
Abstract
Background
Coumarin derivatives exhibit a wide range of biological properties including promising antioxidant activity. Furthermore, microwave-assisted organic synthesis has delivered rapid routes to N- and O-containing heterocycles, including coumarins and thiazoles. Combining these features, the use of microwave-assisted processes will provide rapid access to a targeted coumarin library bearing a hydrazino pharmacophore for evaluation of antioxidant properties
Results
Microwave irradiation promoted 3 of the 4 steps in a rapid, convergent synthesis of a small library of hydrazinyl thiazolyl coumarin derivatives, all of which exhibited significant antioxidant activity comparable to that of the natural antioxidant quercetin, as established by DPPH and ABTS radical assays
Conclusions
Microwave dielectric heating provides a rapid and expedient route to a series of hydrazinyl thiazolyl coumarins to investigate their radical scavenging properties. Given their favourable properties, in comparison with known antioxidants, these coumarin derivatives are promising leads for further development and optimization.

Similar content being viewed by others


Background
The synthesis and biological activities of coumarin derivatives occupy an important position in heterocyclic chemistry as well as in medicinal chemistry. The compounds containing this heterocyclic motif are widely found as additives in food, in cosmetic products, as pharmaceutical agents [1] and as luminescent materials [2]. They have pronounced medicinal value as anticoagulants [3], free radical scavengers [4, 5], and as lipoxygenase [6] and cyclooxygenase inhibitors [7]. Moreover, many coumarins exhibit high antibacterial [8], antifungal [9] and cytotoxic activities [10]. The incorporation of a 3-thiazolyl substituent can further enhance the activity of this pharmacophore: thiazolyl coumarins have been reported to exhibit anticonvulsant [11], anticancer, antimicrobial [12], analgesic and anti-inflammatory properties [13] and display good activity against Mycobacterium tuberculosis[14] and Helicobactor pylori[15]. The pathophysiology of many of the above-mentioned diseases, and others, has been linked with oxidative stress, produced in our body as a result of various oxidation processes essential for life. Although the importance of antioxidants to prevent the progression of age-related diseases, or interfere in the ageing process itself, could be contested [16, 17], their role in enzymatic and non-enzymatic defense mechanisms in both the lipid and aqueous phase is well established [18]. Given that coumarin and its derivatives are well-known as antioxidants [19], we set out to access a series of thiazolyl coumarin derivatives, rapidly and in a convergent manner, to explore their antioxidant properties. Previously we have reported the synthesis and crystalline structure of various thiazolyl coumarin derivatives prepared by conventional methods, including the synthesis of a series of hydrazinyl thiazolyl coumarins [20–24]. Given the versatility and capability of microwave-assisted synthesis for rapid delivery of compounds of biological interest [25–28], our previous success in the use of microwave irradiation for the rapid synthesis of thiazoles of biological interest [29, 30], and the previous application of this technology in the synthesis of coumarins, both on small and large scale [31–34], as well as thiazolyl coumarin Schiff bases [35], we set out to investigate a rapid microwave-assisted synthesis of a hydrazinyl thiazolyl coumarin library for evaluation of in vitro antioxidant activity by DPPH radical and ABTS radical cation assays, using the natural antioxidant quercetin and a synthetic antioxidant (butylatedhydroxytoluene) as reference standards.
Results and discussion
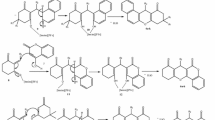
3-(Bromoacetyl)coumarin (4), a key precursor of Hantzsch thiazole synthesis for the hydrazinyl thiazolyl coumarin library, was prepared by a two-step sequence (Scheme 1). Firstly Knoevenagel condensation, with spontaneous α-pyrone 3 formation, was investigated by heterocyclocondensation of salicylaldehyde (1) and ethyl acetoacetate (2) under microwave irradiation using a range of conditions (Table1). It has been reported that the use of microwave heating is beneficial for 3-acetylcoumarin synthesis, allowing for low catalyst loadings and short reaction times to limit the generation of unwanted side products [33, 34]. Although solvent-free conditions have been used for α-pyrone 3 formation under microwave irradiation [31, 32] and conventional heating [36], in the presence of a catalytic amount of piperidine or l-proline, these give rise to widely varying reaction times, operating temperatures and thus chemical yields [31]. In our hands (entry 1), the solvent-free process resulted in a rapid rise of pressure and so was discarded in favour of the more reliable Leadbeater method, carried out in ethanol solvent, which has been reported to proceed in reasonable yield (67–81%) on multigram scale in sealed vessel microwave apparatus [33] and more recently in a large scale batch reactor [34]. In our modified procedure, on 18 mmol scale, with a small excess of carbonyl compound 2 (1.3 equiv), the use of piperidine base (entries 6–8) seemed superior to the l-proline catalyst (entries 2–5) giving 3-acetylcoumarin (3) in 99% isolated yield in a reaction time of only 5 min (entry 8) following purification by recrystallization.

Synthesis of 3-(bromoacetyl)coumarin (4) component for Hantzsch thiazole synthesis.
With a rapid and highly efficient route to acetylcoumarin 3 established, the first of the building blocks for Hantzsch thiazole synthesis, bromoacetylcoumarin 4, was prepared in 68% yield by the electrophilic bromination of acetylcoumarin 3, in CHCl3, according to the method of Gursoy and Karali [14] (Scheme 1). Furthermore the second, thioamide, component was provided by the condensation of thiosemicarbazide 5 and one member of a subset of benzaldehydes 6 or naphthaldehydes 8 in a microwave-assisted condensation. Microwave irradiation of a methanolic solution of a range of these precursors at 120°C for 10 min (Scheme 2) gave thiosemicabazones 7 or 9, respectively, in excellent isolated yield (70–92%) after purification by recrystallization (Table2). The efficiency of the microwave-assisted procedure compared highly favourably, in terms of isolated yield and reaction time, with the condensation carried out using more traditional methods, at reflux in MeOH in the presence of AcOH for 1–4 h, using the same isolation and purification regime.

Synthesis of the thiosemicarbazone (7 or 9) component for Hantzsch thiazole synthesis.
Given that microwave irradiation has been used before in the synthesis of thiazole derivatives of biological interest [29, 30, 37–39] and that, in particular, Hantzsch thiazole synthesis conducted in ethanol was successful in generating thiazoles in good yields for evaluation of antiproliferative activity [40], the Hantzsch synthesis of thiazolyl coumarins 10a-h from corresponding building blocks was investigated under related conditions (Scheme 3). Microwave irradiation of semicarbazone 7 9 and bromoketone 4 in ethanol at 60°C for 10 min (hold time) followed by treatment with ammonium hydroxide (5%) gave coumarin derivatives 10a-h in very good yield (71–80%) (Table3). In all cases, the isolated yield after purification by recrystallization was closely comparable with traditional conductive heating methods (see Table3). The identity and purity was confirmed by analysis of spectroscopic and mass spectrometric data and by comparing the melting point with literature values [20]. The use of microwave irradiation had facilitated and accelerated 3 out of the 4 steps in the convergent synthesis of thiazolyl coumarins 10a-h, including two heterocyclocondensations (α-pyrone formation and Hantzsch thiazole synthesis) and semicabazone formation, to provide an extremely rapid route to a focused library for examination of antioxidant properties.

Hantzsch synthesis of thiazolyl coumarins 10a-h.
Hydrazino-thiazole derivatives have been shown recently to possess radical scavenging ability and some simple structure-activity relationships have been described for a small library of compounds [41]. The antioxidant properties of this motif were evaluated quickly and efficiently using a 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging model [42] in which the decrease in the strong absorption band at ν 517 nm due to the unpaired electron in DPPH decreased stoichiometrically on scavenging an electron or hydrogen atom. Using the DPPH free radical assay, according to a modification of our previously reported procedure [43], the radical scavenging activity of this new series of thiazolyl coumarins 10a-h was determined and expressed as IC50 values [41, 43], which is the concentration of tested compound required to scavenge 50% of the DPPH radical concentration (0.11 mM in this case on dilution). All of the DPPH assays were conducted in triplicate and both a synthetic antioxidant, 2,6-di-tert-butyl-4-methylphenol (BHT), and the natural antioxidant quercetin were used as reference standards. The results from the DPPH radical scavenging assay were validated by 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) (ABTS) assay [42, 44], which established IC50 values for each tested compound to trap the ABTS·+ radical. For consistency, IC50 values in ABTS assay were established in triplicate and compared with quercetin, again, and Trolox, which is a common reference antioxidant for interaction with ABTS·+[42, 44–46]. As expected, DPPH radical assay indicated that radical scavenging activity was dose dependent and increased with the concentration of the tested compound (Figure1). The IC50 values of thiazolyl coumarins 10a-h in the DPPH assay were found to be in the range 16–85 μM (Table4), displaying excellent antioxidant activity above and beyond that of BHT and comparable to that of quercetin (Figure2). It was worthy to note that compounds 10a, 10c, 10d, 10e, 10f and 10h exhibited very high activity against the DPPH radical (at a concentration of 0.11 mM) with IC50 values in the order of 16–30 μM. The activity of this thiazolyl coumarin library compared closely with other known hydrazino-thiazoles (IC50 15–60 μM at a DPPH radical concentration of 0.1 mM) [41] implying that the coumarin and phenolic functions had not adversely affected antioxidant activity. The activity of these compounds was attributed for the most part to the hydrazinothiazole functionality but clearly was modulated and enhanced by the incorporation of other unique groups. The presence of the hydrazino N-H group (Scheme 4) and the phenolic hydroxyl were viewed as important structural features, both having the ability for hydrogen atom transfer (HAT) to the DPPH free radical to give a resonance stabilized radical 11a-f. The mesomeric stabilization of this radical, in particular through the addition of electron donating aromatic units could contribute to radical scavenging ability, with notable improvements observed for 2-hydroxy 10a and 4-hydroxyphenyl 10c analogues over the 3-hydroxy precursor 10b (Table4, entry 2). Alternatively sequential proton loss electron transfer (SPLET) could compete with HAT (Scheme 4c) for phenolic scavengers (ArOH) as has been described for curcumin [42]. In this latter case, synergistic contributions to the activity of phenolic and hydrazino scavengers could not be ruled out but these features were not explored further. The presence of hydroxyl groups in compounds 10a-10f and 10h appeared to contribute to the antioxidant activity of these compounds and when absent resulted in a dramatic loss of radical scavenging ability (Table4, compare lower activity of 10g in entry 7 with entry 8), suggesting the involvement of a SPLET mechanism. The reaction of the DPPH radical with scavengers (Scheme 4a) in general suggests a 1:1 stoichiometry of reaction at high scavenger concentration. When the concentration of the tested compounds is significantly lower than the DPPH radical, as could be the case for compounds with potent IC50 values, the remaining DPPH radical may combine with the hydrazinyl thiazolyl radical 11a-f (Scheme 4b) and thus, the stoichiometry of the reaction could appear higher than 1:1 [41]. Finally, on review it was bromophenyl analogue 10e that exhibited the highest activity in the DPPH assay (Table4, entry 5) even exceeding the activity of the natural antioxidant quercetin (entry 10).

Evaluation of antioxidant properties by DPPH assay.

Comparing antioxidant activities using DPPH and ABTS assays.

The findings from the DPPH assays correlated well with IC50 values derived in ABTS experiments. The ABTS radical was generated by treatment with the strong oxidizing agent K2S2O8 and then reduced by the addition of the antioxidant; this was observed by the suppression of the characteristic long wave absorption of ABTS·+. For the most part, the mean IC50 values were of very similar magnitude with minimal differences between the ABTS and DPPH values (Table4), an observation that has been made before in measuring antioxidant activities of sorghum products [47]. Thus no matter which assay was used, the compounds exhibited the same activity trends and SAR (Figure2), validating the reliability of both techniques. However, it was noted that there appeared to be greater consistency with the DPPH data and greater variability in the ABTS IC50 values according to the observed standard deviations.
Conclusions
The synthesis and antioxidant activity of hydrazinyl thiazolyl coumarin derivatives have been reported. Microwave irradiation promoted 3 out of the 4 steps to establish an extremely rapid, convergent and highly efficient route to the target library. All of the compounds were purified by recrystallization and obtained in good isolated yields, with spectroscopic and physical data that fully supported the proposed structures. DPPH and ABTS assays indicated that almost all of the synthesized hydrazinyl thiazolyl coumarin derivatives (10a, 10c, 10d, 10e, 10f and 10h) had significant radical scavenging activity that was comparable or better than the known antioxidants, quercetin, BHT and Trolox. The thiazole, hydrazino, and phenolic moieties are the likely structural components contributing to free radical scavenging activity. However, the facile incorporation of the coumarin motif offers an opportunity to further optimize radical scavenging activity in the future for hit-to-lead development of new and improved synthetic antioxidants.
Experimental
Preparation of 3-acetyl-2H-chromen-2-one (3) by knoevenagel condensation
Piperidine (0.1 mL) was added dropwise to a mixture of salicylaldehyde (2.0 mL, 18 mmol) and ethyl acetoacetate (3.0 mL, 24 mmol) in EtOH (1.0 mL) The reaction mixture was stirred under microwave irradiation for 5 min (hold time) at 50°C (initial power 20 W) and then was cooled in a stream of compressed air, resulting in a yellow solid. Purification by recrystallization (EtOH) gave the title compound 3 (3.3 g, 99%) as fine yellow needles, mp 118–119°C (Lit. mp 119–121°C [48]); IR (KBr) νmax 2930 (C-H aliphatic), 1742 (C = O), 1677 (O-C = O); 1H NMR (500 MHz, DMSO-d 6 ) δ 8.58 (1H, s), 7.89 (1H, dd, J = 7.6, 1.6 Hz), 7.70 (1H, ddd, J = 8.1, 7.6, 1.6 Hz), 7.40 (1H, d, J = 8.1 Hz), 7.38 (1H, td, J =7.6, 1 Hz), 2.55 (3H, s).
Alternatively l-proline (0.18 g, 16 mmol) was added to a mixture of salicylaldehyde (2.0 mL, 18 mmol) and ethyl acetoacetate (3.0 mL, 24 mmol) in EtOH (1.0 mL). The reaction mixture was stirred under microwave radiation for 1–10 min (hold time) at 50 or 120°C, resulting in the formation of a yellow solid. Purification by recrystallization (EtOH) gave the title compound 3 (2.8 g, 93%) (Table2, entry 4) as fine yellow needles, mp 117–118°C (Lit. mp 119–121°C [48]) with identical spectroscopic properties.
Bromination of acetylcoumarins
3-Acetyl-2H-chromen-2-one (3) (20 g, 0.11 mol), obtained from the combined product of a series of parallel experiments, was dissolved in alcohol-free CHCl3 (20 mL) and a solution of Br2 (5.45 mL, 0.11 mol) in CHCl3 (20 mL) was added dropwise from a dropping funnel with constant stirring at 0–5°C. After 3 h, a dark yellow solid separated. The reaction mixture was heated for 15 min at reflux, cooled and CHCl3 was removed using a rotary evaporator. Purification by recrystallization (glacial AcOH) gave 3-bromoacetylcoumarin 4 (19 g, 68%) as off-white needles, mp 162–165°C (Lit. mp 160–163°C [11]); IR (KBr) νmax 2930 (C-H aliphatic), 1731 (C = O), 1686 (O-C = O); 1H NMR (400 MHz, CDCl3) δ 8.66 (1H, s), 7.69–7.75 (2H, m), 7.39–7.44 (2H, m), 4.79 (2H, s).
General procedure for the preparation of thiosemicarbazones 7a-f or 9a,b from aldehydes 6a-f or 8a,b, respectively
Thiosemicarbazide (5) (0.46 g, 5.00 mmol) was added slowly to a stirred solution of salicylaldehyde (0.11 mL, 5.00 mmol) in hot absolute EtOH (5 mL). The resulting solution was heated at 120°C for 10 min (hold time) under microwave irradiation. The mixture was cooled in a stream of compressed air, then allowed to cool further in an ice bath for 30 min to give a colourless precipitate, which was filtered and washed with cold water. Purification by recrystallization (95% EtOH) gave (E)-2-(2-hydroxybenzylidene)hydrazinecarbothioamide (7a). A similar procedure was employed for the preparation of the other 2-(benzylidene)hydrazinecarbothioamides 7b-f.
( E )-2-(2-Hydroxybenzylidene)hydrazinecarbothioamide (7a) (0.68 g, 70%) was obtained as colourless crystals, mp 208–210°C (Lit. mp 210°C [49]); IR (KBr) νmax 3462, 3415 (NH2), 3374 (OH), 3215 (NH), 1610 (C = N), 1230 (C = S); with identical spectroscopic properties. ( E )-2-(3-Hydroxybenzylidene)hydrazinecarbothioamide (7b) (0.80 g, 82%) was obtained as light brown crystals, mp 166–168°C (Lit. mp 168–170°C [50]); IR (KBr) νmax 3458, 3409 (NH2), 3365 (OH), 3260 (NH), 1614 (C = N), 1225 (C = S); with identical spectroscopic properties. ( E )-2-(4-Hydroxybenzylidene)hydrazinecarbothioamide (7c) (0.69 g, 71%) was obtained as colourless crystals, mp 214–216°C (Lit. mp 218–219°C [46]); IR (KBr) νmax 3469, 3414 (NH2), 3377 (OH), 3235 (NH), 1610 (C = N), 1231 (C = S); with identical spectroscopic properties. ( E )-2-(5-Bromo-2-hydroxybenzylidene)hydrazinecarbothioamide (7e) (1.10 g, 78%) was obtained as a beige solid, mp 234–236°C (Lit. mp 238.5°C [51]); IR (KBr) νmax 3543, 3429 (NH2), 3317 (OH), 3253 (NH), 1612 (C = N), 1217 (C = S); with identical spectroscopic properties. ( E )-2-(2-Hydroxy-3-methoxybenzylidene)hydrazinecarbothioamide (7f) (0.80 g, 72%) was obtained as a colourless solid, mp 222–224°C (Lit. mp 220–222°C [52]); IR (KBr) νmax 3458, 3424 (NH2), 3342 (OH), 3164 (NH), 1594 (C = N), 1140 (C = S); with identical spectroscopic properties.
General procedure for the synthesis of 2-(naphthalen-1-ylmethylene)hydrazinecarbothioamides 9
The experimental procedure employed for the synthesis of (E)-2-(2-hydroxybenzylidene)hydrazinecarbothioamide (7a) was employed using naphthaldehyde 8 (5.0 mmol), thiosemicarbazide (5) (5.0 mmol) and absolute EtOH. Purification by recrystallization, using EtOH–EtOAc (1:2), gave the target compound (9a,b). ( E )-2-(Naphthalen-1-ylmethylene)hydrazinecarbothioamide (9a) (1.05 g, 92%) was obtained as a yellow solid, mp 138–140°C (Lit. mp 126°C [53]); IR (KBr) νmax 3448, 3412 (NH2), 3228 (NH), 1592 (C = N), 1249 (C = S); with identical spectroscopic properties. ( E )-2-[(2-Hydroxynaphthalen-1-yl)methylene]hydrazinecarbothioamide (9b) (0.88 g, 72%) was obtained as a pink-yellow solid, mp 272–274°C (Lit. mp 271°C [54]); IR (KBr) νmax 3550, 3426 (NH2), 3236 (NH), 3148 (OH), 1589 (C = N), 1226 (C = S); with identical spectroscopic properties.
Synthesis of hydrazinyl thiazolyl coumarin derivatives 10a-f from bezylidenethiosemicabazides 7a-f
A stirred solution of 3-bromoacetylcoumarin (4) (107 mg, 0.40 mmol) and benzylidenethiosemicarbazone 7a-f (78 mg, 0.40 mmol) in EtOH was heated at 60°C for 10 min (hold time) under microwave irradiation and then cooled in a stream of compressed air to give a thick yellow precipitate. The reaction mixture was neutralized with aqueous ammonium hydroxide solution (5%) and the precipitated solid was filtered. Purification by recrystallization, using CHCl3–EtOH (1:3), gave (E)-3-(2-(2-(2-hydroxybenzylidene)hydrazinyl)thiazol-4-yl)-2H-chromen-2-one (10a). A similar procedure was employed to prepare the target compounds 10b-h, after purification by recrystallization using CHCl3, EtOH or EtOAc–EtOH.
( E )-3-{2-[2-(2-Hydroxybenzylidene)hydrazinyl]thiazol-4-yl}-2 H -chromen-2-one (10a)
The title compound (10a) (107 mg, 74%) was obtained after purification by recrystallization, using CHCl3–EtOH (1:3), as a yellow solid, mp 270–272°C (Lit. mp 270–272°C [20]); IR (KBr) νmax 3420 (NH), 3212 (OH), 1699 (O-C = O), 1603 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.30 (1H, br s), 10.41 (1H, br s), 8.55 (1H, s), 8.29 (1H, s), 7.87 (1H, dd, J = 7.5, 1.2 Hz), 7.77 (1H, s), 7.65 (1H, dd, J = 8.0, 1.1 Hz), 7.64 (1H, ddd, J = 8.2, 7.5, 1.2 Hz), 7.47 (1H, d, J = 8.2 Hz), 7.40 (1H, t, J = 7.5 Hz), 7.37 (1H, td, J = 8.0, 1.1 Hz), 6.91 (1H, d, J = 8.0 Hz), 6.89 (1H, t, J = 8.0 Hz).
( E )-3-{2-[2-(3-Hydroxybenzylidene)hydrazinyl]thiazol-4-yl}-2 H -chromen-2-one (10b)
The title compound (10b) (116 mg, 80%) was obtained after purification by recrystallization, using EtOAc–EtOH (1:2), as bright yellow crystals, mp 253–254°C (Lit. mp 254–256°C [20]); IR (KBr) νmax 3365 (NH), 3262 (OH), 1698 (O-C = O), 1602 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.18 (1H, br s), 9.63 (1H, br s), 8.55 (1H, s), 7.98 (1H, s), 7.87 (1H, dd, J = 7.6, 0.9 Hz), 7.78 (1H, s), 7.64 (1H, ddd, J = 8.2, 7.6, 0.9 Hz), 7.46 (1H, d, J = 8.2 Hz), 7.40 (1H, t, J = 7.6 Hz), 7.23 (1H, t, J = 7.8 Hz), 7.12 (1H, s), 7.05 (1H, d, J = 7.8 Hz), 6.80 (1H, dd, J = 7.8, 2.4 Hz).
( E )-3-{2-[2-(4-Hydroxybenzylidene)hydrazinyl]thiazol-4-yl}-2 H -chromen-2-one (10c)
The title compound (10c) (103 mg, 71%) was obtained after purification by recrystallization, using CHCl3–EtOH (1:3), as brown crystals, mp: 249–251°C (Lit. mp 249–250°C [20]); IR (KBr) νmax 3424 (NH), 3212 (OH), 1706 (O-C = O), 1605 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.05 (1H, br s), 9.93 (1H, br s), 8.60 (1H, s), 7.93 (1H, s), 7.81 (1H, dd, J = 7.6, 1.0 Hz), 7.72 (1H, s), 7.67 (1H, ddd, J = 8.2, 7.6, 1.0 Hz), 7.55 (2H, d, J = 8.6 Hz), 7.48 (1H, d, J = 8.2 Hz), 7.44 (1H, t, J = 7.6 Hz), 6.88 (2H, d, J = 8.6 Hz).
( E )-3-{2-[2-(2-Hydroxy-5-bromobenzylidene)hydrazinyl]thiazol-4-yl)-2 H -chromen-2-one (10e)
The title compound (10e) (127 mg, 72%) was obtained after purification by recrystallization, using EtOAc–EtOH (3:1), as a yellow solid, mp: 295–296°C (Lit. mp 296–298°C [20]); IR (KBr) νmax 3430 (NH), 3260 (OH), 1701 (O-C = O), 1583 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.30 (1H, br s), 10.45 (1H, br s), 8.57 (1H, s), 8.32 (1H, s), 7.88 (1H, dd, J = 7.5, 1.1 Hz), 7.81 (1H, s), 7.79 (1H, d, J = 2.5 Hz), 7.68 (1H, ddd, J = 8.3, 7.5, 1.1 Hz), 7.50 (1H, d, J = 8.3 Hz), 7.44 (1H, t, J = 7.5 Hz), 7.38 (1H, dd, J = 8.5, 2.5 Hz), 6.87 (1H, d, J = 8.5 Hz).
( E )-3-{2-[2-(2-Hydroxy-3-methoxybenzylidene)hydrazinyl]thiazol-4-yl}-2 H -chromen-2-one (10f)
The title compound (10f) (113 mg, 72%) was obtained after purification by recrystallization, using EtOAc–EtOH (2:1), as a brown solid, mp 266–267°C (Lit. mp 266°C [20]); IR (KBr) νmax 3445 (NH), 3239 (OH), 1704 (O-C = O), 1600 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.18 (1H, br s), 9.45 (1H, br s), 8.56 (1H, s), 8.39 (1H, s), 7.87 (1H, dd, J = 7.5, 0.9 Hz), 7.78 (1H, s), 7.62 (1H, ddd, J = 8.3, 7.5, 0.9 Hz), 7.46 (1H, d, J = 8.3 Hz), 7.41 (1H, t, J = 7.5 Hz), 7.27 (1H, d, J = 8.0 Hz), 6.99 (1H, d, J = 8.0 Hz), 6.84 (1H, t, J = 8.0 Hz), 3.83 (3H, s).
Synthesis of hydrazinyl thiazolyl coumarin derivatives 10g,h from (naphthalenylmethylene)thiosemicabazides 9a,b
The title compound was synthesized by a similar procedure described for the synthesis of 10a-f, using 3-bromoacetylcoumarin (4) with naphthalenylmethylene)thiosemicabazide 9a,b.
( E )-3-{2-[2-(1-Naphthylidene)hydrazinyl]thiazol-4-yl}-2 H -chromen-2-one (10g)
The title compound (10g) (114 mg, 71%) was obtained after purification by recrystallization, using EtOAc–EtOH (3:1), as a yellow solid, mp 262–264°C (Lit. mp 262–265°C [20]; 240–242°C [15]); IR (KBr) νmax 3424 (NH), 1702 (O-C = O), 1594 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.35 (1H, br s), 8.79 (1H, d, J = 8.5 Hz), 8.71 (1H, s), 8.59 (1H, s), 8.01 (2H, t, J = 7.8 Hz), 7.88 (2H, d, J = 7.6 Hz), 7.83 (1H, s), 7.67 (1H, ddd, J = 8.1, 7.5, 0.9 Hz), 7.63 (1H, t, J = 8.5 Hz), 7.62 (2H, dd, J = 7.8, 1.8 Hz), 7.49 (1H, d, J = 8.1 Hz), 7.41 (1H, t, J = 7.5 Hz).
( E )-3-{2-[2-(2-Hydroxynaphthylidene)hydrazinyl]thiazol-4-yl}-2H-chromen-2-one (10h)
The title compound (10h) (119 mg, 79%) was obtained after purification by recrystallization, using EtOAc–EtOH (2:1), as a brown solid, mp 271–273°C, (Lit. mp 272–274°C [20]); IR (KBr) νmax 3439 (NH), 3207 (NH), 1705 (O-C = O), 1602 (C = N); 1H NMR (400 MHz, DMSO-d 6 ) δ 12.25 (1H, br s), 10.92 (1H, br s), 8.99 (1H, s), 8.78 (1H, d, J = 8.6 Hz), 8.59 (1H, s), 7.90-7.81 (3H, m), 7.82 (1H, s), 7.63 (1H, ddd, J = 8.2, 7.5, 1.5 Hz), 7.58 (1H, t, J = 7.5 Hz), 7.48 (1H, d, J = 8.2 Hz), 7.42 (2H, ddd, J = 8.2, 7.2, 4.0 Hz), 7.24 (1H, d, J = 8.4 Hz).
Methods
1H and 13C NMR spectra were obtained using d 6 -dimethyl sulfoxide at 25°C using a Bruker DPX 400 instrument or 500 Avance instrument operating at 400 or 500 MHz for 1H spectra and 100 MHz for 13 C spectra, unless stated otherwise, and were reported in ppm; J values were recorded in Hz and multiplicities were expressed by the usual conventions (s = singlet, d = doublet, t = triplet, app = apparent, m = multiplet). Infra-red (IR) spectra were recorded in the range 4000–600 cm-1 on a Perkin-Elmer 1600 series FTIR spectrometer using KBr disks and are reported in cm-1. All compounds were examined by analytical thin layer chromatography carried out using aluminium-backed plates coated with Merck Kieselgel 60 GF254, eluting with hexane–ethyl acetate (3:1, v/v), that were visualised under UV light (at 254 and/or 360 nm). Microwave-assisted syntheses were carried out at the recorded temperature by the modulation of the initial magnetron power (given in parentheses) in a sealed tube using a CEM Discover single-mode instrument, with magnetic stirring and temperature measurement using the in-built IR sensor. Melting points (mp) were determined on a Kofler hot stage apparatus and are uncorrected. Commercially available reagents were used without further purification; solvents were dried by standard procedures.
DPPH radical-scavenging activity
DPPH radical-scavenging activity of the samples was assessed by our reported method [43] with minor modifications. The 1,1-diphenyl-2-picrylhydrazyl radical solution was prepared by dissolving an appropriate amount of DPPH in MeOH to give a concentration of 1 mM. The DPPH radical solution (1 mM; 0.5 mL) was added to a solution of the compound to be tested in MeOH (4 mL) at various concentrations (to give a final concentration of DPPH of 0.11 mM). The mixture was shaken vigorously and incubated at room temperature in the dark for 30 min. The decrease in the absorbance of the resulting solution was then measured spectrophotometrically at ν 517 nm. All measurements were made in triplicate. Two controls were used for this test: a negative control (blank) consisting of MeOH (4 mL) and the DPPH radical solution (0.5 mL) and a positive control comprising the reference anti-oxidant (quercetin or BHT) in MeOH and DPPH radical solution. Inhibition of free radical DPPH in percentage was calculated as follows:
Where A blank is the absorbance of negative control (containing all reagents except test compounds) and A sample is the absorbance of the test compounds and all the reagents. Sample concentration providing 50% inhibition (IC50) was calculated by plotting the inhibition percentage against sample concentration. All the synthesized compounds were evaluated for DPPH radical scavenging ability and the antioxidant activity of the synthesized compounds was compared with a synthetic antioxidant BHT (butylated hydroxytoluene) and a natural antioxidant quercetin, as the reference standards.
ABTS.+radical cation-scavenging activity
According to a modified method from Arnao et al. [45], a solution of ABTS (7.4 mM) in MeOH and a solution of potassium persulphate (2.6 mM) in MeOH were mixed in equal volumes and allowed to react for 12 hours in the dark at room temperature. The resulting solution (1.0 mL) was diluted (to a volume of 30.0 mL) by the addition of methanol to give an ABTS·+ concentration of 0.12 mM and absorbance of 1.1 ± 0.02 at 734 nm. A portion of the ABTS·+ solution (3.0 mL) was added to a methanolic solution (150 μL) of the compound to be tested at various concentrations and the resulting mixture was incubated in the dark for 2 hours. The absorbance of each solution was recorded at 734 nm. All measurements were made in triplicate and for each assay a fresh ABTS·+ stock solution was prepared. Radical scavenging activity was calculated in a similar fashion to the DPPH radical-scavenging assays. The IC50 value of each compound was calculated by plotting the inhibition percentage against concentration of the tested compounds and the results were expressed in μM.
References
O’Kennedy R, Thornes RD: Coumarins: biology, applications, and mode of action. John Wiley & Sons, Chichester; New York, 1997
Bullock SJ, Felton CE, Fennessy RV, Harding LP, Andrews M, Pope SJA, Rice CR, Riis-Johannessen T: Coumarin-based luminescent ligand that forms helicates with dicationic metal ions. Dalton Trans. 2009, 47: 10570-10573.
Ghazaryan A, Khatchadouria A, Karagyozyan M, Kachatryan A, Sekoyan E, Bdoyan H, Melkumyan H, Karageuzyan K: Peculiarities of the anticoagulant properties for the newly synthesized preparations of coumarin related compounds. Cardiovasc Hematol Disord Drug Targets. 2007, 7: 170-173. 10.2174/187152907781745233.
Kancheva VD, Boranova PV, Nechev JT, Manolov II: Structure-activity relationships of new 4-hydroxy bis-coumarins as radical scavangers and chain breaking antioxidants. Biochimie. 2010, 92: 1138-1146. 10.1016/j.biochi.2010.02.033.
Morabito GDT, Singh B, Prasad AK, Parmar VS, Naccari FM, Saija A, Cristani M, Firuzi O, Saso L: Antioxidant properties of 4-methyl coumarins in in vitro cell-free systems. Biochimie. 2010, 92: 1107-1117.
Grimm E, Brideau C, Chauret N, Chan C, Delorme D, Ducharme Y, Ethier D, Falgueyret J, Friesen R, Guay J: Substituted coumarins as potent 5-lipoxygenase inhibitors. Bioorg Med Chem Lett. 2006, 16: 2528-2531. 10.1016/j.bmcl.2006.01.085.
Marion R, Alois S, Zhong-liang C, Rudolf B: 5-Lipoxygenase and cyclooxygenase inhibitory active compounds from Atractylodes lancea. J Nat Prod. 1998, 61: 347-350. 10.1021/np970430b.
Lafitte D, Lamour V, Tsvetkov P, Makarov A, Klich M, Deprez P, Moras D, Briand C, Gilli R: DNA gyrase interaction with coumarin-based inhibitors: the role of the hydroxybenzoate isopentenyl moiety and the 5′-methyl group of the noviose†. Biochemistry. 2002, 41: 7217-7223. 10.1021/bi0159837.
Montagnev C, de Souza S, Groposoa C, Delle Monache F, Smania E, Smania A: Antifungal activity of coumarins. J Biosci. 2008, 63: 21-28.
Kostova I: Synthetic and natural coumarins as cytotoxic agents. Curr Med Chem Anti Canc Agents. 2005, 5: 29-46. 10.2174/1568011053352550.
Siddiqui N, Arshad M, Khan S: Synthesis of some new coumarin incorporated thiazolyl semicarbazones as anticonvulsants. Acta Pol Pharm Drug Res. 2009, 66: 161-167.
Kamal A, Adil S, Tamboli J, Siddardha B, Murthy U: Synthesis of coumarin linked naphthalimide conjugates as potential anticancer and antimicrobial agents. Lett Drug Des Discov. 2009, 6: 201-209. 10.2174/157018009787847855.
Kalkhambkar R, Kulkarni G, Shivkumar H, Rao R: Synthesis of novel triheterocyclic thiazoles as anti-inflammatory and analgesic agents. Eur J Med Chem. 2007, 42: 1272-1276. 10.1016/j.ejmech.2007.01.023.
Gursoy A, Karali N: Synthesis, characterization and primary antituberculosis activity evaluation of 4-(3-coumarinyl)-3-benzyl-4-thiazolin-2-one benzylidenehydrazones. Turk J Chem. 2003, 27: 545-552.
Chimenti F, Bizzarri B, Bolasco A, Secci D, Chimenti P, Granese A, Carradori S, D’Ascenzio M, Scaltrito MM, Sisto F: Synthesis and anti Helicobacter pylori activity of 4-(coumarin-3-yl)-thiazol-2-ylhydrazone derivatives. J Heterocycl Chem. 2010, 47: 1269-1274. 10.1002/jhet.464.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J: Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007, 39: 44-84. 10.1016/j.biocel.2006.07.001.
Rhee SG: H2O2, a necessary evil for cell signalling. Science. 2006, 312: 1882-1883. 10.1126/science.1130481.
Sies H: Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997, 82: 291-295.
Fylaktakidou K, Hadjipavlou-Litina D, Litinas K, Nicolaides D: Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr Pharm Des. 2004, 10: 3813-3833. 10.2174/1381612043382710.
Arshad A, Osman H, Bagley MC, Chan K, Mohamad S, Zahariluddin ASM: Synthesis and antimicrobial properties of some new thiazolyl coumarin derivatives. Eur J Med Chem. 2011, 46: 3788-3794. 10.1016/j.ejmech.2011.05.044.
Arshad A, Osman H, Chan K, Yeap C, Fun HK: 3-{2-[2-(Diphenylmethylene) hydrazinyl] thiazol-4-yl}-2H-chromen-2-one. Acta Crystallogr Sect E: Struct Rep Online. 2010, 66: o1788-o1789. 10.1107/S1600536810023627.
Arshad A, Osman H, Chan K, Goh JH, Fun HK: 3-{2-[2-(3-Hydroxybenzylidene)-hydrazin-1-yl]-1,3-thiazol-4-yl}-2H-chromen-2-one hemihydrate. Acta Crystallogr Sect E: Struct Rep Online. 2010, 66: o1498-o1499. 10.1107/S1600536810019653.
Arshad A, Osman H, Chan K, Hemamalini M, Fun HK: (E)-3-(2-{2-[1-(3-Hydroxyphenyl)ethylidene]hydrazinyl}-1, 3-thiazol-4-yl)-2H-chromen-2-one. Acta Crystallogr Sect E: Struct Rep Online. 2011, 67: o1072-o1073. 10.1107/S1600536811012189.
Arshad A, Osman H, Chan K, Hemamalini M, Fun HK: (E)-6-Bromo-3-(2-(2-(2-chlorobenzylidene)hydrazinyl)thiazol-5-yl)-2H-chromen-2-one dimethylsulfoxide monosolvate. Acta Crystallogr Sect E: Struct Rep Online. 2011, 67: o1009-o1010. 10.1107/S1600536811011160.
Kappe CO, Stadler A: Microwaves in Organic and Medicinal Chemistry. 2005, Wiley-VCH: Weinheim
Kappe CO, Dallinger D: The impact of microwave synthesis on drug discovery. Nat Rev Drug Discov. 2006, 5: 51-63. 10.1038/nrd1926.
Alcázar J, Oehlrich D: Recent applications of microwave irradiation to medicinal chemistry. Future Med Chem. 2010, 2: 169-176. 10.4155/fmc.09.144.
Collins JM, Leadbeater NE: Microwave energy: a versatile tool for the biosciences. Org Biomol Chem. 2007, 5: 1141-1150. 10.1039/b617084f.
Merritt E, Bagley MC: Holzapfel−Meyers−Nicolaou modification of the Hantzsch thiazole synthesis. Synthesis. 2007, 22: 3535-3541.
Merritt EA, Bagley MC: Synthesis of the central heterocyclic domain of micrococcin P1. Synlett. 2007, 6: 954-958.
Bogdal D: Coumarins: fast synthesis by Knoevenagal condensation under microwave irradiation. J Chem Res-S. 1998, 468-469.
Ajani OO, Nwinyi OC: Microwave-assisted synthesis and evaluation of antimicrobial activity of 3-{3-(s-aryl and s-heteroaromatic)acryloyl}-2H-chromen-2-one derivatives. J Heterocyclic Chem. 2010, 47: 179-187.
Bowman MD, Schmink JR, McGowan CM, Kormos CM, Leadbeater NE: Scale-up of microwave-promoted reactions to the multigram level using a sealed-vessel microwave apparatus. Org Process Res Dev. 2008, 12: 1078-1088. 10.1021/op8001239.
Schmink JR, Kormos CM, Devine WG, Leadbeater NE: Exploring the scope for scale-up of organic chemistry using a large batch microwave reactor. Org Process Res Dev. 2010, 14: 205-214. 10.1021/op900287j.
Venogopala KN, Jayashree BS: Microwave-induced synthesis of Schiff bases of aminothiazolyl bromocoumarins as antibacterials. Indian J Pharm Sci. 2008, 70: 88-91. 10.4103/0250-474X.40338.
Karade NN, Gampawar SV, Shinde SV, Jadhav WN: L-Proline catalyzed solvent-free knoevenagel condensation for the synthesis of 3-substituted coumarins. Chin J Chem. 2007, 25: 1686-1689. 10.1002/cjoc.200790311.
Gaikwad SA, Patil AA, Deshmukh MB: An efficient, uncatalyzed, and rapid synthesis of thiazoles and aminothiazoles under microwave irradiation and investigation of their biological activity. Phosphorus Sulfur. 2010, 185: 103-109. 10.1080/10426500802715163.
Hughes RA, Thompson SP, Alcaraz L, Moody CJ: Total synthesis of the thiopeptide antibiotic amythiamicin D. J Am Chem Soc. 2005, 127: 15644-15651. 10.1021/ja0547937.
Kiryanov AA, Sampson P, Seed AJ: Synthesis of 2-alkoxy-substituted thiophenes, 1,3-thiazoles, and related S-heterocycles via Lawesson’s reagent-mediated cyclization under microwave irradiation: applications for liquid crystal synthesis. J Org Chem. 2001, 66: 7925-7929. 10.1021/jo016063x.
Romagnoli R, Baraldi PG, Lopez OC, Carrion MD, Cara CL, Preti D, Tabrizi MA, Balzarini J: Microwave-assisted synthesis of substituted 2,4-diarylthiazoles and their evaluation as anticancer agents. Lett Drug Des Discov. 2007, 4: 464-466. 10.2174/157018007781788507.
Shih M, Su Y, Wu C: Syntheses of aromatic substituted hydrazino-thiazole derivatives to clarify structural characterization and antioxidant activity between 3-arylsydnonyl and aryl substituted hydrazino-thiazoles. Chem Pharm Bull. 2007, 55: 1126-1135. 10.1248/cpb.55.1126.
Liu Z-Q: Chemical methods to evaluate antioxidant ability. Chem Rev. 2010, 110: 5675-5691. 10.1021/cr900302x.
Rahim A, Rocca E, Steinmetz J, Kassim MJ, Ibrahim MS, Osman H: Antioxidant activity of mangrove Rhizophora apiculata bark extracts. Food Chem. 2008, 107: 200-207. 10.1016/j.foodchem.2007.08.005.
Huang D, Ou B, Prior RL: The chemistry behind antioxidant capacity assays. J Agric Food Chem. 2005, 53: 1841-1856. 10.1021/jf030723c.
Arnao MB, Cano A, Acosta M: The hydrophilic and lipophilic contribution to total antioxidant activity. Food Chem. 2001, 73: 239-244. 10.1016/S0308-8146(00)00324-1.
Arts MJTJ, Haenen GRMM, Voss H-P, Bast A: Antioxidant capacity of reaction products limits the applicability of the Trolox Equivalent Antioxidant Capacity (TEAC) assay. Food Chem Toxicol. 2004, 42: 45-49. 10.1016/j.fct.2003.08.004.
Awika JM, Rooney LW, Wu X, Prior RL, Cisneros-Zevallos L: Screening methods to measure antioxidant activity of sorghum (Sorghum bicolor) and Sorghum products. J Agric Food Chem. 2003, 51: 6657-6662. 10.1021/jf034790i.
Valizadeh H, Gholipur H, Shockravi A: Microwave assisted synthesis of coumarins via potassium carbonate catalyzed knoevenagel condensation in 1 n butyl 3 methylimidazolium bromide ionic liquid. J Heterocyclic Chem. 2007, 44: 867-870. 10.1002/jhet.5570440419.
Kostas ID, Andreadaki FJ, Kovala-Demertzi D: Suzuki-Miyaura cross-coupling reaction of aryl bromides and chlorides with phenylboronic acid under aerobic conditions catalyzed by palladium complexes with thiosemicarbazone ligands. Tetrahedron Lett. 2005, 46: 1967-1970. 10.1016/j.tetlet.2005.02.003.
Yi W, Cao RH, Chen ZY, Yu L, Ma L, Song HC: Design, synthesis and biological evaluation of hydroxy-or methoxy-substituted phenylmethylenethiosemicarbazones as tyrosinase inhibitors. Chem Pharm Bull. 2009, 57: 1273-1277. 10.1248/cpb.57.1273.
Güveli S, Bal-Demirci T, Özdemir N, Ülküseven B: Nickel(II) complexes of ONS and ONN chelating thiosemicarbazones with triphenylphosphine co-ligands. Transition Met Chem. 2009, 34: 383-388. 10.1007/s11243-009-9206-z.
Kumar AP, Reddy KPPRM, Reddy TV, Reddy PR, Reddy VK: Synthesis and characterisation of 2-hydroxy-3-methoxy benzaldehyde thiosemicarbazone (HMBATSC) and its application of simultaneous second order derivative spectrophotometric method for determination of cobalt(II), nickel(II) and vanadium(V). Main Group Chem. 2006, 5: 141-151. 10.1080/13547500601100201.
Jensen KA, Jensen CL: Thiohydrazides and thiohydrazones: a new class of antibacterial substances. Acta Chem Scand. 1952, 6: 957-958.
Yilmaz I, Çukurovali A: Cobalt(II), copper(II), nickel(II), and zinc(II) complexes of naphthaldehyde thiazolyl hydrazones. Pol J Chem. 2004, 78: 663-672.
Acknowledgements
This work was supported by the Universiti Sains Malaysia (USM) for a fellowship award to HO, the Universiti Sains Malaysia fellowship for financial support of AA, and grants from the SPARC initiative (award to MCB) and RU grant [1001/PKimia/811133] to conduct antioxidant research work (award to AA).
Author details
1School of Chemical Sciences, Universiti Sains Malaysia, 11800, Penang, Malaysia. 2School of Pharmaceutical Sciences, Universiti Sains Malaysia, 11800, Penang, Malaysia. 3Department of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HO and AA participated in study design and coordination, manuscript preparation and carried out the synthetic experiments, MCB and KCL participated in study design and coordination and manuscript preparation. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Osman, H., Arshad, A., Lam, C.K. et al. Microwave-assisted synthesis and antioxidant properties of hydrazinyl thiazolyl coumarin derivatives. Chemistry Central Journal 6, 32 (2012). https://doi.org/10.1186/1752-153X-6-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-6-32