
Abstract
Nanotechnology in its widest sense seeks to exploit the special biophysical and chemical properties of materials at the nanoscale. While the potential technological, diagnostic or therapeutic applications are promising there is a growing body of evidence that the special technological features of nanoparticulate material are associated with biological effects formerly not attributed to the same materials at a larger particle scale. Therefore, studies that address the potential hazards of nanoparticles on biological systems including human health are required. Due to its large surface area the lung is one of the major sites of interaction with inhaled nanoparticles. One of the great challenges of studying particle-lung interactions is the microscopic visualization of nanoparticles within tissues or single cells both in vivo and in vitro. Once a certain type of nanoparticle can be identified unambiguously using microscopic methods it is desirable to quantify the particle distribution within a cell, an organ or the whole organism. Transmission electron microscopy provides an ideal tool to perform qualitative and quantitative analyses of particle-related structural changes of the respiratory tract, to reveal the localization of nanoparticles within tissues and cells and to investigate the 3D nature of nanoparticle-lung interactions.
This article provides information on the applicability, advantages and disadvantages of electron microscopic preparation techniques and several advanced transmission electron microscopic methods including conventional, immuno and energy-filtered electron microscopy as well as electron tomography for the visualization of both model nanoparticles (e.g. polystyrene) and technologically relevant nanoparticles (e.g. titanium dioxide). Furthermore, we highlight possibilities to combine light and electron microscopic techniques in a correlative approach. Finally, we demonstrate a formal quantitative, i.e. stereological approach to analyze the distributions of nanoparticles in tissues and cells.
This comprehensive article aims to provide a basis for scientists in nanoparticle research to integrate electron microscopic analyses into their study design and to select the appropriate microscopic strategy.
Similar content being viewed by others


1. Introduction
Each day a human inhales and exhales more than 10,000 litres of air. With an epithelial surface area of approximately 140 m2 [1], the internal surface of the human lungs is destined to interact with an enormous number of airborne particles with each breath. After inhalation particles encounter several protective structural and functional barriers of the respiratory tract which include the surfactant film [2–4], the aqueous lining layer with the mucociliary escalator [5], airway and alveolar macrophages [6–8], the epithelium with the underlying basement membrane [9, 10], and dendritic cells residing in or underneath the epithelial layer [11].
Particles can be classified according to their size which predominantly defines to which compartments of the lungs they gain access [12]. In recent years, particulate matter at least in one dimension smaller than 100 nm has become a focus of pulmonary particle research [13–19]. For the purpose of this review, particles < 100 nm at least in one dimension will be referred to as nanoparticles (NP) although the authors are aware that several practical subclassifications of particles exist [20], e.g. with respect to their shape (tubes, rods etc.) or their origin (combustion-derived nano-sized particles are usually referred to as ultrafine particles to distinguish them from synthetic NP). The growing interest in NP has several obvious reasons. First of all, there is epidemiological evidence that the nano-sized fraction of particles associated with air pollution is a major contributor to adverse health effects attributed to air pollution [21, 22]. Additionally, a growing number of experimental studies have focussed on the enhanced toxicological potential of synthetic NP in contrast to larger sized particles of the same material [23, 24]. While the progress of nanotechnology basically relies on the fact that NP may have different physicochemical properties than larger sized particles of the same material, it has been recognized that these different features may also be accompanied by a different biological reaction of the cells of the respiratory tract upon exposure [25, 26]. Although this fact clearly poses a challenge to researchers involved in pulmonary toxicology, it is also of particular interest for respiratory medicine since inhaled NP may offer an innovative approach for an improved medical treatment [27].
Where then is the justification to employ different transmission electron microscopic (TEM) tools for the analysis of the interaction of NP with cells of the respiratory system? First of all, it is of principal interest to investigate whether the morphology of the tissues and cells of interest changes following NP exposure. Usually, conventional light and electron microscopic methods will suffice to address these questions. Second, however, the localization and distribution of particles within tissues and cells needs to be studied to understand how and why NP cause cellular responses and whether a targeted particle has reached its target cell compartment [28, 29]. Unfortunately, NP may not always be distinguishable from cellular organelles by conventional TEM which evokes the requirement of analytical microscopic methods such as energy filtered TEM (EFTEM) [16, 30]. On the other hand, when the entering mechanisms of particles into cells are under investigation it may be necessary to distinguish between cellular compartments that cannot be identified by their morphological appearance alone (e.g. early and late endosomes). Immunogold labeling of the compartments may help to overcome these limitations. Since all structures including NP present as a two-dimensional profile on a tissue section, a high resolution 3D microscopic approach by electron tomography is desirable to study the interaction of NP with cellular organelles as well as their 3D shape and size after contact with the cells [31]. Importantly, it will be necessary to analyze the morphological alterations of the lung (e.g. investigate potential emphysema development upon long-term NP exposure) and the distribution characteristics of NP in pulmonary tissues and cells in appropriate quantitative terms for which stereology offers a great number of unbiased and efficient methods [32–34].
Though the importance of TEM analyses in NP research is unquestionable the use of advanced imaging techniques and appropriate quantification is still rare. We are convinced that this may partly result from the unawareness of the pitfalls when applying conventional TEM to NP research and the possibilities to overcome these problems. Despite the great efforts to standardize and optimize the generation and exposure systems of NP [35] the appropriate application of microscopic techniques requires optimization. Therefore, this review aims to provide a comprehensive overview of advanced TEM techniques and a critical appraisal of their potentials and limitations in order to stimulate the implementation of more advanced and quantitative TEM methods for NP-related research in the respiratory tract.
2. Focus A: Transmission electron microscopic methods
2.1. Fixation and embedding
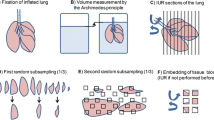
A prerequisite for transmission electron microscopy is that all material entering the microscope has to be fixed in one way or another. Fixation of cells and tissues aims to preserve the specimens as close to the living state as possible. As further outlined, different electron microscopic techniques as well as specific questions of a particular study significantly influence the choice of the fixation and embedding method. Currently, there are two major approaches to fix biological samples, viz. chemical or physical fixation. For the lung as an entire organ, there is no routine alternative approach to chemical fixation to date but for restricted tissue samples, such as larger airways or cell cultures, physical fixation offers an excellent alternative. The following paragraphs as well as Figure 1 provide an overview on different possible methods, their impact for the different TEM techniques and relevant references. Nevertheless, one will have to evaluate the usefulness of a specific protocol for each particular study. For this reason, Weibel et al. [36] have introduced a number of very instructive external and internal standards. In Figure 2, we provide a chemically and a physically fixed specimen for comparison between both methods.

Survey on several transmission electron microscopy strategies. The figure provides help to chose a specific strategy for TEM preparation based on the scientific purpose of the study. It shows the crucial decisions in specimen preparation from the fixation level to the investigation at the TEM. Abbreviations: GA = glutaraldehyde; PFA = paraformaldehyde; CTEM = conventional TEM; EFTEM = energy-filtered TEM; ET = electron tomography.

Chemical and physical fixation of the lung. Alveolar epithelial type II cells were studied by cTEM either after chemical (A and B) or after physical (C and D) fixation. The overview in A shows a well-preserved type II cell from a newborn rat lung fixed by instillation of 1.5% GA, 1.5% PFA in Hepes buffer and processed according to Table 1. Lamellar bodies (LB), nucleus (Nu) and mitochondria (Mt) are well preserved. At a higher magnification, details of the endoplasmic reticulum (ER) as well as an ER related multivesicular transport vesicle (MvTV) can be visualized. The overview in C shows a well-preserved type II cell from an adult rat lung. A small piece of tissue was cut from the whole lung, put in a syringe with 1-hexadecene and air was extracted from the tissue block by negative pressure. Afterwards, the specimen was high-pressure frozen (Leica EMPact 2.0, Leica, Vienna, Austria), freeze-substituted with acetone containing 1% osmium tetroxide (AFS 2.0, Leica, Vienna, Austria) and embedded in epoxy resin. Most likely due to the lack of uranyl acetate during freeze-substitution the lamellar bodies are not well preserved, with almost complete loss of the surfactant material, only the limiting membrane can be seen. However, the ultrastructure of other organelles like multivesicular bodies (MvB) is highly increased (D) due to the excellent preservation of the membrane structures (Me). Since this is the first description of high-pressure frozen lung tissue, systematic studies are needed to determine the ideal processing both for conventional and immuno TEM. Bars = 1 μm (A, C), 250 nm (B, D).
2.1.1. Chemical fixation
Two basic routes exist for the delivery of chemical fixative to the lung: instillation via the airways [1, 37] and perfusion through the vasculature [38, 39]. Instillation fixation via a cannula introduced into the trachea is a straightforward method to fix the lungs, however, it disturbs the surface lining ultrastructure of airway or alveoli including mucus, surfactant, macrophages and foreign material as NP. The lungs need to be collapsed prior to instillation of the fixative at a pressure of approximately 20–25 cm of water. Afterwards the trachea is clamped to prevent fluid outflow, the lung is removed from the chest cavity and stored in cold fixative, e.g. for at least 24 hours [40]. These procedures guarantee that the lungs are fixed at around two thirds to three fourths of total lung capacity [41].
Vascular perfusion fixation via a cannula/catheter introduced into the pulmonary trunk fixes the lungs very quickly and thoroughly [42], but its performance is far more complex and demanding than instillation [43]. Air and perfusate inflow pressures need to be in a correct relation to each other, controlled and possibly adjusted throughout the fixation procedure in order to obtain the tissue of the whole lung properly fixed. This is in itself technically demanding. Particular attention must be paid to the question of which effective perfusion pressure should be used. Since the intrapulmonary pressure can be adjusted the lung can be fixed at a nearly arbitrary fraction of total lung capacity with excellent results at approximately 60% of total lung capacity. A detailed instruction and discussion of both methods is provided by Gil [39] and Gehr et al. [41].
First, the preferred method of fixation for NP research depends on the actual scientific question. If the uptake of particles by alveolar macrophages or their interaction with surfactant is the subject of interest one will have to use vascular perfusion fixation. However, if the translocation of NP to the blood circulation is investigated it will be desirable not to wash out all the blood from the pulmonary circulation and one would therefore prefer to use instillation fixation. Second, however, the choice of the fixation method is also influenced by the experience and skills of a laboratory. If one is not familiar with the different aspects of vascular perfusion fixation and their interpretation this technique may lead to false conclusions because the relationship between the airway and vascular pressures determines the structural appearance of the parenchyma [43].
Fixation of lung samples by immersion in the fixative is by far the least preferable technique because the lung tissue is collapsed and artefactual changes occur, which can not be controlled for [44]. It should therefore only be applied when neither perfusion nor instillation fixation can be used (human biopsy material or specimens sampled from one individual over a sequence of time). In cell cultures, immersion fixation is the method of choice.
The fixative consists of a fixing agent in a suitable vehicle [45]. The most widely used fixatives contain paraformaldehyde (PFA) and glutaraldehyde (GA) often combined with a post-fixative, mostly molecules including heavy atoms such as uranium or osmium [44, 46]. PFA is a higher polymer of formaldehyde and is sold as a white powder. Formaldehyde solutions made of PFA do not contain methanol in contrast to the 10% formalin solutions frequently used in light microscopy [47]. There is a large number of protocols using either PFA or GA individually or a combination of PFA and GA, in varying concentrations and with different buffers. Often, the choice of the fixation solution is not based on scientific reasons but on lab traditions. Detailed reviews of the literature on this topic are available [48–50]. The choice of the fixative as well as the further processing of the samples will depend on the actual purpose of the study because chemical fixation always means a compromise between preservation of cellular ultrastructure and maintenance of antigenicity. High concentrations of GA as well as osmium containing post-fixatives often delete the antigenicity and make samples unfeasible for immuno TEM studies [48] whereas very low GA concentrations as well as the lack of exposure to uranyl salts fail to sufficiently preserve pulmonary ultrastructure [51]. In Table 1, we recommend two protocols for chemical fixation and embedding for conventional and immuno TEM of the lung routinely used in our lab though we are aware that many other suitable protocols may exist.
2.1.2. Physical fixation
Physical fixation is often referred to as physical immobilization, cryo-immobilization or cryofixation. The purpose is to solidify the water in the sample, thereby arresting the biological machinery. However, the formation of ice crystals induces severe damage to the cell [52]. Cryo-immobilization has a long-standing history in light microscopy since segregation artefacts, induced by ice crystals reside in a size range of about 100 nm, well below the resolution limit of light microscopic techniques.
In order to be useful for TEM studies, these segregation artefacts need to be prevented, or reduced below the resolution limit of the TEM. This can be achieved by vitrification: a phase transition of liquid water to the vitrified state, which is an amorphous (i.e. lacking crystalline structures), solid state of water [53–57]. Such a state of water can only be achieved by extremely fast extraction of heat from the sample surface [58]. As water is a poor thermal conductor and the most abundant molecule in most biological samples, sufficient heat can be eliminated only from small samples [59]. In fact, after plunging the average biological sample in a cryogen (e.g. liquid nitrogen), a superficial layer of about 10–20 μm is vitrified but the ultrastructure of deeper structures will be damaged by ice crystal growth [60]. Therefore, applications involving small particles (e.g. viruses or protein complexes) or single cells were particularly attracted by this technique [61–63].
It has been recognized that high pressure (roughly 200 MPa) influences the physics of water in a way cryoprotectants do [64] which led to the development of high pressure freezing (HPF) procedures [58, 65, 66]. Application of 200 MPa pressure during the cooling process increases the depth of vitrification ten-fold [50, 67, 68], thereby making physical immobilization available up to 200 μm and suitable for larger biological objects, such as tissues [69–72] and even entire multicellular organisms [73]. The impact of 200 MPa on the survival rate of cellular organisms was shown to be minimal [65, 74] but not absent [75, 76].
The vitrification of the water in the sample results in a highly increased preservation of the fine structure and an improved retention of the structural components compared to chemical fixation [61, 66, 77–79] and is therefore often superior for antigenicity studies at the TEM level [80–82].
After successful vitrification, a variety of follow-up procedures are available (Figure 1). The fully hydrated specimen can be cryosectioned and investigated under a cryoTEM [56, 69, 83]. As no dehydration step takes place, this approach will give the closest impression of the native biology of the sample. The topology of the sample can be studied by freeze fracturing methods [84, 85].
Alternatively, the water can be removed from the sample either by freeze drying [86, 87] or by the much more widely used technique of freeze substitution [88]. Dehydration of the sample implies that the threat of ice crystal damage does not longer exist. Low temperature embedding became available after the development of resins with a low enough viscosity to allow infiltration and polymerization at temperatures below 0°C [82, 89, 90]. However, since the sample can be returned to room temperature conditions, the techniques established for chemically fixed samples apply as well.
We here for the first time present evidence that lung tissue can be fixed by HPF (Figure 2). However, as air-filled spaces in the lung cannot withstand the pressure induced (about 200 MPa) an approach is required in which the airways must be filled with an inert, uncompressible fluid, e.g. perfluocarbon, prior to freezing.
2.2. Conventional TEM
Conventional TEM in NP research is frequently used to characterize particle structure [18, 91–93], to demonstrate the intracellular localization of NP [28, 94–96] and less frequently to assess the morphology of tissue or cell samples [28, 96]. Its popularity is partly explained by the high resolution and because it is established in many laboratories. However, particles that are shown in TEM figures are often agglomerated structures with diameters of far more than 100 nm. For this reason, too low magnifications are used, which do not allow the identification of particles in the range of 10–20 nm or even less. Furthermore, it is often ignored that NP can sometimes be indistinguishable from cellular structures in the same size range. For example, electron dense particles (e.g. titanium dioxide) may have similarity with glycogen granules, mitochondrial matrix granules or ribosomes whereas electron lucent particles (e.g. polystyrene) may be confused with spherical vesicular structures [97]. Therefore, there is a potential risk that a technical bias is present either due to the fact that cellular structures are mistaken for NP or vice versa. Using only conventional TEM this bias cannot be overcome (Figure 3).

Conventional TEM of polystyrene nanoparticles. This figure demonstrates the impossibility to distinguish between NP and cellular structures by conventional TEM unambiguously. In A, five polystyrene NP (NP!) with a mean diameter of 78 nm are observed next to an A549 cell. Once taken up by the cells, they may have an appearance as shown in B. It is very likely that the spherical structures in B (NP?) are not NP but vesicular structures like caveolae. CC = Clathrin coated vesicle; PM = Plasma membrane; AJ = Adherens junction. Chemical fixation, Epon embedding, 40–70 nm sections. Bar = 1 μm (A and B are at identical magnification).
Despite its wide use in NP research, conventional TEM as a sole tool for the qualitative or quantitative evaluation of the cellular localization of NP needs to be carefully evaluated from case to case. With few exceptions (e.g. colloidal gold particles), it needs to be accompanied by methods that increase the identification of every NP, independent of its size, shape or electron transmissibility. Nevertheless, the application of conventional TEM is justified by its suitability to detect potential effects of NP on cellular ultrastructure and to characterize NP structure.
2.3. Immuno TEM
Once a specimen has undergone all fixation and embedding procedures, it has maintained its antigenicity, and preserved its ultrastructure, immuno TEM offers some useful applications for NP research: visualization, co-localization and quantification of NP and antigens [98–101]. However, the authors are not aware of any study related to NP that used immunocytochemistry and TEM. The purpose of immunogold labeling is to localize pools of proteins and lipids or to identify structures that cannot be clearly identified or cannot be seen at all at high resolution. For example, a primary antibody binding to the antigen of interest is visualized by a secondary antibody carrying a colloidal gold particle of defined size. This gold particle is visible under the TEM due to its electron density. In contrast to light microscopy, this method allows to determine the localization of antigens in association with subcellular structures at the nano-meter range.
If NP could be made visual by immunogold labeling their identification would be straightforward and unambiguous. The binding specificity could then be cross-checked by the co-localization of the particle (which cannot be performed with immunogold labeling of proteins). Once particles are undoubtedly detectable by TEM, it is possible to determine their exact localization within cellular organelles and to quantitatively assess their tissue or cellular distribution in an unbiased way (see below, stereology). This approach requires that the particles have a surface coating which can be recognized by a specific antibody. As the surface coating is likely to influence the biological effects of the NP, this approach for NP detection might not be advantageous in toxicological studies. However, particles targeted to specific tissues, cells or organelles are designed with a specific coating that guides them their way. In these cases, immunogold labeling for detection may be feasible and very useful.
If coated NP are labeled by immunogold particles it may be interesting to determine their exact localization. For example, early and late endosomes as well as lysosomes cannot be distinguished from each other by their morphology alone. The double-labeling technique (e.g. using 5 nm golds for the NP and 15 nm golds for the organelle) may represent an interesting approach to perform co-localization studies. In those cases where NP can be identified by EFTEM (see below) immunogold labeling of organelles or cytoskeletal proteins offers a detailed analysis of how NP are associated with cellular proteins. Finally, exposure of cells to NP may result in alterations of the cellular distribution of a particular antigen [102] which can be studied by quantitative immuno TEM [103]. An example of sufficient immunogold labeling for an organelle suspicious of being involved in active cellular NP uptake is shown in Figure 4.

Immuno TEM of rat lung labeled for caveolin-1. Caveolae are cholesterol-rich regions of the plasma membrane involved in endocytosis. One of the constituting proteins of caveolae is caveolin-1 which was labeled here using newborn rat lung tissue fixed by instillation of 4% PFA, 0.1% GA in 0.2 M Hepes buffer. After freeze-substitution and embedding in acrylic resin (Table 1), ultrathin sections (40–70 nm) were cut and mounted on formvar-coated Ni mesh grids. Immunogold labeling was performed according to standard protocols [99]. The primary antibody was a rabbit anti-caveolin-1 antibody (BD Biosciences, Pharmingen, Germany) diluted 1:50. The secondary antibody was a goat-anti-rabbit antibody coupled to 10 nm gold particles (British Biocell, Cardiff, United Kingdom). A strong signal is found for caveolae in capillary endothelium and alveolar epithelium. Unspecific background labeling was weak (note the gold particle in the interstitium) but not completely absent. Immunogold labeling requires good knowledge about the biology of the target antigen and the specificity of the antibody. Before going to the TEM level, one is well advised to perform pilot light microscopic experiments. CL = Capillary lumen; EC = Endothelial cell; IC = Interstitial cell; AEI = Alveolar epithelial type I cell; AL = Alveolar lumen. Bar = 1 μm.
2.4. Energy filtered TEM
As shown above, the major problem in the investigation of interactions between NP and the lung is the fact that it is often not possible to distinguish unambiguously between cellular structures and NP, a fact that is often neglected or even unrecognized. Energy filtered TEM combines the high resolution of the TEM with the analytical capabilities of electron energy loss spectroscopy and imaging [104]. This allows the analysis and visualization of the spatial distribution of the elemental composition of a particular structure. During transmission of the beam, inelastic scattering events between beam electrons and the specimen occur, resulting in a specific loss of energy of the electrons in the beam. In such a way, the atoms in the sample leave their fingerprint in the transmitted electron beam. By analysing the energy spectra of the transmitted electrons, the elemental composition of the atoms in the specimen can be retrieved. However, EFTEM requires the use of sections with a thickness of about 30 nm in order to avoid multiple scattering [105].
NP often contain elements such as titanium, zinc or cadmium with no or low abundance in biological systems. This differential atomic composition allows for the distinguishing potential of elemental mapping, and therefore of the NP. If lungs or cell cultures are exposed to an aerosol containing NP of a known composition, it is possible to test whether and which cells or organelles contain the elements the NP are composed of.
EFTEM is an advanced imaging technique that requires substantial technical know-how and a suitable microscope, which explains why it is not widely established in life sciences. If the elemental composition of the particles is not known beforehand, the analysis by electron energy loss spectroscopy may turn out cumbersome and frustrating. Furthermore, analysing NP within biological preparations using EFTEM may also be frustrating if the NP consists of elements that occur in large amounts in biological systems as well although recent progress in the EFTEM technique might improve this situation [106]. Electron spectroscopy images should always be accompanied by electron energy loss spectra of the region of interest and a negative control region (e.g. pure embedding media).
So far, the use of EFTEM in pulmonary research is limited to a few studies, e.g. analysing plasma membrane integrity by lanthanide tracers [107] or phosphorous spreading in alveolar type II cells during ischemia [108, 109]. Research involving the intracellular localization of NP at high resolution should adopt more frequently the use of this methodology. Although the authors are aware that EFTEM requires a great deal of resources (highly trained personnel and infrastructure) the effort seems justified by the fact that there is no real alternative, especially if particles are subject to quantification of their distribution.
Fortunately, EFTEM has witnessed a more frequent use in recent years for the study of NP localization in vitro [16, 95], in vivo [14, 30, 110, 111] and in sputum macrophages [112]. Yet, the majority of the ultrastructural studies still relies on only conventional TEM images without additional proof of the particle identity. In Figure 5, we provide an example of titanium dioxide NP visualized by EFTEM.

Titanium detection by ESI and pEELS. A and B show an energy loss imaging (ESI) series of the L3 orbital of Ti at 440 eV (background) and 464 eV (signal) in an erythrocyte culture. In C, the resulting difference is calculated, revealing the distribution of Ti in the sample. Not shown, but used in the three-window calculation of the Ti distribution is a second background window recorded at 390 eV. The occurrence of Ti is confirmed by parallel electron energy loss spectroscopy (pEELS), shown in the graph. The energy a beam electron looses when interacting with the sample is representative for the atom and orbital it is interacting with. The graph shows a peak intensity around 460 eV, indicative for the interaction with the L3 orbital of Ti (the background follows a negative exponential progression). The zeroloss overview and detail images show a dark particle near a red blood cell. With pEELS confirming the occurrence and ESI revealing the distribution, the particle can be appointed as containing Ti. Bars = 1 μm (D), 250 nm (A to F except D). Titanium dioxide particles were incubated with erythrocytes and fixed and embedded conventionally. The section was placed on a Ni-grid. No supportive film and no staining were used. Zeroloss measuring of the relative thickness of the section at 10 different positions revealed a t/λ of 0.38 (+/- 0.04).
2.5. Electron tomography (ET)
Understanding the interactions of NP with cellular structures, especially with complex membrane-bound organelles requires information about the three-dimensional (3D) arrangement of the morphological components. Before the development of ET, high resolution 3D reconstruction was based on a sequence of serial sections [113–115], which was hampered by the fact that delicate membranous structures would require the production of sections as thin as 5–10 nm, which is not feasible due to limitations in ultramicrotomy. Since in TEM the entire thickness of the section is constantly in focus, the z-resolution of 3D reconstructions from serial sections is limited by the section thickness. In NP research, the use of serial sections is even less sufficient because the diameter of the particles under investigation is often smaller than 50–70 nm, the usual thickness of an ultrathin section.
The development of ET has made it possible to overcome these problems: A rotational relationship between detector (camera) and object is created by tilting the section inside the electron microscope at user defined increment intervals, usually 1° or 2°. This gives rise to a tilt series: a sequence of projections of the object, taken from different angles [116]. Subsequently, the tilt series undergoes several automated digital processing steps including alignment and backprojection [117] by which the object of interest is reconstructed in its three dimensions. The result is a stack of parallel slices, with an axial resolution (distance between two subsequent slices) in the range of 2 to 10 nm, i.e. several times thinner than the thinnest section one could possibly generate physically. Additionally, volume and surface rendering software may be applied to obtain an enhanced 3D impression [118]. In a process known as segmentation [119], particular structures of interest can be extracted, either automated or manually, for further analysis and visualization. Importantly, the segmentation process is influenced by the observer interpretation of the ET slices and therefore the result of a selective cognitive process.
Due to the nature of the electron beam and the increasing relative thickness of the section at increased tilting angles, the section can only be inclined between about ±70°. The remainder, between ±70° and ±90° (the so-called missing wedge) cannot be recovered from the sample and will result in artefact formation during digital reconstruction [120].
For the moment, ET is not widely established for several reasons. First, the electron microscope must be equipped with a motorized goniometer and specialized software, and such instrumentation is resourceful, which, in turn currently limits their purchase to a number of central TEM labs worldwide. Second, ET requires advanced experience and know-how in electron microscopy, specimen preparation and interpretation of the TEM images. The principles and problems of ET are discussed in a number of excellent reviews [121–124].
ET is only emerging to be part of the methodological repertoire in NP research and has already been applied on nanomaterials [125] as well as initial studies on the interactions of NP with biological structures [31, 126].
Visualization by ET allows the analysis of NP shape, volume and surface in 3D. This greatly helps with the characterization of NP and the discrimination between genuine particles and agglomerated NP. Furthermore, ET is projected to become a useful tool to study contact sites between NP and macromolecules in detail. Such insights will potentially improve our understanding of NP entry mechanisms into cells, processing by the cellular machinery and where NP related toxicity originates. One exciting topic also relates to the fact that ET extends the application of a number of stereological tools (e.g. the optical disector, see below) to the electron microscopic level [127]. Particularly, for stereological purposes using ET no segmentation process is needed making the quantification independent of the observer preference. Finally, it needs to be emphasized that ET is independent of sample processing: it can be carried out on materials embedded in epoxy or methacrylate resins as well as cryosections and fully hydrated cryosections and allows to be combined with immuno TEM. Figure 6 provides an example of a 3D reconstruction of a NP in its cellular environment.

Electron tomography of a 250 nm thick section. A tilt series (three stills are shown in A, B and C) between +/- 60° with an increment of 1° provides the information for a volume reconstruction by weighted backprojection (D) of an in vitro grown alveolar epithelial cell (cell line A549), exposed to polystyrene NP prior to chemical fixation. The 2 nm thin slice (E, at a depth of 58 nm in the section) reveals a crisper and clearer depiction of the polystyrene NP than the 250 nm thick Epon section (F). Arbitrary digital slices can be made (G, position shown by the two arrowheads in D) in order to provide unquestionable recognition of the NP. Bars = 250 nm in A-D, 50 nm in E-G. Software based 3D rendering offers a way for segmentation according to the interpretation of the user and allows full perspective freedom (H and I). Moreover, clipping planes can partially dissect the scene (J), segmented objects can be omitted (the membrane surrounding the NP in K) and specific quantitative information on rendered objects obtained. Blue: nanoparticle, shades of green: rough endoplasmic reticulum (RER), orange: plasma membrane, transparent white: membrane surrounding the NP, red: ribosomes (Ri) on the RER. Me = membrane, NP = nanoparticle, PM = plasma membrane.
3. Focus B: Correlative light and electron microscopy
In an ideal world, it would be possible to investigate the interactions of cells with NP combining the benefits of light microscopy, especially live cell microscopy, with the high resolution of transmission electron microscopy. However, in laser scanning microscopy the resolution is limited to 200 nm and 500–900 nm in the lateral and axial directions, respectively, although deconvolution algorithms may increase the resolution 2–3 fold [16, 128, 129]. On the other hand, with TEM only fixed specimens can be analyzed. A combination of light and electron microscopy gives the most complete and accurate picture. Correlative light and electron microscopy is particularly useful for the interpretation of light microscopic data because the low resolution often limits the conclusions that can be drawn from light microscopic investigations alone [130]. To overcome the gap between light and electron microscopy several approaches have been developed which might prove to be very helpful to correlate light and electron microscopic observations in NP research.
Two highly promising ways for correlative light and electron microscopy have been described in detail recently. Using a linker molecule, the fluorescent signal recorded by the optical system of the light microscope is converted into a signal that can be picked up by the optical system of the TEM. In a first approach, a biotinylated antibody specifically directed against a particular antigen is applied and visualized with a streptavidin labeled quantum dot that contains fluorescent molecules at the surface and an electron dense core (CdSe) which can unambiguously be detected by EFTEM [131, 132]. Nisman et al. [132] demonstrated this method by localization studies of the nuclear promyelocytic leukaemia protein in the nucleus of cultured cells. Quantum dots can be quantified similarly to immunogold particles but they are less distinct than colloidal gold particles. A second approach has been described by Griffin et al. [133] and makes use of a short tetracysteine hairpin motif that can be integrated into a large number of proteins. The cells can then be exposed to a biarsenical fluorescent dye which is able to permeate through cell membranes and spread a strong fluorescence signal upon contact with the tetracysteine motif. If diaminobenzidine is added and strong illumination is applied, the biarsenical group reacts with the diaminobenzidine, a process known as photo-oxidation. The diaminobenzidine reaction product is strongly osmiophilic and can easily be visualized by TEM. Gaietta et al. [134] have used this method to visualize the gap junction protein connexin 43 in HeLa cells at light and electron microscopic level.
Direct ways denounce the use of linker molecules between the two microscopic modes. The idea rests on the comparison of images obtained by light and and electron microscopy of the same region of interest. In the case of labeling of specific probes, the signal is obtained by light microscopy (either a precipitation or a fluorescent signal) and superimposed on the high resolution micrographs generated by TEM. One simple way is to use a low-temperature embedded or frozen specimen and cut consecutive ultrathin sections, which can be used for immunofluorescence imaging and immunogold labeling, respectively [135, 136]. This approach allows to visualize "almost the same" region within a specimen, which, however, is not sufficient for studying the "spot-like" interactions of NP with cellular structures. It is therefore desirable to view exactly the same structure of interest at light and electron microscopic level. An interesting approach has been described by Biel et al. [137]. Human skin samples were fixed by HPF, labeled with fluorescent dyes during freeze-substitution, and embedded in Epoxy resins. The entire resin-embedded specimen was studied by confocal laser scanning microscopy, a region of interest was chosen and marked at the surface of the resin block and consequently sectioned into ultrathin sections [138]. Alternatively, one could take a live cell sample exposed to NP, fix it by HPF and use it for further TEM imaging. Specific instrumentation, consumables and protocols were developed for this purpose [83]. For the whole lung, one might think of exposing the lung to NP (ideally by aerosol inhalation) to study the subsequent interactions by real-time lung microscopy [139] and fix the whole lung afterwards by vascular perfusion for further TEM investigations.
A solution of striking simplicity has surfaced recently. Based on the analysis of specific conjugated π-electrons, fluorochromes can be detected by improved EFTEM instrumentation. In this manner, Mhawi et al. [106] could reveal the fluorescent signal of doxorubicin in human breast cancer cells directly at the EM level without the need for further detection systems.
The potential of correlative light and electron microscopic approaches is enormous and their application in NP research may greatly enhance our understanding of the entering mechanisms, the intracellular trafficking and their downstream beneficial or toxic properties. The combination of the different advanced techniques described above offers a methodological armamentarium suited to answer many of the inherent questions currently discussed in NP research.
4. Focus C: Stereology
The significance of microscopic evaluations is directly associated with the extent and representativeness of the observations. Evaluation of the extent of a microscopic finding requires the use of unbiased quantitative methods which describe the structures in terms of number, length, area or volume. The representativeness of the observations is critical because the actual material that enters the microscopic stage is infinitesimally small in relation to what it is thought to represent. Therefore, the aim is to give every part of an organ or a cell culture an equal chance of being selected for the analysis. Stereology offers a wide range of unbiased methods that fulfil these requirements and are frequently used in pulmonary research, but their use in NP research is rare. There are, however, three basic scenarios that justify the use of stereology in NP research: (1) First, it is necessary to estimate the morphological changes that occur upon exposure to NP, e.g. the volume of fibrotic lesions in the lung or the number of inflammatory cells. (2) Second, changes in protein localization due to NP exposure can be estimated by relating immunogold particles to the relative volume of the organelles which results in relative labeling indices. (3) Third, the preferential distribution of NP within tissues or cells needs to be estimated and evaluated using appropriate statistical methods that are closely correlated to the methods for quantification of immunogold labeling. All of these methods require the use of sampling procedures that need to be followed consequently from the whole organ down to the microscopic test field under investigation. These methods include randomization of localization by systematic uniform random sampling [140] or fractionator sampling [141, 142] and randomization of orientation using the isector [143] or the orientator [144]. The basic principles for applying unbiased stereology to lung research have been described in detail in some recent review articles [32, 33].
Unfortunately, morphological changes associated with NP exposures are usually reported by micrographs showing "representative" lesions or are not investigated at all, a consequence from the fact that most studies focus on acute toxicity of NP rather than on chronic exposures. However, it will be necessary to assess these changes both in acute and chronic situations to evaluate the significance of the reported toxicological responses for pulmonary structural remodeling. To give some examples, stereology offers tools to investigate the number of cells or alveoli [145–147], the surface area of endocytosis-related cellular membranes (e.g. caveolae) or the gas exchange region [148], the volume of intracellular or intraalveolar surfactant components [149–151] and many more. For an overview of these methods, the reader may refer to the reviews of Ochs [32] and Weibel et al. [33].
The approaches to analyze immunogold or NP distributions using the comparison of observed and expected particle distributions in terms of indices (relative labelling index, RLI, [103], relative deposition index, RDI, [34]) and with chi-squared analysis have made it possible to distinguish between random and non-random particle distributions. Since RLI and RDI rely on the same basic principle they are only explained for the RDI and the reader may refer to Mayhew et al. [103] for further information on the RLI. The first step in the analysis of NP distribution within a certain tissue or cell is the definition of compartments within the structure of interest. For the lung, one may want to distinguish between airway lumen and wall, alveoli, epithelium, interstitium or endothelium of alveolar septa etc. For intracellular distributions different cellular compartments may be distinguished for the purpose of analysis. The second step is the gathering of a sufficient number of test fields by systematic uniform random sampling which serves to provide two sources of information: 1) The observed number of NP associated with each compartment can be estimated by counting the NP. It needs to be mentioned at this point that the probability a NP is contained in an ultrathin section increases with increasing particle size. If particles with a wide size distribution are used this may lead to a size-dependent bias due to oversampling of the larger sized particles. An unbiased approach of counting particles independent of their size is the use of disectors [145]. However, due to the small size of NP in relation to the thickness of the section it is usually not possible to employ physical disectors. The optical disector using ET may offer an alternative but for NP with a small size distribution and diameter it may be justified to count the particle profiles keeping in mind the chance of introducing a bias. 2) The relative volume of the compartments can be estimated by counting the number of points of a randomly superposed point grid hitting each compartment. This gives rise to the observed number of points. If the NP were distributed randomly within the tissue or cell then the observed number of NP associated with each compartment would be closely correlated with compartment size. This means that the observed number of points includes information about a hypothetical random NP distribution. This can be calculated from the numbers of observed particles and observed points and is called the expected number of particles. The relationship between the observed and expected number of particles can be expressed in terms of an index, the RDI, obtained by dividing the observed number by the expected number of particles for each compartment. If the RDI > 1 for a given compartment, it contains a greater number of NP than would be expected from the compartment size. If RDI < 1, a compartment contains less NP than would be expected from compartment size. And if RDI = 1 a compartment contains as many NP as would be expected from its size. The final step is to compare the observed and expected NP distributions in a statistically valid way using the chi-squared test. By using this test, we test the null-hypothesis of random distribution: "The observed and expected NP distributions are equal." For each compartment a partial chi-squared value is obtained. The larger the difference between the observed and expected number of particles the greater the partial chi-squared value will be and thus, the contribution of a compartment to the overall difference between observed and expected particle distribution as expressed by the sum of all partial chi-squared values, the total chi-squared value. The total chi-squared value indicates whether the null-hypothesis is rejected or accepted. If it is rejected, we can identify the preferential target of the NP when two criteria are met: RDI > 1 and the partial chi-squared value accounts for a substantial proportion of the total chi-squared value (say, 10% or more). For worked examples on synthetic and real sets of data the reader may refer to references [34, 152] and to Table 2. Furthermore, a detailed discussion of the preconditions that have to be met is given in the original description [34].
5. Concluding remarks
This review article has shown that advanced TEM technology, ranging from specimen preparation to various analytical modes as well as to qualitative and quantitative interpretations of the ultrastructural NP localization is challenging and requires considerable resources and experience. Unawareness of the potentials and limitations of the applicability of a particular method contains the risk to lead to false conclusions. Therefore, the infrastructure required for TEM is often centralized in EM core facilities. If possible, one is well advised to ask for assistance at his/her local EM core facility at the earliest stage of planning. For example, with high-pressure freezing it is not possible to perform a proper systematic uniform random sampling as would be necessary for stereological purposes. On the other hand, fixation with high glutaraldehyde fixations might render a specimen unsuitable for immuno TEM purposes. Thus, the choice of the strategy for TEM analysis determines the mode of specimen fixation and sampling.
We have shown that advanced TEM holds great potential for NP research: High-resolution visualization of NP in a biological environment and quantitative evaluation of its localization will greatly enhance our understanding of the NP-cell interactions. Immunocytochemical TEM approaches combined with stereology will help to determine the effects of NP down to the protein level. Tomographic TEM provides the opportunity to increase our understanding of the three-dimensional interactions between NP and structures of the respiratory tract. Correlative light and electron microscopic approaches may combine live cell observations following NP exposure with subsequent detailed ultrastructral analyses.
We hope that this review contributes to a widespread application of advanced TEM techniques in NP research of the respiratory tract.
Abbreviations
- NP:
-
Nanoparticles
- TEM:
-
Transmission electron microscopy
- EFTEM:
-
Energy filtered transmission electron microscopy
- PFA:
-
Paraformaldehyde
- GA:
-
Glutaraldehyde
- HPF:
-
High-pressure freezing
- ET:
-
Electron tomography
- RLI:
-
Relative labelling index
- RDI:
-
Relative deposition index
References
Gehr P, Bachofen M, Weibel ER: The normal human lung: ultrastructure and morphometric estimation of diffusion capacity. Respir Physiol 1978, 32: 121–140. 10.1016/0034-5687(78)90104-4
Gil J, Weibel ER: Extracellular lining of bronchioles after perfusion-fixation of rat lungs for electron microscopy. Anat Rec 1971, 169: 185–199. 10.1002/ar.1091690205
Gehr P, Schurch S, Berthiaume Y, Im Hof V, Geiser M: Particle retention in airways by surfactant. J Aerosol Med 1990, 3: 27–43.
Schurch S, Gehr P, Im Hof V, Geiser M, Green F: Surfactant displaces particles toward the epithelium in airways and alveoli. Respir Physiol 1990, 80: 17–32. 10.1016/0034-5687(90)90003-H
Kilburn KH: A hypothesis for pulmonary clearance and its implications. Am Rev Respir Dis 1968, 98: 449–463.
Brain JD: Lung macrophages: how many kinds are there? What do they do? Am Rev Respir Dis 1988, 137: 507–509.
Lehnert BE: Pulmonary and thoracic macrophage subpopulations and clearance of particles from the lung. Environ Health Perspect 1992, 97: 17–46. 10.2307/3431327
Warheit DB, Hartsky MA: Role of alveolar macrophage chemotaxis in pulmonary clearance responses to inhaled particles: comparisons among rodent species. Microsc Res Tech 1993, 26: 412–422. 10.1002/jemt.1070260509
Godfrey RW: Human airway epithelial tight junctions. Microsc Res Tech 1997, 38: 488–499. 10.1002/(SICI)1097-0029(19970901)38:5<488::AID-JEMT5>3.0.CO;2-E
Maina JN, West JB: Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev 2005, 85: 811–844. 10.1152/physrev.00022.2004
Holt PG, Schon-Hergard MA, McMenamin PG: Dendritic cells in the respiratory tract. Int Rev Immunol 1990, 6: 139–149. 10.3109/08830189009056625
Schulz H, Brand P, Heyder J: Particle deposition in the respiratory tract. In Particle-lung interactions. Edited by: Gehr P, Heyder J. New York Basel: Marcel Dekker; 2000.
Kendall M, Tetley TD, Wigzell E, Hutton B, Nieuwenhuijsen M, Luckham P: Lung lining liquid modifies PM 2.5 in favour of particle aggregation: a protective mechanism. Am J Physiol Lung Cell Mol Physiol 2002, 282: L109-L114.
Geiser M, Rothen-Rutishauser B, Kapp N, Schürch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P: Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environ Health Perspect 2005, 113: 1555–1560.
Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ, Potapovich , Tyurina YY, Gorelik O, Arepalli S, Schwegler-Berry D, Hubbs AF, Antonini J, Evans DE, Ku B-K, Ramsey D, Maynard A, Kagan VE, Castranova V, Baron P: Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. Am J Physiol Lung Cell Mol Physiol 2005, 289: L698-L708. 10.1152/ajplung.00084.2005
Rothen-Rutishauser BM, Schurch S, Haenni B, Kapp N, Gehr P: Interaction of fine particles and nanoparticles with red blood cells visualized with advanced microscopic techniques. Environ Sci Technol 2006, 40: 4353–4359. 10.1021/es0522635
Sydlik U, Bierhals M, Abel J, Schins RPF, Unfried K: Ultrafine carbon particles induce apoptosis and proliferation in rat lung epithelial cells via specific signalling pathways both using EGF-R. Am J Physiol Lung Cell Mol Physiol 2006, 291: L725-L733. 10.1152/ajplung.00131.2006
Nemmar A, Al-Maskari S, Ali BH, Al-Amri IS: Cardiovascular and lung inflammatory effects induced by systematically administered diesel exhaust particles in rats. Am J Physiol Lung Cell Mol Physiol 2007, 292: L664-L670. 10.1152/ajplung.00240.2006
Brown DM, Hutchison L, Donaldson K, Stone V: The effects of PM10 particles and oxidative stress on macrophages and lung epithelial cells: Modulating effects of calcium signalling antagonists. Am J Physiol Lung Cell Mol Physiol 2007, 292: L1444-L1451. 10.1152/ajplung.00162.2006
Oberdörster G, Oberdörster E, Oberdörster J: Nanotoxicology: An emerging discipline evolving from studies on ultrafine particles. Environ Health Perspect 2005, 113: 823–839.
Peters A, Wichmann HE, Tuch T, Heinrich J, Heyder J: Respiratory effects are associated with the number of ultrafine particles. Am J Respir Crit Care Med 1997, 155: 1376–83.
Schulz H, Harder V, Ibald-Mulli A, Khandoga A, Koenig W, Krombach F, Radykewicz R, Stampfl A, Thorand B, Peters A: Cardiovascular effects of fine and ultrafine particles. J Aerosol Med 2005, 18: 1–22. 10.1089/jam.2005.18.1
de Haar C, Hassing I, Bleumink R, Pieters R: Ultrafine but not fine particulate matter causes airway inflammation and allergic airway sensitization to co-administered antigen in mice. Clin Exp All 2006, 36: 1469–1479. 10.1111/j.1365-2222.2006.02586.x
Renwick LC, Brown D, Clouter A, Donaldson K: Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types. Occup Environ Med 2004, 61: 442–447. 10.1136/oem.2003.008227
Kipen HM, Laskin DL: Smaller is not always better: nanotechnology yields nanotoxicology. Am J Physiol Lung Cell Mol Physiol 2005, 289: L696-L697. 10.1152/ajplung.00277.2005
Maynard AD: Nanotechnology: The next big thing, or much ado about nothing? Ann Occup Hyg 2007, 51: 1–12. 10.1093/annhyg/mel071
Ebbesen M, Jensen TG: Nanomedicine: Techniques, potentials, and ethical implications. J Biomed Biotechnol 2006,2006(5):51516.
Takenaka S, Karg E, Kreyling WG, Lentner B, Möller W, Behnke-Semmler M, Jennen L, Walch A, Michalke B, Schramel P, Heyder J, Schulz H: Distribution pattern of inhaled ultrafine gold particles in the rat lung. Inhal Toxicol 2006, 18: 733–740. 10.1080/08958370600748281
Panyam J, Labhasetwar V: Targeting intracellular targets. Curr Drug Deliv 2004, 1: 235–47. 10.2174/1567201043334768
Kapp N, Kreyling W, Schulz H, Im Hof V, Gehr P, Semmler M, Geiser M: Electron energy loss spectroscopy for analysis of inhaled ultrafine particles in rat lungs. Microsc Res Tech 2004, 63: 298–305. 10.1002/jemt.20044
Porter AE, Muller K, Skepper J, Midgley P, Welland M: Uptake of C60 by human monocyte macrophages, its localization and implications for toxicity: studied by high resolution electron microscopy and electron tomography. Acta Biomater 2006, 2: 409–419. 10.1016/j.actbio.2006.02.006
Ochs M: A brief update on lung stereology. J Microsc 2006, 222: 188–200. 10.1111/j.1365-2818.2006.01587.x
Weibel ER, Hsia C, Ochs M: How much is there really? Why stereology is essential in lung morphometry. J Appl Physiol 2007, 102: 459–467. 10.1152/japplphysiol.00808.2006
Mühlfeld C, Mayhew TM, Gehr P, Rothen-Rutishauser B: A novel quantitative method for analysing the distributions of nanoparticles between different tissue and intracellular compartments. J Aerosol Med, in press.
Oberdörster G, Maynard A, Donaldson K, Castranova V, Fitzpatrick J, Ausman K, Carter J, Karn B, Kreyling W, Lai D, Olin S, Monteiro-Riviere N, Warheit D, Yang Hong , ILSI Research Foundation/Risk Science Institute Nanomaterial Toxicity Screening Work Group: Principles for characterizing the potential human health effects from exposure to nanomaterials: elements of a screening strategy. Part Fibre Toxicol 2005, 2: 8. 10.1186/1743-8977-2-8
Weibel ER, Limacher W, Bachofen H: Electron microscopy of rapidly frozen lungs: evaluation on the basis of standard criteria. J Appl Physiol 1982, 53: 516–527.
Weibel ER: Morphometry of the human lung. New York: Academic Press; 1963.
Gil J, Weibel ER: Improvements in demonstration of lining layer of lung alveoli by electron microscopy. Respir Physiol 1969, 8: 13–36. 10.1016/0034-5687(69)90042-5
Gil J: Controlled and reproducible fixation of the lung for correlated studies. In Models of lung diseases. Microscopy and structural methods. Volume 47. Edited by: Gil J. New York: Marcel Dekker; 1990:3–22.
Weibel ER: Morphometric and stereological methods in respiratory physiology, including fixation techniques. In Techniques in the life sciences: respiratory physiology. Edited by: Otis AB. County Clare: Elsevier; 1984:1–35.
Gehr P, Geiser M, Stone KC, Crapo JD: Morphometric analysis of the gas exchange region of the lung. In Toxicology of the lung. 2nd edition. Edited by: Gardner DE, Crapo JD, McClellan RO. New York: Raven Press; 1993:111–154.
Coalson J: A simple method of lung perfusion fixation. Anat Rec 1983, 205: 233–238. 10.1002/ar.1092050214
Bachofen H, Ammann A, Wangensteen D, Weibel ER: Perfusion fixation of lungs for structure-function analysis: credits and limitations. J Appl Physiol 1982, 53: 528–533.
Hayat M: Fixation for electron microscopy. New York: Academic Press; 1981.
Glauert AM: The high voltage electron microscope in biology. J Cell Biol 1974, 63: 717–748. 10.1083/jcb.63.3.717
Glauert AM, Lewis PR: Biological specimen preparation for transmission electron microscopy. Princeton: Princeton University Press; 1998.
Kiernan JA: Formaldehyde, formalin, paraformaldehyde and glutaraldehyde: What they are and what they do. Microsc Today 2000, 8: 1–12.
Griffiths G: Fine Structure immunocytochemistry. Berlin Heidelberg: Springer; 1993.
Hayat M: Principles and Techniques of Electron Microscopy. Cambridge: Cambridge University Press; 2000.
Skepper JN: Immunocytochemical strategies for electron microscopy: choice or compromise. J Microsc 2000, 199: 1–36. 10.1046/j.1365-2818.2000.00704.x
Fehrenbach H, Ochs M: Studying lung ultrastructure. In Methods in Pulmonary Research. Edited by: Uhlig S, Taylor AE. Basel: Birkhäuser; 1998:429–454.
Robards AW, Sleytr UB: Low temperature methods in biological electron microscopy. New York: Elsevier; 1985.
Burton EF, Oliver WF: The crystal structure of ice at low temperature. Proc R Soc London A 1935, 153: 166–172.
Brüggeler P, Mayer E: Complete vitrification in pure liquid water and dilute aqueous solutions. Nature 1980, 288: 569–571. 10.1038/288569a0
Dubochet J, McDowall AW: Vitrification of pure water for electron microscopy. J Microsc 1981, 124: RP3-RP4.
Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P: Cryo-electron microsopcy of vitrified specimens. Q Rev Biophys 1988, 21: 129–228.
Angell CA: Amorphous water. Annu Rev Phys Chem 2004, 55: 559–583. 10.1146/annurev.physchem.55.091602.094156
Moor H: Theory and practice of high pressure freezing. In Cryothechniques in biological electron microscopy. Edited by: Steinbrecht RA, Zierold K. Berlin: Springer; 1987:175–191.
Studer D, Michel M, Wohlwend M, Hunziker EB, Buschmann MD: Vitrification of articular-cartilage by high-pressure freezing. J Microsc 1995, 179: 321–332.
Escaig J: New instruments which facilitate rapid freezing at 83 K and 6 K. J Microsc 1982, 126: 221–229.
Adrian M, Dubochet J, Lepault J, McDowall AW: Cryoelectron microscopy of viruses. Nature 1984, 308: 32–36. 10.1038/308032a0
Frank J, Penczek P, Grassucci R, Srivastava S: Three-dimensional reconstruction of the 70S Escherichia coli ribosome in ice: The distribution of ribosomal RNA. J Cell Biol 1991, 115: 597–605. 10.1083/jcb.115.3.597
Leger-Silvestre I, Trumtel S, Noaillac-Depeyre J, Gas N: Functional compartmentalization of the nucleus in the budding yeast Saccharomyces cerevisiae. Chromosoma 1999, 108: 103–113. 10.1007/s004120050357
Moor H, Riehle U: Snap-freezing under high pressure: a new fixation technique for freeze-etching. Proceedings of the 4th European Conference on Electron Microscopy 1968, 2: 33–34.
Riehle U, Hoechli M: The theory and technique of high-pressure freezing. In Freeze-etching, Techniques and Applications. Edited by: Benedetti EL, Favard P. Paris: Societé Francaise de Microscopie Electronique. Paris; 1973:32–61.
Studer D, Graber W, Al-Amoudi A, Eggli P: A new approach for cryofixation by high-pressure freezing. J Microsc 2001, 203: 285–294. 10.1046/j.1365-2818.2001.00919.x
Sartori N, Richter K, Dubochet J: Vitrification depth can be increased more than 10-fold by high-pressure freezing. J Microsc 1993, 172: 55–61.
Shimoni E, Muller M: On optimizing high-pressure freezing: from heat transfer theory to a new microbiopsy device. J Microsc 1998, 192: 236–247. 10.1046/j.1365-2818.1998.00389.x
Al-Amoudi A, Norlen LPO, Dubochet J: Cryo-electron microscopy of vitreous sections of native biological cells and tissues. J Struct Biol 2004, 148: 131–135. 10.1016/j.jsb.2004.03.010
Hohenberg H, Tobler M, Muller M: High-pressure freezing of tissue obtained by fine-needle biopsy. J Microsc 1996, 183: 133–139. 10.1046/j.1365-2818.1996.820642.x
Hunziker EB, Michel M, Studer D: Ultrastructure of adult human articular cartilage matrix after cryotechnical processing. Microsc Res Tech 1997, 37: 271–284. 10.1002/(SICI)1097-0029(19970515)37:4<271::AID-JEMT3>3.0.CO;2-O
Vanhecke D, Graber W, Herrmann G, Al-Amoudi A, Eggli P, Studer D: A rapid microbiopsy system to improve the preservation of biological samples prior to high-pressure freezing. J Microsc 2003, 212: 3–12. 10.1046/j.1365-2818.2003.01226.x
Muller-Reichert T, Hohenberg H, O'Toole ET, McDonald K: Cryoimmobilization and three-dimensional visualization of C-elegans ultrastructure. J Microsc 2003, 212: 71–80. 10.1046/j.1365-2818.2003.01250.x
Suppes GJ, Egan S, Casillan AJ, Chan KW, Seckar B: Impact of high pressure freezing on DH5 alpha Escherichia coli and red blood cells. Cryobiology 2003, 47: 93–101. 10.1016/S0011-2240(03)00072-5
Semmler K, Wunderlich J, Richter W, Meyer HW: High-pressure freezing causes structural alterations in phospholipid model membranes. J Microsc 1998, 190: 317–327. 10.1046/j.1365-2818.1998.00328.x
Leforestier A, Richter K, Livolant F, Dubochet J: Comparison of slam-freezing and high-pressure freezing effects on the DNA cholesteric liquid crystalline structure. J Microsc 1996, 184: 4–13. 10.1046/j.1365-2818.1996.1090666.x
Kandasamy MK, Parthasarathy MV, Nasrallah ME: High-pressure freezing and freeze substitution improve immunolabeling of s-locus specific glycoproteins in the stigma papillae of Brassica. Protoplasma 1991, 162: 187–191. 10.1007/BF02562562
Studer D, Hennecke H, Muller M: High-pressure freezing of soybean nodules leads to an improved preservation of ultrastructure. Planta 1992, 188: 155–163. 10.1007/BF00216809
von Schack ML, Fakan S, Villiger W, Müller M: Cryofixation and cryosubstitution: a useful alternative in the analyses of cellular fine structure. Eur J Histochem 1993, 37: 5–18.
Monaghan P, Perusinghe N, Müller M: High-pressure freezing for immunocytochemistry. J Microsc 1998, 192: 248–258. 10.1046/j.1365-2818.1998.00387.x
Claeys M, Vanhecke D, Couvreur M, Tytgat T, Coomans A, Borgonie G: High-pressure freezing and freeze substitution of gravid Caenorhabditis elegans (Nematoda: Rhabditida) for transmission electron microscopy. Nematology 2004, 6: 319–327. 10.1163/1568541042360564
Mühlfeld C, Richter J: High-pressure freezing and freeze substitution of rat myocardium for immunogold labelling of connexin 43. Anat Rec A Discov Mol Cell Evol Biol 2006, 288: 1059–67.
Vanhecke D, Studer L, Studer D: Cryoultramicrotomy: cryoelectron microscopy of vitreous sections. Methods Mol Biol 2007, 369: 175–197.
Walther P: Recent progress in freeze fracturing of high-pressure frozen samples. J Microsc 2003, 212: 34–43. 10.1046/j.1365-2818.2003.01236.x
Lopez O, Lopez-Iglesias C, Cocera M, Walther P, Parra JL, De La Maza A: Influence of chemical and freezing fixation methods in the freeze-fracture of stratum corneum. J Struct Biol 2004, 146: 302–309. 10.1016/j.jsb.2004.01.003
Edelmann L: Simple freeze-drying technique for preparing biological tissue without chemical fixation for electron-microscopy. J Microsc 1978, 112: 243–248.
Edelmann L: Freeze-dried and resin-embedded biological material is well suited for ultrastructure research. J Microsc 2002, 207: 5–26. 10.1046/j.1365-2818.2002.01033.x
Steinbrecht RA, Muller M: Freeze-substitution and freeze drying. In Cryotechniques in biological electron microscopy. Edited by: Steinbrecht RA, Zierold K. Berlin: Springer; 1987:149–172.
Carlemalm E, Garavito RM, Villiger W: Resin development for electron microscopy and an analysis for embedding at low temperature. J Microsc 1982, 126: 123–143.
Giddings TH: Freeze-substitution protocols for improved visualization of membranes in high-pressure frozen samples. J Microsc 2002, 212: 53–61. 10.1046/j.1365-2818.2003.01228.x
Penn A, Murphy G, Barker S, Henk W, Penn L: Combustion-derived ultrafine particles transport organic toxicants to target respiratory cells. Environ Health Perspect 2005, 113: 956–963.
Mills NL, Amin N, Robinson SD, Anand A, Davies J, Patel D, de la Fuente JM, Cassee FR, Boon NA, MacNee W, Millar AM, Donaldson K, Newby DE: Do inhaled carbon nanoparticles translocate directly into the circulation in humans? Am J Respir Crit Care Med 2006, 173: 426–431. 10.1164/rccm.200506-865OC
Muller J, Huaux F, Moreau N, Misson P, Heilier J-F, Delos M, Arras M, Fonseca A, Nagy JB, Lison D: Respiratory toxicity of multi-wall carbon nanotaubes. Toxicol Appl Pharmacol 2005, 207: 221–231.
Beck-Speier I, Dayal N, Karg E, Maier KL, Schumann G, Schulz H, Semmler M, Takenaka S, Stettmaier K, Bors W, Ghio A, Samet JM, Heyder J: Oxidative stress and lipid mediators induced in alveolar macrophages by ultrafine particles. Free Radic Biol Med 2005, 38: 1080–1092. 10.1016/j.freeradbiomed.2005.01.004
Stearns RC, Paulauskis JD, Godleski JJ: Endocytosis of ultrafine particles by A549 cells. Am J Respir Cell Mol Biol 2001, 24: 108–115.
Pulskamp K, Diabaté S, Krug H: Carbon nanotubes show no sign of acute toxicity but induce intracellular reactive oxygen species in dependence on contaminants. Toxicol Lett 2007, 168: 58–74. 10.1016/j.toxlet.2006.11.001
Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetvar V: Fluorescence and electron microscopy probes for cellular and tissue uptake of poly (D, L-lactide-co-glycolide) nanoparticles. Int J Pharm 2003, 262: 1–11. 10.1016/S0378-5173(03)00295-3
Ridsdale R, Post M: Surfactant lipid synthesis and lamellar body formation in glycogen-laden type II cells. Am J Physiol Lung Cell Mol Physiol 2004, 287: L743-L751. 10.1152/ajplung.00146.2004
Brasch F, Johnen G, Winn-Brasch A, Guttentag SH, Schmiedl A, Kapp N, Suzuki Y, Muller KM, Richter J, Hawgood S, Ochs M: Surfactant protein B in type II pneumocytes and intra-alveolar surfactant forms of human lungs. Am J Respir Cell Mol Biol 2004, 30: 449–458. 10.1165/rcmb.2003-0262OC
Brasch F, Schimanski S, Mühlfeld C, Barlage S, Langmann T, Aslanidis C, Boettcher A, Dada A, Schroten H, Mildenberger E, Prueter E, Ballmann M, Ochs M, Johnen G, Griese M, Schmitz G: Alteration of the pulmonary surfactant system in full-term infants with hereditary ABCA3 deficiency. Am J Respir Crit Care Med 2006, 174: 571–80. 10.1164/rccm.200509-1535OC
Ochs M, Johnen G, Muller KM, Wahlers T, Hawgood S, Richter J, Brasch F: Intracellular and intraalveolar localization of surfactant protein A (SP-A) in the parenchymal region of the human lung. Am J Respir Cell Mol Biol 2002, 26: 91–98.
Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, Karlsson JM, Watkins SC, Kim HP, Wang X, Lee JS, Postma DS, Kauffman HF, Choi AMK: Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol 2007, 36: 409–417. 10.1165/rcmb.2006-0214OC
Mayhew TM, Lucocq JM, Griffiths G: Relative labelling index: a novel stereological approach to test for non-random immunogold labelling of organelles and membranes on transmission electron microscopy thin sections. J Microsc 2002, 205: 153–164. 10.1046/j.0022-2720.2001.00977.x
Ottensmeyer FP, Andrew JW: High-resolution microanalysis of biological specimens by electron energy loss spectroscopy and by electron spectroscopic imaging. J Ultrastruct Res 1980, 72: 336–348. 10.1016/S0022-5320(80)90069-6
Simon GT: Electron spectroscopic imaging. Ultrastruct Pathol 1987,11(5–6):705–710. 10.3109/01913128709048457
Mhawi AA, Fernandes AB, Ottensmeyer FP: Low-energy-loss electron microscopy of doxorubicin in human breast cancer MCF-7 cells: localization by color. J Struct Biol 2007, 158: 80–92. 10.1016/j.jsb.2006.10.024
Fehrenbach H, Schmiedl A, Brasch F, Richter J: Evaluation of lanthanide tracer methods in the study of mammalian pulmonary parenchyma and cardiac muscle by electron energy-loss spectroscopy. J Microsc 1994, 174: 207–223.
Ochs M, Fehrenbach H, Richter J: Electron spectroscopic imaging (ESI) and electron energy loss spectroscopy (EELS) of multilamellar bodies and multilamellar body-like structures in tannic acid-treated alveolar septal cells. J Histochem Cytochem 1994, 42: 805–809.
Ochs M, Fehrenbach H, Richter J: Ultrastructure of canine type II pneumocytes during hypothermic ischemia of the lung: a study by means of conventional and energy filtering transmission electron microscopy and stereology. Anat Rec 2001, 263: 118–126. 10.1002/ar.1084
Bordat C, Sich M, Réty F, Bouet O, Cournot G, Cuénoud CA, Clément O: Distribution of iron oxide nanoparticles in rat lymph nodes studied using electron energy loss spectroscopy (EELS) and electron spectroscopic imaging (ESI). J Magn Reson Imaging 2000, 12: 505–509. 10.1002/1522-2586(200009)12:3<505::AID-JMRI18>3.0.CO;2-A
Kapp N, Studer D, Gehr P, Geiser M: Electron energy-loss spectroscopy as a tool for elemental analysis in biological specimens. Methods Mol Biol 2007, 369: 431–447.
Alexis NE, Lay JC, Zeman KL, Geiser M, Kapp N, Bennett WD: In vivo particle uptake by airway macrophages in healthy volunteers. Am J Respir Cell Mol Biol 2006, 34: 305–313. 10.1165/rcmb.2005-0373OC
Young SL, Fram EK, Craig BL: Three-dimensional reconstruction and quantitative analysis of rat lung type II cells: a computer-based study. Am J Anat 1985, 174: 1–14. 10.1002/aja.1001740102
Young SL, Fram EK, Larson EW: Three-dimensional reconstruction of tubular myelin. Exp Lung Res 1992, 18: 497–504. 10.3109/01902149209064342
Kayar SR, Hoppeler H, Mermod L, Weibel ER: Mitochondrial size and shape in equine skeletal muscle: a three-dimensional reconstruction study. Anat Rec 1988, 222: 333–339. 10.1002/ar.1092220405
Baumeister W, Grimm R, Walz J: Electron tomography of molecules and cells. Trends Cell Biol 1999, 9: 81–85. 10.1016/S0962-8924(98)01423-8
Radermacher M: Weighted back-projection methods. In Electron Tomography. 2nd edition. Edited by: Frank J. New York: Springer; 2005:245–274.
Kremer JR, Mastronarde DN, McIntosh JR: Computer visualization of three-dimensional image data using IMOD. J Struct Biol 1996, 116: 71–76. 10.1006/jsbi.1996.0013
Frangakis AS, Hegerl R: Segmentation of three-dimensional electron tomographic images. In Electron Tomography. 2nd edition. Edited by: Frank J. New York: Springer; 2005:353–370.
Frank J: Electron Tomography. 2nd edition. New York, Springer; 2005.
Marco S, Boudier T, Messaoudi C, Rigaud J-L: Electron Tomography of biological samples. Biochemistry (Mosc.) 2004, 69: 1219–1225. 10.1007/s10541-005-0067-6
Frey TG, Perkins GA, Ellisman MH: Electron tomography of membrane-bound cellular organelles. Annu Rev Biophys Biomol Struct 2006, 35: 199–224. 10.1146/annurev.biophys.35.040405.102039
McDonald KL, Auer M: High-pressure freezing, cellular tomography, and structural cell biology. Biotechniques 2006, 41: 137–143.
Leapman RD: Novel techniques in electron microscopy. Curr Opin Neurobiol 2004, 14: 591–598. 10.1016/j.conb.2004.08.004
Thomas JM, Midgley PA: High-resolution transmission electron microscopy: the ultimate nanoanalytical technique. Chem Commun 2004, 11: 1253–1267. 10.1039/b315513g
Porter AE, Gass M, Muller K, Skepper JN, Midgley P, Welland M: Visualizing the uptake of C60 to the cytoplasm and nucleus of human monocyte-derived macrophage cells using energy-filtered transmission electron microscopy and electron tomography. Environ Sci Technol 2007, 41: 3012–3017. 10.1021/es062541f
Vanhecke D, Studer D, Ochs M: Stereology meets electron tomography: towards quantitative 3D electron microscopy. J Struct Biol, in press.
van der Voort HTM, Strasters KC: Restoration of confocal images for quantitative image analysis. J Microsc 1995, 178: 165–181.
Shaw P: Deconvolution in 3D optical microscopy. Histochem J 1994, 26: 687–694. 10.1007/BF00158201
Griffiths G, Parton RG, Lucocq J, van Deurs B, Brown D, Slot JW, Geuze HJ: The immunofluorescent era of membrane traffic. Trends Cell Biol 1993, 3: 214–219. 10.1016/0962-8924(93)90114-G
Ren Y, Kruhlak MJ, Bazett-Jones DP: Same serial section correlative light and energy-filtered transmission electron microscopy. J Histochem Cytochem 2003, 51: 605–612.
Nisman R, Dellaire G, Ren Y, Li R, Bazett-Jones DP: Application of quantum dots as probes for correlative fluorescence, conventional, and energy-filtered transmission electron microscopy. J Histochem Cytochem 2004, 52: 13–18.
Griffin BA, Adams SR, Tsien RY: Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281: 269–272. 10.1126/science.281.5374.269
Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH: Multicolor and electron microscopic imaging of connexion trafficking. Science 2002, 296: 503–507. 10.1126/science.1068793
van der Wel N, Fluitsma DM, Dascher CC, Brenner MB, Peters PJ: Subcellular localization of mycobacteria in tissues and detection of lipid antigens in organelles using cryo-techniques for light and electron microscopy. Curr Opin Microbiol 2005, 8: 323–330. 10.1016/j.mib.2005.04.014
Schwarz H, Humbel BM: Correlative light and electron microscopy using immunolabeled resin sections. Methods Mol Biol 2007, 369: 229–256.
Biel SS, Kawaschinski K, Wittern K-P, Hintze U, Wepf R: From tissue to cellular ultrastructure: closing the gap between micro- and nanostructural imaging. J Microsc 2003, 212: 91–99. 10.1046/j.1365-2818.2003.01227.x
Vanhecke D, Bellmann R, Baum O, Graber W, Eggli P, Keller HU, Studer D: Pseudovacuoles – immobilized by high pressure freezing – are associated with blebbing in Walker carcinosarcoma cells. J Microsc, in press.
Kuebler WM, Parthasarathi K, Lindert J, Bhattacharya J: Real-time lung microscopy. J Appl Physiol 2007, 102: 1255–1264. 10.1152/japplphysiol.00786.2006
Mayhew TM: The new stereological methods for interpreting functional morphology from slices of cells and organs. Exp Physiol 1991, 76: 639–665.
Gundersen HJ: Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc 1986, 143: 3–45.
Gundersen HJ: The smooth fractionator. J Microsc 2002, 207: 191–210. 10.1046/j.1365-2818.2002.01054.x
Nyengaard JR, Gundersen HJG: The isector: a simple and direct method for generating isotropic, uniform random sections from small specimens. J Microsc 1992, 165: 427–431.
Mattfeld T, Mall G, Gharehbaghi H, Moller P: Estimation of surface area and length with the orientator. J Microsc 1990, 159: 301–317.
Sterio DC: The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc 1984, 134: 127–136.
Hyde DM, Tyler NK, Putney LF, Singh P, Gundersen HJ: Total number and mean size of alveoli in mammalian lung estimated using fractionator sampling and unbiased estimates of the Euler characteristic of alveolar openings. Anat Rec A Discov Mol Cell Evol Biol 2004, 277: 216–226. 10.1002/ar.a.20012
Ochs M, Nyengaard JR, Jung A, Knudsen L, Voigt M, Wahlers T, Richter J, Gundersen HJ: The number of alveoli in the human lung. Am J Respir Crit Care Med 2004, 169: 120–124. 10.1164/rccm.200308-1107OC
Weibel ER: Stereological Methods. Practical Methods for Biological Morphometry. Volume 1. London: Academic Press; 1979.
Ochs M, Nenadic I, Fehrenbach A, Albes JM, Wahlers T, Richter J, Fehrenbach H: Ultrastructural alterations in intraalveolar surfactant subtypes after experimental ischemia and reperfusion. Am J Respir Crit Care Med 1999, 160: 718–724.
Ochs M, Fehrenbach H, Nenadic I, Bando T, Fehrenbach A, Schepelmann D, Albes JM, Wahlers T, Richter J: Preservation of intraalveolar surfactant in a rat lung ischaemia/reperfusion injury model. Eur Respir J 2000, 15: 526–531. 10.1034/j.1399-3003.2000.15.16.x
Mühlfeld C, Liakopoulos OJ, Schaefer IM, Schöndube FA, Richter J, Dörge H: Methylprednisolone Fails to Preserve Pulmonary Surfactant and Blood-Air Barrier Integrity in a Porcine Cardiopulmonary Bypass Model. J Surg Res 2007, in press. [Epub ahead of print]
Mühlfeld C, Geiser M, Kapp N, Gehr P, Rothen-Rutishauser B: Re-evaluation of pulmonary titanium dioxide nanoparticle distribution using the "relative deposition index": Evidence for clearance through microvasculature. Part Fibre Toxicol 2007, 4: 7. 10.1186/1743-8977-4-7
Acknowledgements
The rat lung tissue used for figures 2 and 4 was obtained from experiments performed on newborn and adult rat lungs in a different context. Specifically, the authors gratefully acknowledge the help of Dr. Hendrik Tevaearai, Bern, Switzerland, who kindly provided the lung tissue for high-pressure freezing during their rat heart experiments. The instillation-fixed newborn rat lungs were taken from experiments on myocardial ischemia tolerance performed by one of the authors (CM). The experimental procedures were indicated to and permitted by the respective governmental authorities.
The authors are thankful to H. Hühn and S. Freese (both Göttingen), S. Frank, B. Haenni, F. Graber, A. Stokes, B. Tschirren and C. Haller (all Bern) for expert technical assistance. The authors also thank Dr. Christophe von Garnier, Bern, Switzerland, for correcting the English language in this manuscript. This work was supported by the Swiss National Foundation (grant No. 310000-107628 and No. 326000-113159) and by the Swiss Federal Office for the Environment (FOEN/BAFU).
Finally, we wish to thank those of our scientific teachers that made us become interested in electron microscopy and stereology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
CM planned the work presented in this article, performed parts of the experiments and electron microscopic analyses, and wrote major parts of the manuscript. BRR performed parts of the experiments, reviewed the manuscript critically and provided important intellectual content. DV performed parts of the experiments and electron microscopic analyses, wrote major parts of the manuscript and provided essential intellectual content. FB performed parts of the experiments and reviewed the manuscript critically. PG supervised the work on this article, made important suggestions, reviewed the manuscript critically and provided important intellectual content. MO participated in the development of this work, made important suggestions, reviewed the manuscript critically and provided essential intellectual content. All authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mühlfeld, C., Rothen-Rutishauser, B., Vanhecke, D. et al. Visualization and quantitative analysis of nanoparticles in the respiratory tract by transmission electron microscopy. Part Fibre Toxicol 4, 11 (2007). https://doi.org/10.1186/1743-8977-4-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-8977-4-11