139–141. 10.1016/S0304-3940(00)01716-X
Grunfeld JF, Barhum Y, Blondheim N, Rabey JM, Melamed E, Offen D: Erythropoietin delays disease onset in an amyotrophic lateral sclerosis model. Exp Neurol 2007, 204: 260–263. 10.1016/j.expneurol.2006.11.002
Mennini T, De Paola M, Bigini P, Mastrotto C, Fumagalli E, Barbera S, Mengozzi M, Viviani B, Corsini E, Marinovich M, Torup L, Van Beek J, Leist M, Brines M, Cerami A, Ghezzi P: Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med 2006, 12: 153–160. 10.2119/2006-00045.Mennini
Kanaan NM, Collier TJ, Marchionini DM, McGuire SO, Fleming MF, Sortwell CE: Exogenous erythropoietin provides neuroprotection of grafted dopamine neurons in a rodent model of Parkinson's disease. Brain Res 2006, 1068: 221–229. 10.1016/j.brainres.2005.10.078
Gil JM, Leist M, Popovic N, Brundin P, Petersen A: Asialoerythropoietin is not effective in the R6/2 line of Huntington's disease mice. BMC Neurosci 2004, 5: 17. 10.1186/1471-2202-5-17
Assaraf MI, Diaz Z, Liberman A, Miller WH Jr, Arvanitakis Z, Li Y, Bennett DA, Schipper HM: Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J Neuropathol Exp Neurol 2007, 66: 389–398. 10.1097/nen.0b013e3180517b28
Springborg JB, Sonne B, Frederiksen HJ, Foldager N, Poulsgaard L, Klausen T, Jorgensen OS, Olsen NV: Erythropoietin in the cerebrospinal fluid of patients with aneurysmal subarachnoid haemorrhage originates from the brain. Brain Res 2003, 984: 143–148. 10.1016/S0006-8993(03)03124-X
Koehne P, Hochhaus F, Felderhoff-Mueser U, Ring-Mrozik E, Obladen M, Buhrer C: Vascular endothelial growth factor and erythropoietin concentrations in cerebrospinal fluid of children with hydrocephalus. Childs Nerv Syst 2002, 18: 137–141. 10.1007/s00381-002-0567-2
Hassan K, Gross B, Simri W, Rubinchik I, Cohen H, Jacobi J, Shasha SM, Kristal B: The presence of erythropoietin receptors in the human peripheral nervous system. Clin Nephrol 2004, 61: 127–129.
Scarlato M, Previtali SC, Carpo M, Pareyson D, Briani C, Del Bo R, Nobile-Orazio E, Quattrini A, Comi GP: Polyneuropathy in POEMS syndrome: role of angiogenic factors in the pathogenesis. Brain 2005, 128: 1911–1920. 10.1093/brain/awh519
Brettschneider J, Widl K, Ehrenreich H, Riepe M, Tumani H: Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci Lett 2006, 404: 347–351. 10.1016/j.neulet.2006.06.011
Widl K, Brettschneider J, Schattauer D, Sussmuth S, Huber R, Ludolph AC, Tumani H: Erythropoietin in cerebrospinal fluid: age-related reference values and relevance in neurological disease. Neurochem Res 2007, 32: 1163–1168. 10.1007/s11064-007-9286-0
Cariou A, Claessens YE, Pene F, Marx JS, Spaulding C, Hababou C, Casadevall N, Mira JP, Carli P, Hermine O: Early high-dose erythropoietin therapy and hypothermia after out-of-hospital cardiac arrest: a matched control study. Resuscitation 2008, 76: 397–404. 10.1016/j.resuscitation.2007.10.003
Hassan K, Simri W, Rubenchik I, Manelis J, Gross B, Shasha SM, Kristal B: Effect of erythropoietin therapy on polyneuropathy in predialytic patients. J Nephrol 2003, 16: 121–125.
Ehrenreich H, Hinze-Selch D, Stawicki S, Aust C, Knolle-Veentjer S, Wilms S, Heinz G, Erdag S, Jahn H, Degner D, Ritzen M, Mohr A, Wagner M, Schneider U, Bohn M, Huber M, Czernik A, Pollmacher T, Maier W, Sirén AL, Klosterkotter J, Falkai P, Ruther E, Aldenhoff JB, Krampe H: Improvement of cognitive functions in chronic schizophrenic patients by recombinant human erythropoietin. Mol Psychiatry 2007, 12: 206–220. 10.1038/sj.mp.4001907
Springborg JB, Moller C, Gideon P, Jorgensen OS, Juhler M, Olsen NV: Erythropoietin in patients with aneurysmal subarachnoid haemorrhage: a double blind randomised clinical trial. Acta Neurochir (Wien) 2007, 149: 1089–1101. discussion 1101 10.1007/s00701-007-1284-z
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
NB drafted the manuscript and designed the Figure. ALS corrected and wrote the final manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Byts, N., Sirén, AL. Erythropoietin: a multimodal neuroprotective agent. Exp & Trans Stroke Med 1, 4 (2009). https://doi.org/10.1186/2040-7378-1-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2040-7378-1-4