
Abstract
The past decade has witnessed an explosion of genome sequencing and mapping in evolutionary diverse species. While full genome sequencing of mammals is rapidly progressing, the ability to assemble and align orthologous whole chromosome regions from more than a few species is still not possible. The intense focus on building of comparative maps for companion (dog and cat), laboratory (mice and rat) and agricultural (cattle, pig, and horse) animals has traditionally been used as a means to understand the underlying basis of disease-related or economically important phenotypes. However, these maps also provide an unprecedented opportunity to use multispecies analysis as a tool for inferring karyotype evolution. Comparative chromosome painting and related techniques are now considered to be the most powerful approaches in comparative genome studies. Homologies can be identified with high accuracy using molecularly defined DNA probes for fluorescence in situ hybridization (FISH) on chromosomes of different species. Chromosome painting data are now available for members of nearly all mammalian orders. In most orders, there are species with rates of chromosome evolution that can be considered as 'default' rates. The number of rearrangements that have become fixed in evolutionary history seems comparatively low, bearing in mind the 180 million years of the mammalian radiation. Comparative chromosome maps record the history of karyotype changes that have occurred during evolution. The aim of this review is to provide an overview of these recent advances in our endeavor to decipher the karyotype evolution of mammals by integrating the published results together with some of our latest unpublished results.
Similar content being viewed by others


Mammalian Phylogenomics
Modern mammals (Class Mammalia) are divided into three distinct groups (Figure 1). The subclass Prototheria (monotremes) comprises three species of egg-laying mammals: platypus and two echidna species. The infraclasses Metatheria (marsupials) and Eutheria (placentals) together form the subclass Theria. Over the last decade our understanding of the relationships among eutherian mammals has experienced a virtual revolution. Molecular phylogenomics, new fossils finds and innovative morphological interpretations now group the more than 4600 extant species of eutherians into four major super-ordinal clades: Euarchontoglires (including Primates, Dermoptera, Scandentia, Rodentia, and Lagomorpha), Laurasiatheria (Cetartiodactyla, Perissodactyla, Carnivora, Chiroptera, Pholidota, and Eulipotyphla), Xenarthra, and Afrotheria (Proboscidea, Sirenia, Hyracoidea, Afrosoricida, Tubulidentata, and Macroscelidea) [1]. This modern phylogenetic tree serves as a useful scaffold for combining the various parts of a puzzle in comparative mammalian cytogenetics.

An evolutionary tree of mammals. The tree depicts historic divergence relationships among the living orders of mammals. The phylogenetic hierarchy is a consensus view of several decades of molecular genetic, morphological and fossil inference (see for example [98, 99]. Double rings indicate mammalian supertaxa, numbers indicate the possible time of divergences.
Karyotypes: a global view of the genome
Genes provide instructions to build living organisms and each gene maps to the same chromosome in every cell. Linkage is provided by the co-localization of two or more loci on the same chromosome and the largest linkage group is an entire chromosome. The entire chromosome set of a species is known as a karyotype, which can be thought of as a global map of the nuclear genome.
A seemingly logical consequence of descent from common ancestors is that more closely related species should have more similar chromosomes. However, it is now widely appreciated that species may have phenetically similar karyotypes because they are genomically conservative. Therefore in comparative cytogenetics, phylogenetic relationships should be determined on the basis of the polarity of chromosome differences (derived traits).
Historical development of Comparative Cytogenetics
Mammalian comparative cytogenetics, an indispensable part of phylogenomics, has evolved in a series of steps from a purely descriptive science to a heuristic science of the genomic era. Technical advances have marked the various developmental steps of cytogenetics.
Classical Phase of Cytogenetics
It can be argued that the first step of the Human Genome Project took place when Tjio and Levan in 1956 finally reported the correct diploid number of humans as 2n = 46 [2].
During this phase of cytogenetics, data on the karyotypes of literally hundreds of mammalian species (including information on diploid numbers, relative length and morphology of chromosomes, presence of B-chromosomes) were described (Figure 2). Diploid numbers (2n) were found to vary from 2n = 6-7 in the Indian muntjac [3] to over 100 in some rodents [4].

Examples of mammalian chromosomes. a. Metaphase spread of the Indian muntjac (Muntiacus muntjak vaginalis, 2n = 6, 7), the species with the lowest chromosome number. b. Metaphase spread of the Viscacha rat (Tympanoctomys barrerae, 2n = 102), the species with the highest chromosome number. c. Metaphase spread of the Siberian Roe deer (Capreolus pygargus, 2n = 70 + 1-14 B's). The species with additional, or B- chromosomes. d. Metaphase spread of the Transcaucasian mole vole female (Ellobius lutescens, 2n = 17, X0 in both sexes).
Chromosome banding
The second step derived from the invention of C-, G-, R- and other banding techniques and was marked by the Paris Conference (1971) which lead to a standard nomenclature to recognize and classify each human chromosome [5].
G-and R- banding
The most widely used banding methods are G-banding (Giemsa-banding) and R-banding (Reverse-banding). These techniques produce a characteristic pattern of contrasting dark and light transverse bands on the chromosomes. Banding made it possible to identify homologous chromosomes and construct chromosomal nomenclatures for many species. With banding homologous chromosomes, chromosome segments and rearrangements could be identified. The banded karyotypes of 850 mammalian species were summarized in the Atlas of Mammalian Chromosomes [6]. These basic data present an invaluable resource for the contemporary comparative genomics era, and will assist in selection of new mammalian species for detailed study.
C-banding and heterochromatin
One important source of karyotype variability in mammals is related to heterochromatin. Once the amount of heterochomatin is subtracted from total genome content all mammals have very similar genome sizes.
Species of mammals differ considerably in the heterochromatin content and its location (Figure 3). Heterochromatin is most often detected using C-banding [7] and early studies using C-banding showed that differences in the fundamental number (i.e., the number of chromosome arms) could be entirely due to the addition of heterochromatic chromosome arms. It is well documented that heterochromatin may consists of different types of repetitive DNA, not all seen with C-banding, and it can vary greatly between karyotypes of even closely related species. The differences of heterochromatin amount among congeneric rodent species may reach 33% of nuclear DNA in Dipodomys species [8], 36% in Peromyscus species [9], 42% in Ammospermophilus [10] and 60% in Thomomys species where C-value (haploid DNA content) ranges between 2.1 and 5.6 pg [11, 12]. The red viscacha rat (Tympanoctomys barrerae) has a record C-value among mammals - 9.2 pg [13]. Although tetrapoidy was first proposed to be a reason for its high genome size and diploid chromosome number, Svartman et al [14] showed that the high genome size was due to the enormous amplification of heterochromatin. Although one single copy number gene was found to be duplicated in the red viscacha rat genome [15], our data on absence of large genome segment duplications (single paints of most Octodon degu probes) and repetitive DNA hybridization evidence rules against tetraploidy. The study of heterochromatin composition, repeated DNA amount and its distribution on chromosomes of octodontids is absolutely necessary to define exactly what heterochromatin fraction is responsible for the large genomes of the red viscacha rat.

Examples of distribution of C-heterochromatin in mammalian chromosomes. a. C-banded chromosomes of the Eurasian shrew (Sorex araneus, 2n = 21). Example of the smallest amount of heterochromatic bands in mammalian genome. b. C-banded chromosomes of the Ground squirrel (Spermophilus erythrogenys, 2n = 36) with very large centomeric C-bands. c. C-banded chromosomes of the marbled polecat (Vormela peregusna, 2n = 38) with the largest additional heterochromatic arms on some autosomes. d. C-banded chromosomes of the Amur hedgehog (Erinaceus amurensis, 2n = 48) with the very large telomeric C-bands on autosomes. e. C-banded chromosomes of the Eversmann's hamster (Allocricetulus eversmanni, 2n = 26) with pericentomeric C-bands on the X and Y chromosomes. f. C-banded chromosomes of the Southern vole (Microtus rossiaemeridionalis, 2n = 54) with very large C-bands on both sex chromosomes.
In comparative cytogenetics, chromosome homology between species was proposed on the basis of similarities in banding patterns. Closely related species often had very similar banding pattern and after 40 years of comparing bands it seems safe to generalize that karyotype divergence in most taxonomic groups follows their phylogenetic relationship although there are notable exceptions (see [16] and reviews in [6]).
The conservation of large chromosome segments makes comparison between species possible and worthwhile. On the whole chromosome banding has been a reliable indicator of chromosome homology, i.e. that the chromosome identified on the basis of banding actually carry the same genes. However, this is not always the case especially when phylogenetically distant species or species that have experienced extremely rapid chromosome evolution are compared. Banding after all is still morphology and is not always a foolproof indicator of DNA content.
Comparative molecular cytogenetics
The third step occurred when molecular techniques were incorporated into cytogenetics. These techniques use DNA probes of diverse sizes to compare chromosomes directly at the DNA level. Therefore homology was more confidently compared even between phylogenetically distant species or highly rearranged species (gibbons). Using cladistic analysis rearrangements that have diversified the mammalian karyotype were then more precisely mapped and placed in a phylogenomic perspective. "Comparative chromosomics" - is a new term that was used to define the field of cytogenetics dealing with recent molecular approaches [17], although "chromosomics" was originally introduced to define the research of chromatin dynamics and morphological changes in interphase chromosome structures [18].
Chromosome painting or Zoo-FISH was the first techniques to have a wide ranging impact [19–23]. With this method the homology of chromosome regions between different species are identified by hybridizing DNA probes of individual, whole chromosomes of one species to metaphase chromosomes of another species (Figure 4). Comparative chromosome painting allows a rapid and efficient comparison of many species and the distribution of homologous regions makes it possible to track the translocation scenario of chromosomal evolution. When many species covering different mammalian orders are compared, the analysis provides information on trends and rates of chromosomal evolution in different branches.

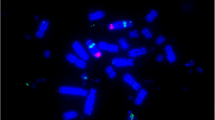
Examples of chromosome painting. Human probes HSA1 (green) and HSA19 (red) onto (a) manatee, Trichechus manatus latirostris, and (b) aardvark, Orycteropus afer. HSA 1/19 syntenic association is evident on manatee chromosome 2 and aardvark chromosome 3. Image courtesy of F. Yang, The Wellcome Trust Sanger Institute, Cambridge, UK.
However, homology is only detected qualitatively, and resolution (about 4 Mb, according to our data) is limited by the size of visualized regions, thus the method does not detect all tiny homologous regions resulted from multiple rearrangements (as between mouse and human). Besides, the method fails to report internal inversions within large segments. Another limitation is that painting across great phylogenetic distance often results in a decreased efficiency. Nevertheless, use of painting probes derived form different species combined with comparative sequencing projects helps to increase the resolution of the method. Chromosome painting sets were made from about 100 vertebrate species (mostly mammals) and the results are summarized in table 1.
In addition to sorting, microdissection of chromosomes and chromosome regions was used to obtain probes for chromosome painting. Impressive results were obtained when a series of microdissection probes covering the total human genome was localized on anthropoid primate chromosomes via multicolor banding (MCB) [24, 25]. A limitation of MCB is that it can only be used within a group of closely related species ("phylogenetic" resolution is too low). Spectral karyotyping (SKY) and MFISH - the ratio labeling and simultaneous hybridization of a complete chromosome set have similar drawbacks and have had little application outside of clinical cytogenetics.
All new comparative genomics data including chromosome painting confirmed the high extent of conservation for mammalian chromosomes [23] (Figures 5, 6). Total human chromosomes or their arms can efficiently paint extended chromosome regions in many placentals down to Afrotheria and Xenarthra. Humans are most commonly used as a reference species in chromosome comparisons. Gene localization data on human chromosomes can be extrapolated to the homologous chromosome regions of other species with high reliability. Another surprising feature that facilitates the use of the human genome in comparative studies is that humans are a species with a conserved syntenic chromosome organization that is not so distant from the ancestral condition of all placentals. In Figure 5 we present chromosomal maps of chicken, opossum, and some species of placentals with homologies to human chromosomes and putative mammalian ancestor chromosomes.

A comparative chromosome map of birds and mammals inferred human homologies (right numbers) on chromosome idiograms. a. Reconstructed karyotype of the ancestral Eutherian genome [61]. Each chromosome is assigned a specific color. These colors are used for mark homologies in idiograms of chromosomes of other species (Figure 5b-5i) b. Idiogram of chicken (Gallus gallus domesticus, 2n = 78) chromosomes. The reconstruction is based on alignments of chicken and human genome sequences [100]. c. Idiogram of short-tailed opossum (Monodelphis domestica, 2n = 18) chromosomes. The reconstruction is based on alignments of opossum and human genome sequences [100]. d. Idiogram of aardvark (Orycteropus afer, 2n = 20) chromosomes. The reconstruction is based on painting data [61] e. Idiogram of mink (Mustela vison, 2n = 30) chromosomes. The reconstruction is based on painting data [101][97] f. Idiogram of the Red fox (Vulpes vulpes vison, 2n = 34 + 0-8 B's) chromosomes. The reconstruction is based on painting and mapping data [77][48] g. Reconstructed karyotype of the ancestral Sciuridae (Rodentia) genome, based on painting data (Li et al., 2004). h. Idiogram of the House mouse (Mus musculus, 2n = 40) chromosomes. The reconstruction is based on alignments of Mus and human genome sequences [100]. i. Idiogram of human (Homo sapiens, 2n = 46) chromosomes.

Conservation of chromosome banding pattern between mammals. A. Conservation of chromosomes between opossum (Metatheria) and some eutherians based on alignments and painting data. The high degree of conservation in G-banding patterns between the homologous segments of opossum and placental mammals. MDO - short-tailed opossum, Monodelphis domestica (Metatheria, Marsupialia); HSA - human, Homo sapiens (Eutheria, Euarchontoglires, Primates); OAF - aardvark, Orycteropus afer (Eutheria, Afrotheria, Tubulidentata); LAF - African Savannah elephant, Loxodonta africana (Eutheria, Afrotheria, Proboscidea); FCA - domestic cat, Felis catus (Eutheria, Laurasiatheria, Carnivora); GMA - short-finned pilot whale, Globicephala macrorhynchus (Eutheria, Laurasiatheria, Cetartiodactyla); OCU - Old World rabbit. Oryctolagus cuniculus (Eutheria, Euarchontoglires, Lagomorpha); CBA- Bactrian camel, Camelus bactrianus (Eutheria, Laurasiatheria, Cetartiodactyla); SSC - domestic pig, Sus scrofa (Eutheria, Laurasiatheria, Cetartiodactyla); CFA - domestic dog, Canis familiaris (Eutheria, Laurasiatheria, Carnivora); MVI - American mink, Mustela vison (Eutheria, Laurasiatheria, Carnivora); SVU - European squirrel, Sciurus vulgaris (Eutheria, Euarchontoglires, Rodentia). B. Banding patterns for some characteristic eutherian signatures. a. HSA 19/3/21 signature of Carnivora. MVI -mink, Mustela vison; AFU - lesser panda, Ailurus fulgens b. HSA 9/11 and 10p/1q signatures of Glires. OCU - Old World rabbit. Oryctolagus cuniculus; TSI - Siberian chipmunk, Tamias sibiricus c. HSA 1/19 signature of Afrotheria. LAF - African savannah elephant, Loxodonta africana; OAF - aardvark, Orycteropus afer d. Putative HSA 10/14/15 signature of Pegasoferae taxa. EAS - donkey, Equus asinus; MAL- Szechwan myotis, Myotis altarium e. Conservation and putative inversions of HSA4/8p mammalian signature, inferred by localizations of some human and dog chromosomes painting probes. HSA - human, Homo sapiens; CFA - domestic dog, Canis familiaris; OCU - Old World rabbit, Oryctolagus cuniculus; FCA - domestic cat, Felis catus; MVI- American mink, Mustela vison; OAF - aardvark, Orycteropus afer; CCR - spotted hyena, Crocuta crocuta; PLA - masked palm civet, Paguma larvata; AFU - lesser panda, Ailurus fulgens
Post-genomic time and comparative chromosomics
After the Human Genome Project was completed, researchers focused on evolutionary comparisons of the genome structures of different species. The whole genome of any species can be sequenced completely and repeatedly to obtain a comprehensive, single-nucleotide map. The method makes it possible to compare genomes for any two species regardless of their taxonomic distance. Genome assemblies are available for about 93 fungi; 38 protozoa; 13 plants; more than 40 invertebrates; a few fish, reptiles, and birds; and 38 mammalian species (http://www.genome.gov, http://genome.ucsc.edu, http://www.ncbi.nlm.nih.gov/).
Sequencing efforts provided a host of products that were put to good use in molecular cytogenetics. Fluorescence in situ hybridization (FISH) with DNA clones (BAC and YAC clones, cosmids) allowed the construction of chromosome maps at a resolution of several megabases which could detect relatively small chromosome rearrangements. A resolution of several kilobases can be achieved on interphase chromatin. A limitation is that hybridization efficiencies drop off with increasing phylogenetic distance.
Radiation hybrid (RH) genome mapping is another efficient approach. This method includes the irradiation of cells to disrupt the genome into the desired number of fragments that are subsequently fused with Chinese hamster cells. The resulting somatic cell hybrids contain individual fragments of the genome of interest. Then, 90-100 (sometimes, more) clones covering the total genome are selected, and the sequences of interest are localized on the cloned fragments via the polymerase chain reaction (PCR) or direct DNA-DNA hybridization. To compare the genomes and chromosomes of two species, RHs should be obtained for both of them.
Sex Chromosome Evolution
In contrast to many other taxa, therian mammals and birds are characterized by highly conserved systems of genetic sex determination that lead to special chromosomes, i.e. the sex chromosomes. Although the XX/XY sex chromosome system is the most common among eutherian species, it is not universal. In some species X-autosomal translocations result in the appearance of "additional Y" chromosomes (for example, XX/XY1Y2Y3 systems in Black munjac [26, 27]). In other species Y-autosomal translocations lead to appearance of additional X chromosomes (for example, in some New World primates such as howler monkeys). In this respect rodents again represent a peculiar, derived group, comprising the record number of species with non-classical sex chromosomes such as the wood lemming, the collared lemming, the creep vole, the spinous country rat, the Akodon and the bandicoot rat (reviewed in [28]). One of the most intriguing and enigmatical cases represents the genus of mole voles where Ellobius lutescens has X0/X0 constitution in both sexes (Figure 2) [29], and - E. alaicus, E. talpinus, E. tancrei have XX/XX system [30].
Novel methods of genome study have revealed some new interesting data concerning sex chromosome evolution. In monotremes, the most basal mammalian clade, there are multiple sex chromosomes consisting of blocks that are autosomal in therians [31–33]. The homologous region to marsupial and eutherian X chromosomes is located on a pair of autosomes in both platypus and echidna [27]. Strikingly there are blocks homologous to the avian Z chromosome, thus presuming a recent origin of therian X [29] and more ancestral mode of avian Z-homologous sex determination as presumed in [34]. This theory is in contrast to comparative painting studies in reptiles and recent lizard genome sequencing project, where most sex chromosomes were found to have no homology with avian Z chromosomes [35, 36].
Unfortunately, most current genome sequencing projects ignore Y and W chromosomes. Only the human and chimpanzee Y chromosomes have been sequenced completely [37] and new approaches and studies are necessary to trace the evolution of this essential element of the karyotype in various lineages.
Diploid number polymorphism
Most mammalian species are characterized by a particular chromosome number, but sometimes variation of diploid numbers within a species results from polymorphisms for centric fusions (Robertsonian translocations) involving acrocentric chromosomes. These "Robertsonian fans", were found in many species, including Mus musculus, where all diploid numbers range from 22 to 40 [38]. Another Robertsonian fan was revealed in Sorex araneus with 2n varying from 20 to 33. In both these taxa the number of different karyotypes reaches 60 [39–41]. Twenty four different karyotypes were found in Akodon cursor [42] and twenty were found in Gerbillus nigeriae [43].
B-chromosomes
B-chromosomes or dispensable, supernumerary chromosomes were found in certain mammalian species. The number of B-chromosomes (Bs) per cell may vary among different tissues, individuals, and populations. They do not pair and recombine with any of the standard A-chromosomes at meiosis. The Bs occur in about 1.5% of mammalian species, at least two thirds are rodents, mostly from the superfamily Muroideae ([44], our data). Here we give only two spectacular examples of B-chromosome variation, that were found in the collared lemming Dicrostonyx torquatus, 2N = 40 plus 1 to 42 Bs [45] and in Apodemus peninsulae with 2n = 48 plus 0 to 24 Bs, some of which may be larger than the largest A chromosomes [44]. Figure 2c presents a karyotype of the Siberian Roe deer Capreolus pygargus with eight B-chromosomes [46].
Application of comparative painting has shown that in addition to different heterochromatic blocks [47] B-chromosomes of many mammalian species may contain rather large duplicated segments from autosomes genes and gene segments [48–50]. Although transcription and of these genes has not been demonstrated, this finding has led to a change in our view of B-chromosomes and suggests a new role of these elements as harboring genome segment duplications thus giving raw material for appearance of new genes and new combinations of genetic material.
Evolutionary new centromeres
Comparative cytogenetic studies have demonstrated that centromeres are cytogenetic hot spots and that new centromeres occasionally arise. In clinical cytogenetics these events are called "neocentromeres" and "evolutionary new centromeres" or "ENC" in comparative cytogenetics. The first human neocentromere was discovered in 1993 [51]. Later the use of FISH of DNA clones and chromosome painting revealed multiple ENCs in primates [52–55], perissodactyls [56], rodents [57], marsupials [58] and even birds [59]. ENCs represent a phenomenon that is almost always detected cytogenetically because the centromere is a black hole to most genome sequencing methods.
Reconstructing the Ancestral Mammalian Genome
Comparative painting revealed that particular human chromosomal blocks were often adjacent in a particular phylogenetic array of species. These chromosome associations, often termed evolutionary signatures or landmarks, make it possible to identify the genomic characteristics that are thought to have been present in the common ancestor of the taxons considered. For instance human chromosome signatures 4/8p, 3/21, 14/15b, 10p/12a/22a, 16q/19q, 7a/16p, and 12b/22b occur in the genomes of most Boreoeutheria mammals and thus were considered to be characteristic for the genome of their common ancestor. Signature 1/19p and 5/21 are found in all Afrotheria and are considered characteristic for this group [60–62] (Figure 4). Some workers have suggested that the 1/19 signature, also found in some anteaters (Edentata), was present in the ancestral placental genome [63]. A variant of the ancestral placental genome is shown in Figure 5 [61]. Association HSA1/19 was also found in marsupials on Monodelphis chromosome 4 according to sequencing data (http://www.ensembl.org/Monodelphis_domestica), but the associations are not homologous because reciprocal painting shows that the Afrotheria association is 1p/19p while that of the marsupial is 1p36/19q13. Other variants do not include signatures 1/19p and 10p/12a/22, associating the ancestral placental genome mostly with Boreoeutheria [64, 65].
Anaylsis of the genome assemblies of mammalian genomes can serve as a test for hypotheses about the content of the ancestral eutherian genome. We expect that the structure of the putative ancestral mammalian genome will be further refined due to new information derived from the genome assemblies of additional mammalian genomes become available. Although purely bioinformatic approaches with a limited number of species [66] has proved unreliable [65], it is clear that a an integration of the two approaches holds promise [67–69].
The homologous nature of syntenic associations should be confirmed on a high-resolution basis. Convergent events can be established if the syntenic associations originate from segments derived by different breakpoints. Breakpoints can be established at the cytogenetic level by FISH with cloned DNA such as BACs or at even higher levels of resolution by sequencing. However, breakpoints are often located in duplicated and repeat rich regions of the genome where sequencing is both costly and time consuming. Further, breakpoint reuse may be an additional confounding factor [70]. For example, Robertsonian rearrangements and simple fissions may contribute to homoplasy in cytogenetic analyses.
In spite of these limitations common syntenic associations are still considered as a useful category for phylogenomics. Data on associations of conserved syntenic blocks have been accumulated for all orders of mammals, where each block is identified on the basis of its location on human (HSA) chromosomes. Table 2 lists syntenic associations in a range of animals based on homologies with human chromosomes.
It is extremely important to note, that many of ancestral Placentalia chromosomal associations are present not only in eutherians, but also in marsupials and even in birds (Figure 5). Genome sequencing studies have shown that many ancestral blocks can be found in fish, insects and even in cnidarians [71]. This situation confirms the general rule of high evolutionary genome conservation as first proposed on the basis of chromosome banding up to current comparisons of genome assemblies (Figures 5, 6).
The most reliable conclusions on the content of ancestral genomes, the pathways and rates of chromosome evolution are made when data is available for the widest possible phylogenetic array of species. Different rates of chromosomal evolution in groups have lead to errors in interpreting phylogenetic relationships. For example, although gibbons are closely related to humans and are included in the Hominoidea, the high number of chromosome rearrangements in these taxa makes them phenetically more distant from humans than human are from some species outside the primate order such as cats [72–74]. The reasons for high rates of genome reshuffling are far from being clearly understand, still some authors hypothesize that such factors as population structure and the repetitive fraction of DNA content may increase the rate of karyotype evolution [75, 76].
In the following paragraphs we will concentrate on particular mammalian orders that were studied by comparative painting which provide particularly informative examples of karyotype evolution.
Canidae
A great number of species has been examined by chromosome painting in the order Carnivora, which now, quite naturally, includes pinnipeds (walruses and seals) as a sister group to mustelids. Human chromosome probes detected 30-35 homologous regions on the chromosomes of cats, weasels, lesser panda, pinnipeds, civets, and hyenas; 43-45 regions in the karyotypes of bears and giant panda; and over 70 regions in the canine karyotype. Almost all conserved regions that are characteristic for mammalian ancestral genome and, in particular, for carnivores, are disrupted in the canine genome (Figures 5, 6B) [77].
It should be noted that the high-quality flow sorted canine chromosome probes [77] proved to be extremely useful for genome mapping. Due to their evolutionary fragmented character, these probes allowed the identification of rearrangements (inversions) within the regions that seem conservative when studied by human chromosome probes (Figure 6B). As a whole, the use of dog paints has shown that inversions inside of the conservative regions are not frequent. Therefore it is possible that a proportion of the high number of inversions found in many species in mammalian Genome Projects may result from assembly mistakes [78].
Rodentia
Unequal rates of genome evolution have been observed for different mammalian groups; rodents are most remarkable in this respect. The mechanisms that triggered such increased rates of genome reshuffling remain unknown. The order Rodentia comprises more than 40% of all mammalian species. It is the most numerous and evolutionarily diverse taxon of mammals. About one-third of rodent species belong to the superfamily Muroidea (mice, rats, and hamsters). It is muroid rodents that are the champions in the great evolutionary competition, to the shame of other mammalian orders. Comparative reciprocal painting with chromosome probes of mouse and rat showed that the rate of chromosome rearrangements differentiating these extremely close species was tenfold higher than between human and cat, which are rather distant [79]. Yet the most impressive finding was the structure of the mouse genome. After human, mouse Mus musculus is the most thoroughly studied mammal. Early integrative data on mouse chromosome mapping suggested that there were a large number of chromosome rearrangements differentiating the mouse and human genomes [80]. Later, attempts to localize human chromosome probes on mouse chromosomes were, mostly, unsuccessful: the size of many regions homologous in the mouse and human genomes proved lower than the resolution of chromosome painting, confirming that the mouse genome is much more rearranged than that of most other taxa [22]. It is remarkable that the mouse genome includes unusual chromosomes such as chromosome 17, which appears as a "genome dustbin," combining fragments of many chromosomes occurring intact even in other species of the genus Mus (Figure 5H).
It should be noted that the great number of rearrangements found between humans and the mouse also applies to other Muroidea species, including another well-studied species, the rat, Rattus norvegicus. Thus, in terms of comparative chromosomics, Muroidea appear to have experienced a genomic revolution that sets them apart from the other placental mammals.
Muroid rodents present a particular challenge, considering the high number of species and high rates of chromosome reshuffling. Various techniques are needed to sort out their karyotypic relationships. In addition to flow sorted chromosome paints [81–83], a set of chromosome region specific microdissection derived murine probes were used [84]. The hybridization of microdissected murine probes provided a multicolor banding pattern which was particularly useful to identifying new evolutionary breakpoints, previously unrecognized small homologous segments, inversions, and evolutionary new centromeres (discussed above) (Figure 7).

Some examples of using mcb probe set for murine chromosome 6. a-c)Results obtained after application of mcb probe set for chromosome 6 on mouse (MMU), rat (RNO) and striped field mouse (APE). The figures show: mcb pseudocolor banding, partial chromosome paints labeled in SpectrumGreen, SpectrumOrange, TexasRed, Cyanine5 and diethylaminocoumarine, the fluorochrome profile along the chromosome and the inverted DAPI-banding pattern of the studied chromosome. d)Similarity of banding patterns of mouse chromosome 6, rat chromosome 4 and field mouse chromosome 2. e)Localization of partial chromosome painting probes of murine chromosome 6 and the labeling scheme.
It is important to place on the evolutionary tree the triggering of a mechanism that allowed a considerable increase in the rate of chromosome evolution in rodents. This event took place after Sciuridae (tree squirrels, chipmunks, marmots, and ground squirrels) split from the main lineage of Rodentia. Detailed localization of human chromosome probes on chromosomes of many squirrels and reciprocal painting showed that the squirrel genomes are highly conserved, are similar to the human and ancestral genomes, and have several signatures suggesting a common origin for rodents and lagomorphs [85, 86].
The putative karyotype of the rodent ancestor (Figure 5) is close to the karyotype of placentals and is very distant from that of the Muridae ancestor [81, 82, 87]. It should be noted that this proposal of rodent ancestral genome organization differs fundamentally from other proposals based on different methods [66]. Bioinformatics approaches up to now are limited because their database is restricted to mouse and rat. The result is a rodent "ancestral genome" intermediate between the human and mouse genomes, which is totally without merit. Only a phylogenetically rich and appropriate array of species may eventually reveal at the bioinformatics level the main regularities of the organization and evolution of mammals and, in particular, rodents.
Perissodactyla
The odd-toed ungulates are a good example to illustrate the potential of chromosome painting for the reconstruction of evolutionary events [88]. This order includes three extant families that have different modes of chromosome evolution. Tapirs and rhinoceroses were found to be extremely conserved and had hardly undergone any rearrangements for millions of years. On the other hand, equids underwent an explosion of karyotype reshuffling accompanied by rapid species divergence. Our data obtained from study of almost all extant representatives of the order (excluding only two Asian rhinoceroses) was subjected to PAUP analysis and resulted in the phylogenetic tree that turned out to be identical to those obtained with sequence analysis data. The phylogeny of equids was particularly easy to reconstruct and non-controversial, probably due to relatively recent fixation of multiple rearrangements.
Cetartiodactyla and chiroptera
The most controversial phylogenies are usually obtained from species, whose divergence occurred long ago and was accompanied by small number of rearrangements. Another problem comes from the appearance of parallelisms or homoplasies. Thus many convergent events were probably characteristic for cetartiodactyls, hampering the reconstruction of non-controversial phylogenetic trees [89]. Convergence and homoplasy was found to be frequent in bats. It was difficult to resolve the phylogeny of the main families in spite of many species involved [90]. Later scrutiny revealed a single association that may reflect the closer relationship of bat families Pteropodidae and Rhinolopoidea [91].
Chromosome painting in resolving superordinal clades
Chromosome-derived characters turned out to be very useful in resolving or supporting some problematic superordinal clades. An example was the support of afrotherian clade at the cytogenetic level. The grouping of Afrotheria was originally based on molecular data [92] [1] [93] while paleontological and morphological data did not support the clade. Importantly, independent support came from cytogenetics when two synapomorphic associations, 1/19 and 5/21, were found in all afrotherian species studied [60] [62] [94]. Within Afrotheria such clades as Paenungulata (Hyracoidea, Sirenia and Proboscidea) [94, 95] and Afroinsectiphillia (aardvark, golden mole, elephant-shrew) [62] were supported by painting data.
Within the cohort Euarchontoglires (Primates, Dermoptera, Scadentia, Rodentia, Lagomorpha) the superorder Glires (Rodentia+Lagomorpha) was supported by human syntenic associations 1/10p and 9/11 [85, 86] and the superorder Sundatheria (Dermoptera+Scadentia) by human association 2q/21 [96].
Although comparative chromosome painting did not reveal any association uniting all orders of the Laurasiatheria clade (Eulipothypla, Carnivora, Pholidota, Cetartiodactyla, Perissodactyla, Chiroptera), the order Pinnipedia was placed within Carnivora as sister clade to Mustelidae [97], Cetacea was nested within Artiodactyla [89] and Perissodactyla and Cetartiodactyla were found to be sister clades [63] [88]. All these findings are consistent with most modern phylogenies obtained using molecular data.
Conclusions
The Postgenomic research in mammalian cytogenetics has confirmed the previously established general tendencies of karyotype evolution, brought new data for finalizing phylogenetic trees and allowed a detail analysis of genome evolution in various branches. New molecular approaches led to a precise characterization of breakpoints in evolution and altered our understanding of sex chromosome and B-chromosome evolution.
Studies of mammalian genome evolution are set to take a quantum leap as ever more completely sequenced multiple genomes become available. The previously studied karyotypes characterized from techniques ranging from classical staining and banding to molecular cytogenetic approaches from chromosome paints to cloned DNA will serve as basis for high resolution maps construction for hundreds of mammalian and vertebrate species. A newly proposed Genome 10 K project presumes whole genome sequencing of 10,000 vertebrate species in the near future (G10KCOS 2009), which will provide a foundation for the next generation of postgenomic studies.
References
Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O'Brien SJ: Molecular phylogenetics and the origins of placental mammals. Nature 2001, 409: 614–618. 10.1038/35054550
Tjio HJ LA: The chromosome numbers of man. Hereditas 1956, 42: 1–6.
Wurster DH, Benirschke K: Indian muntjac, Muntiacus muntjak: a deer with a low diploid chromosome number. Science 1970, 168: 1364–1366. 10.1126/science.168.3937.1364
Contreras LC, Torresmura JC, Spotorno AE: The Largest Known Chromosome-Number for a Mammal, in a South-American Desert Rodent. Experientia 1990, 46: 506–508. 10.1007/BF01954248
Paris Conference (1971): Standardization in human cytogenetics Cytogenetics 1972, 11: 317–362.
S.J. O'Brien WGN, Menninger JC: Atlas of Mammalian Chromosomes. John Wiley and Sons Publishers; 2006.
Hsu TC, Arrighi FE: Distribution of constitutive heterochromatin in mamallian chromosomes. Chromosoma 1971, 34: 243–253.
Hatch FT, Bodner AJ, Mazrimas JA, Moore DH: Satellite DNA and cytogenetic evolution. DNA quantity, satellite DNA and karyotypic variations in kangaroo rats (genus Dipodomys). Chromosoma 1976, 58: 155–168. 10.1007/BF00701356
Deaven LL, Vidal-Rioja L, Jett JH, Hsu TC: Chromosomes of Peromyscus (rodentia, cricetidae). VI. The genomic size. Cytogenet Cell Genet 1977, 19: 241–249. 10.1159/000130816
Mascarello JT MJ: Chromosomes of antelope squirrels (genus Ammospermophilus): a systematic banding analysis of four species with unusual constitutive heterochromatin. Chromosoma 1977, 64: 207–217. 10.1007/BF00328078
Patton JL, Sherwood SW: Genome evolution in pocket gophers (genus Thomomys). I. Heterochromatin variation and speciation potential. Chromosoma 1982, 85: 149–162. 10.1007/BF00294962
Sherwood SW, Patton JL: Genome evolution in pocket gophers (genus Thomomys). II. Variation in cellular DNA content. Chromosoma 1982, 85: 163–179. 10.1007/BF00294963
Gallardo MH, Bickham JW, Honeycutt RL, Ojeda RA, Kohler N: Discovery of tetraploidy in a mammal. Nature 1999, 401: 341. 10.1038/43815
Svartman M, Stone G, Stanyon R: Molecular cytogenetics discards polyploidy in mammals. Genomics 2005, 85: 425–430. 10.1016/j.ygeno.2004.12.004
Gallardo MH, Gonzalez CA, Cebrian I: Molecular cytogenetics and allotetraploidy in the red vizcacha rat, Tympanoctomys barrerae (Rodentia, Octodontidae). Genomics 2006, 88: 214–221. 10.1016/j.ygeno.2006.02.010
Graphodatsky AS: Conserved and variable elements of mammalian chromosomes. In Cytogenetics of animals. Edited by: CRE H. Oxon, UK: CAB International Press; 2006:95–124.
Grafodatskii AS: [Comparative chromosomics]. Mol Biol (Mosk) 2007, 41: 408–422.
Claussen U: Chromosomics. Cytogenet Genome Res 2005, 111: 101–106. 10.1159/000086377
Wienberg J, Jauch A, Stanyon R, Cremer T: Molecular cytotaxonomy of primates by chromosomal in situ suppression hybridization. Genomics 1990, 8: 347–350. 10.1016/0888-7543(90)90292-3
Telenius H, Pelmear AH, Tunnacliffe A, Carter NP, Behmel A, Fergusonsmith MA, Nordenskjold M, Pfragner R, Ponder BAJ: Cytogenetic Analysis by Chromosome Painting Using Dop-Pcr Amplified Flow-Sorted Chromosomes. Gene Chromosome Canc 1992, 4: 257–263. 10.1002/gcc.2870040311
Scherthan H, Cremer T, Arnason U, Weier HU, Limadefaria A, Fronicke L: Comparative Chromosome Painting Discloses Homologous Segments in Distantly Related Mammals. Nature Genetics 1994, 6: 342–347. 10.1038/ng0494-342
Ferguson-Smith MA: Genetic analysis by chromosome sorting and painting: phylogenetic and diagnostic applications. Eur J Hum Genet 1997, 5: 253–265.
Ferguson-Smith MA, Trifonov V: Mammalian karyotype evolution. Nat Rev Genet 2007, 8: 950–962.
Mrasek K, Heller A, Rubtsov N, Trifonov V, Starke H, Rocchi M, Claussen U, Liehr T: Reconstruction of the female Gorilla gorilla karyotype using 25-color FISH and multicolor banding (MCB). Cytogenetics and Cell Genetics 2001, 93: 242–248. 10.1159/000056991
Mrasek K, Heller A, Rubtsov N, Trifonov V, Starke H, Claussen U, Liehr T: Detailed Hylobates lar karyotype defined by 25-color FISH and multicolor banding. International Journal of Molecular Medicine 2003, 12: 139–146.
Yang F, Carter NP, Shi L, Ferguson-Smith MA: A comparative study of karyotypes of muntjacs by chromosome painting. Chromosoma 1995, 103: 642–652. 10.1007/BF00357691
Huang L, Chi J, Wang J, Nie W, Su W, Yang F: High-density comparative BAC mapping in the black muntjac (Muntiacus crinifrons): molecular cytogenetic dissection of the origin of MCR 1p+4 in the X1X2Y1Y2Y3 sex chromosome system. Genomics 2006, 87: 608–615. 10.1016/j.ygeno.2005.12.008
Fredga K: Aberrant sex chromosome mechanisms in mammals. Evolutionary aspects. Differentiation 1983, 23 Suppl: S23–30.
Romanenko SA, Sitnikova NA, Serdukova NA, Perelman PL, Rubtsova NV, Bakloushinskaya IY, Lyapunova EA, Just W, Ferguson-Smith MA, Yang F, Graphodatsky AS: Chromosomal evolution of Arvicolinae (Cricetidae, Rodentia). II. The genome homology of two mole voles (genus Ellobius), the field vole and golden hamster revealed by comparative chromosome painting. Chromosome Res 2007, 15: 891–897. 10.1007/s10577-007-1171-9
Vorontsov NN, Lyapunova EA, Borissov YM, Dovgal VE: Variability of Sex-Chromosomes in Mammals. Genetica 1980, 52–3: 361–372.
Rens W, Grutzner F, O'Brien PC, Fairclough H, Graves JA, Ferguson-Smith MA: Resolution and evolution of the duck-billed platypus karyotype with an X1Y1X2Y2X3Y3X4Y4X5Y5 male sex chromosome constitution. Proc Natl Acad Sci USA 2004, 101: 16257–16261. 10.1073/pnas.0405702101
Grutzner F, Rens W, Tsend-Ayush E, El-Mogharbel N, O'Brien PCM, Jones RC, Ferguson-Smith MA, Graves JAM: In the platypus a meiotic chain of ten sex chromosomes shares genes with the bird Z and mammal X chromosomes. Nature 2004, 432: 913–917. 10.1038/nature03021
Rens W, O'Brien PC, Grutzner F, Clarke O, Graphodatskaya D, Tsend-Ayush E, Trifonov VA, Skelton H, Wallis MC, Johnston S, et al.: The multiple sex chromosomes of platypus and echidna are not completely identical and several share homology with the avian Z. Genome Biol 2007, 8: R243. 10.1186/gb-2007-8-11-r243
Veyrunes F, Waters PD, Miethke P, Rens W, McMillan D, Alsop AE, Grutzner F, Deakin JE, Whittington CM, Schatzkamer K, et al.: Bird-like sex chromosomes of platypus imply recent origin of mammal sex chromosomes. Genome Res 2008, 18: 965–973. 10.1101/gr.7101908
Pokorna M, Giovannotti M, Kratochvil L, Kasai F, Trifonov VA, O'Brien PC, Caputo V, Olmo E, Ferguson-Smith MA, Rens W: Strong conservation of the bird Z chromosome in reptilian genomes is revealed by comparative painting despite 275 million years divergence. Chromosoma 2011.
Alfoldi J, Di Palma F, Grabherr M, Williams C, Kong L, Mauceli E, Russell P, Lowe CB, Glor RE, Jaffe JD, et al.: The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011.
Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SK, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al.: Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 2010, 463: 536–539. 10.1038/nature08700
Capanna E: Robertsonian numerical variation in animal speciation: Mus musculus, an emblematic model. Prog Clin Biol Res 1982, 96: 155–177.
Bauchau V: Phylogenetic Analysis of the Distribution of Chromosomal Races of Mus-Musculus Domesticus Rutty in Europe. Biol J Linn Soc 1990, 41: 171–192. 10.1111/j.1095-8312.1990.tb00828.x
Král B RS: Banding patterns and Robertsonian fusion in the Western Siberian population of Sorex araneus (Insectivora, Soricidae). Zool Listy 1974, 23: 217–227.
Volobouev VT: Phylogenetic-Relationships of the Sorex-Araneus-Arcticus Species Complex (Insectivora, Soricidae) Based on High-Resolution Chromosome Analysis. Journal of Heredity 1989, 80: 284–290.
Fagundes V, Vianna-Morgante AM, Yonenaga-Yassuda Y: Telomeric sequences localization and G-banding patterns in the identification of a polymorphic chromosomal rearrangement in the rodent Akodon cursor (2n = 14,15 and 16). Chromosome Res 1997, 5: 228–232. 10.1023/A:1018463401887
Volobouev V, Vogt N, Viegas-Pequignot E, Malfoy B, Dutrillaux B: Characterization and chromosomal location of two repeated DNAs in three Gerbillus species. Chromosoma 1995, 104: 252–259. 10.1007/BF00352256
Volobujev VT: B-Chromosomes System of the Mammals. Caryologia 1981, 34: 1–23.
Cernyavsky FB, Kozlovsky AI: Species Status and History of the Arctic Lemmings (Dicrostonyx, Rodentia) of the Wrangel Island. Zool Zh 1980, 59: 266–273.
AS G: Karyotypical relationships between Cervidae. J Zool 1990, 69: 101–114.
Trifonov VA, Perelman PL, Kawada SI, Iwasa MA, Oda SI, Graphodatsky AS: Complex structure of B-chromosomes in two mammalian species: Apodemus peninsulae (Rodentia) and Nyctereutes procyonoides (Carnivora). Chromosome Res 2002, 10: 109–116. 10.1023/A:1014940800901
Graphodatsky AS, Kukekova AV, Yudkin DV, Trifonov VA, Vorobieva NV, Beklemisheva VR, Perelman PL, Graphodatskaya DA, Trut LN, Yang FT, et al.: The proto-oncogene C-KIT maps to canid B-chromosomes. Chromosome Research 2005, 13: 113–122. 10.1007/s10577-005-7474-9
Yudkin DV, Trifonov VA, Kukekova AV, Vorobieva NV, Rubtsova NV, Yang F, Acland GM, Ferguson-Smith MA, Graphodatsky AS: Mapping of KIT adjacent sequences on canid autosomes and B chromosomes. Cytogenet Genome Res 2007, 116: 100–103. 10.1159/000097424
Trifonov VA, Dement'eva PV, Beklemisheva VR, Iudkin DV, Vorob'eva NV, Grafodatskii AS: [Supernumerary chromosomes, segmental duplications, and evolution]. Genetika 2010, 46: 1234–1236.
Voullaire LE, Slater HR, Petrovic V, Choo KHA: A Functional Marker Centromere with No Detectable Alpha-Satellite, Satellite-Iii, or Cenp-B Protein - Activation of a Latent Centromere. Am J Hum Genet 1993, 52: 1153–1163.
Rocchi M, Stanyon R, Archidiacono N: Evolutionary new centromeres in primates. Prog Mol Subcell Biol 2009, 48: 103–152. 10.1007/978-3-642-00182-6_5
Carbone L, Ventura M, Tempesta S, Rocchi M, Archidiacono N: Evolutionary history of chromosome 10 in primates. Chromosoma 2002, 111: 267–272. 10.1007/s00412-002-0205-5
Montefalcone G, Tempesta S, Rocchi M, Archidiacono N: Centromere repositioning. Genome Research 1999, 9: 1184–1188. 10.1101/gr.9.12.1184
Ventura M, Antonacci F, Cardone MF, Stanyon R, D'Addabbo P, Cellamare A, Sprague LJ, Eichler EE, Archidiacono N, Rocchi M: Evolutionary formation of new centromeres in macaque. Science 2007, 316: 243–246. 10.1126/science.1140615
Carbone L, Nergadze SG, Magnani E, Misceo D, Francesca Cardone M, Roberto R, Bertoni L, Attolini C, Francesca Piras M, de Jong P, et al.: Evolutionary movement of centromeres in horse, donkey, and zebra. Genomics 2006, 87: 777–782. 10.1016/j.ygeno.2005.11.012
Trifonov VA, Kosyakova N, Romanenko SA, Stanyon R, Graphodatsky AS, Liehr T: New insights into the karyotypic evolution in muroid rodents revealed by multicolor banding applying murine probes. Chromosome Res 2010, 18: 265–275. 10.1007/s10577-010-9110-6
Ferreri GC, Liscinsky DM, Mack JA, Eldridge MD, O'Neill RJ: Retention of latent centromeres in the Mammalian genome. J Hered 2005, 96: 217–224. 10.1093/jhered/esi029
Kasai F, Garcia C, Arruga MV, Ferguson-Smith MA: Chromosome homology between chicken (Gallus gallus domesticus) and the red-legged partridge (Alectoris rufa); evidence of the occurrence of a neocentromere during evolution. Cytogenetic and Genome Research 2003, 102: 326–330. 10.1159/000075770
Fronicke L, Wienberg J, Stone G, Adams L, Stanyon R: Towards the delineation of the ancestral eutherian genome organization: comparative genome maps of human and the African elephant (Loxodonta africana) generated by chromosome painting. Proc Biol Sci 2003, 270: 1331–1340. 10.1098/rspb.2003.2383
Yang F, Alkalaeva EZ, Perelman PL, Pardini AT, Harrison WR, O'Brien PCM, Fu B, Graphodatsky AS, Ferguson-Smith MA, Robinson TJ: Reciprocal chromosome painting among human, aardvark, and elephant (superorder Afrotheria) reveals the likely eutherian ancestral karyotype. P Natl Acad Sci USA 2003, 100: 1062–1066. 10.1073/pnas.0335540100
Robinson TJ, Fu B, Ferguson-Smith MA, Yang F: Cross-species chromosome painting in the golden mole and elephant-shrew: support for the mammalian clades Afrotheria and Afroinsectiphillia but not Afroinsectivora. Proc Biol Sci 2004, 271: 1477–1484. 10.1098/rspb.2004.2754
Yang FT, Graphodatsky AS, Li TL, Fu BY, Dobigny G, Wang JH, Perelman PL, Serdukova NA, Su WT, O'Brien PCM, et al.: Comparative genome maps of the pangolin, hedgehog, sloth, anteater and human revealed by cross-species chromosome painting: further insight into the ancestral karyotype and genome evolution of eutherian mammals. Chromosome Research 2006, 14: 283–296. 10.1007/s10577-006-1045-6
Svartman M, Stone G, Stanyon R: The ancestral Eutherian karyotype is present in Xenarthra. Plos Genet 2006, 2: 1006–1011.
Froenicke L, Caldes MG, Graphodatsky A, Muller S, Lyons LA, Robinson TJ, Volleth M, Yang F, Wienberg J: Are molecular cytogenetics and bioinformatics suggesting diverging models of ancestral mammalian genomes? Genome Res 2006, 16: 306–310. 10.1101/gr.3955206
Bourque G, Zdobnov EM, Bork P, Pevzner PA, Tesler G: Comparative architectures of mammalian and chicken genomes reveal highly variable rates of genomic rearrangements across different lineages. Genome Res 2005, 15: 98–110. 10.1101/gr.3002305
Murphy WJ, Larkin DM, Everts-van der Wind A, Bourque G, Tesler G, Auvil L, Beever JE, Chowdhary BP, Galibert F, Gatzke L, et al.: Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science 2005, 309: 613–617. 10.1126/science.1111387
Ma J, Zhang LX, Suh BB, Raney BJ, Burhans RC, Kent WJ, Blanchette M, Haussler D, Miller W: Reconstructing contiguous regions of an ancestral genome. Genome Research 2006, 16: 1557–1565. 10.1101/gr.5383506
Rocchi M, Archidiacono N, Stanyon R: Ancestral genomes reconstruction: An integrated, multi-disciplinary approach is needed. Genome Research 2006, 16: 1441–1444. 10.1101/gr.5687906
Robinson TJ, Ruiz-Herrera A, Castresana J: Is mammalian chromosomal evolution driven by regions of genome fragility? Genome Biology 2006., 7:
Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al.: Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 2007, 317: 86–94. 10.1126/science.1139158
Muller S, Hollatz M, Wienberg J: Chromosomal phylogeny and evolution of gibbons (Hylobatidae). Human Genetics 2003, 113: 493–501. 10.1007/s00439-003-0997-2
Roberto R, Capozzi O, Wilson RK, Mardis ER, Lomiento M, Tuzun E, Cheng Z, Mootnick AR, Archidiacono N, Rocchi M, Eichler EE: Molecular refinement of gibbon genome rearrangements. Genome Res 2007, 17: 249–257. 10.1101/gr.6052507
Misceo D, Capozzi O, Roberto R, Dell'oglio MP, Rocchi M, Stanyon R, Archidiacono N: Tracking the complex flow of chromosome rearrangements from the Hominoidea Ancestor to extant Hylobates and Nomascus Gibbons by high-resolution synteny mapping. Genome Res 2008, 18: 1530–1537. 10.1101/gr.078295.108
Wichman HA, Payne CT, Ryder OA, Hamilton MJ, Maltbie M, Baker RJ: Genomic distribution of heterochromatic sequences in equids: implications to rapid chromosomal evolution. J Hered 1991, 82: 369–377.
Bush GL, Case SM, Wilson AC, Patton JL: Rapid speciation and chromosomal evolution in mammals. Proc Natl Acad Sci USA 1977, 74: 3942–3946. 10.1073/pnas.74.9.3942
Yang F, O'Brien PC, Milne BS, Graphodatsky AS, Solanky N, Trifonov V, Rens W, Sargan D, Ferguson-Smith MA: A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. Genomics 1999, 62: 189–202. 10.1006/geno.1999.5989
Roberto R, Misceo D, D'Addabbo P, Archidiacono N, Rocchi M: Refinement of macaque synteny arrangement with respect to the official rheMac2 macaque sequence assembly. Chromosome Res 2008, 16: 977–985. 10.1007/s10577-008-1255-1
Stanyon R, Yang F, Cavagna P, O'Brien PCM, Bagga M, Ferguson-Smith MA, Wienberg J: Reciprocal chromosome painting shows that genomic rearrangement between rat and mouse proceeds ten times faster than between humans and cats. Cytogenetics and Cell Genetics 1999, 84: 150–155. 10.1159/000015244
Nadeau JH, Taylor BA: Lengths of Chromosomal Segments Conserved since Divergence of Man and Mouse. P Natl Acad Sci-Biol 1984, 81: 814–818. 10.1073/pnas.81.3.814
Romanenko SA, Perelman PL, Serdukova NA, Trifonov VA, Biltueva LS, Wang JH, Li TL, Nie WH, O'Brien PCM, Volobouev VT, et al.: Reciprocal chromosome painting between three laboratory rodent species. Mamm Genome 2006, 17: 1183–1192. 10.1007/s00335-006-0081-z
Romanenko SA, Volobouev VT, Perelman PL, Lebedev VS, Serdukova NA, Trifonov VA, Biltueva LS, Nie W, Brien PCMO, Bulatova NS, et al.: Karyotype evolution and phylogenetic relationships of hamsters (Cricetidae, Muroidea, Rodentia) inferred from chromosomal painting and banding comparison. Chromosome Research 2007, 15: 283–297.
Sitnikova NA, Romanenko SA, O'Brien PC, Perelman PL, Fu B, Rubtsova NV, Serdukova NA, Golenishchev FN, Trifonov VA, Ferguson-Smith MA, et al.: Chromosomal evolution of Arvicolinae (Cricetidae, Rodentia). I. The genome homology of tundra vole, field vole, mouse and golden hamster revealed by comparative chromosome painting. Chromosome Res 2007, 15: 447–456. 10.1007/s10577-007-1137-y
Trifonov V, Karst C, Claussen U, Mrasek K, Michel S, Avner P, Liehr T: Microdissection-derived murine mcb probes from somatic cell hybrids. J Histochem Cytochem 2005, 53: 791–792. 10.1369/jhc.4B6598.2005
Stanyon R, Stone G, Garcia M, Froenicke L: Reciprocal chromosome painting shows that squirrels, unlike murid rodents, have a highly conserved genome organization. Genomics 2003, 82: 245–249. 10.1016/S0888-7543(03)00109-5
Li TL, O'Brien PCM, Biltueva L, Fu BY, Wang JH, Nie WH, Ferguson-Smith MA, Graphodatsky AS, Yang FT: Evolution of genome organizations of squirrels (Sciuridae) revealed by cross-species chromosome painting. Chromosome Research 2004, 12: 317–335.
Graphodatsky AS, Yang F, Dobigny G, Romanenko SA, Biltueva LS, Perelman PL, Beklemisheva VR, Alkalaeva EZ, Serdukova NA, Ferguson-Smith MA, et al.: Tracking genome organization in rodents by Zoo-FISH. Chromosome Research 2008, 16: 261–274. 10.1007/s10577-007-1191-5
Trifonov VA, Stanyon R, Nesterenko AI, Fu B, Perelman PL, O'Brien PC, Stone G, Rubtsova NV, Houck ML, Robinson TJ, et al.: Multidirectional cross-species painting illuminates the history of karyotypic evolution in Perissodactyla. Chromosome Res 2008, 16: 89–107. 10.1007/s10577-007-1201-7
Kulemzina AI, Trifonov VA, Perelman PL, Rubtsova NV, Volobuev V, Ferguson-Smith MA, Stanyon R, Yang F, Graphodatsky AS: Cross-species chromosome painting in Cetartiodactyla: reconstructing the karyotype evolution in key phylogenetic lineages. Chromosome Res 2009, 17: 419–436. 10.1007/s10577-009-9032-3
Volleth M, Heller KG, Pfeiffer RA, Hameister H: A comparative ZOO-FISH analysis in bats elucidates the phylogenetic relationships between Megachiroptera and five microchiropteran families. Chromosome Research 2002, 10: 477–497. 10.1023/A:1020992330679
Ao L, Mao X, Nie W, Gu X, Feng Q, Wang J, Su W, Wang Y, Volleth M, Yang F: Karyotypic evolution and phylogenetic relationships in the order Chiroptera as revealed by G-banding comparison and chromosome painting. Chromosome Res 2007, 15: 257–267.
Stanhope MJ, Waddell VG, Madsen O, de Jong W, Hedges SB, Cleven GC, Kao D, Springer MS: Molecular evidence for multiple origins of Insectivora and for a new order of endemic African insectivore mammals. Proc Natl Acad Sci USA 1998, 95: 9967–9972. 10.1073/pnas.95.17.9967
Springer MS, Cleven GC, Madsen O, de Jong WW, Waddell VG, Amrine HM, Stanhope MJ: Endemic African mammals shake the phylogenetic tree. Nature 1997, 388: 61–64. 10.1038/40386
Pardini AT, O'Brien PC, Fu B, Bonde RK, Elder FF, Ferguson-Smith MA, Yang F, Robinson TJ: Chromosome painting among Proboscidea, Hyracoidea and Sirenia: support for Paenungulata (Afrotheria, Mammalia) but not Tethytheria. Proc Biol Sci 2007, 274: 1333–1340. 10.1098/rspb.2007.0088
Kellogg ME, Burkett S, Dennis TR, Stone G, Gray BA, McGuire PM, Zori RT, Stanyon R: Chromosome painting in the manatee supports Afrotheria and Paenungulata. BMC Evol Biol 2007, 7: 6. 10.1186/1471-2148-7-6
Nie WH, Fu BY, O'Brien PCM, Wang JH, Su WT, Tanomtong A, Volobouev V, Ferguson-Smith MA, Yang F: Flying lemurs - The 'flying tree shrews'? Molecular cytogenetic evidence for a Scandentia-Dermoptera sister clade. Bmc Biol 2008., 6:
Graphodatsky AS, Yang F, Perelman PL, O'Brien PCM, Serdukova NA, Milne BS, Biltueva LS, Fu B, Vorobieva NV, Kawada SI, et al.: Comparative molecular cytogenetic studies in the order Carnivora: mapping chromosomal rearrangements onto the phylogenetic tree. Cytogenetic and Genome Research 2002, 96: 137–145. 10.1159/000063032
Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W: Using genomic data to unravel the root of the placental mammal phylogeny. Genome Research 2007, 17: 413–421. 10.1101/gr.5918807
Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A: The delayed rise of present-day mammals. Nature 2007, 446: 507–512. 10.1038/nature05634
Ensembl Genome Browser [http://www.ensembl.org]
Hameister H, Klett C, Bruch J, Dixkens C, Vogel W, Christensen K: Zoo-FISH analysis: the American mink (Mustela vison) closely resembles the cat karyotype. Chromosome Res 1997, 5: 5–11. 10.1023/A:1018433200553
Acknowledgements
Authors are grateful to F. Yang for Figure 4 and to W. Nie for helpful comments. AG and VT were supported by MCB and SB RAS Programs and research grants of Russian Fund for Basic Research. RS was supported by grants from MUIR (Italian Ministry for Universities and Research).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ASG, VAT and RS wrote and edited the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Graphodatsky, A.S., Trifonov, V.A. & Stanyon, R. The genome diversity and karyotype evolution of mammals. Mol Cytogenet 4, 22 (2011). https://doi.org/10.1186/1755-8166-4-22
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-4-22