
Abstract
Glioblastoma multiforme (GBM) is the most common primary brain tumor in adults. The development of this malignant glial lesion involves a multi-faceted process that results in a loss of genetic or epigenetic gene control, un-regulated cell growth, and immune tolerance. Of interest, atopic diseases are characterized by a lack of immune tolerance and are inversely associated with glioma risk. One cell type that is an established effector cell in the pathobiology of atopic disease is the eosinophil. In response to various stimuli, the eosinophil is able to produce cytotoxic granules, neuromediators, and pro-inflammatory cytokines as well as pro-fibrotic and angiogenic factors involved in pathogen clearance and tissue remodeling and repair. These various biological properties reveal that the eosinophil is a key immunoregulatory cell capable of influencing the activity of both innate and adaptive immune responses. Of central importance to this report is the observation that eosinophil migration to the brain occurs in response to traumatic brain injury and following certain immunotherapeutic treatments for GBM. Although eosinophils have been identified in various central nervous system pathologies, and are known to operate in wound/repair and tumorstatic models, the potential roles of eosinophils in GBM development and the tumor immunological response are only beginning to be recognized and are therefore the subject of the present review.
Similar content being viewed by others


Introduction
Glioblastoma (GBM), atopy, and the immune response
Cancer is a multi-faceted cellular process involving the exploitation of genetic and/or epigenetic DNA modifications by a microenvironment that endows immune privilege. This process has been characterized as occurring in three stages known as initiation, promotion and progression where each stage respectively relates to the biological events required for the formation, proliferation and migration of altered immortal cells [1]. Each of these developmental stages may be influenced by immune cells [2]. Although the brain has historically been considered an organ of immune privilege [3], recent research indicates that immune cells may play a pivotal role in both the development of and host defense against brain tumors [4–6].
In U.S. adults, primary brain tumors account for 2% of all cancers, yielding approximately 22,000 diagnoses and 13,000 deaths annually [7]. The developmental pathologies of brain tumors are diverse and may be influenced by age, gender, environmental factors and/or genetic predispositions. These tumors may be classified as glioma (astrocytoma, oligodendroglioma, ependymomas) or non-glioma (meningiomas, pituitary tumors and medulloblastomas) [8]. Gliomas account for approximately 30% of all brain tumors and 80% of malignant brain tumors [9]. Glioblastoma multiforme (GBM) is the most common malignant glioma, and is generally lethal within one year after diagnosis [10].
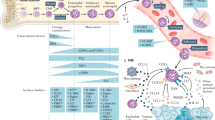
Treatment of GBM is confounded by the complex nature of the tumor and the tumor microenvironment. GBM tumor cells have been indicated to evade surgical, radiotherapeutic, chemotherapeutic and immunotherapeutic interventions by respectively infiltrating into the surrounding brain tissue, down-regulating tumor suppressor proteins, up-regulating DNA repair enzymes, and producing immunosuppressive cytokines [11]. Tumor evasiveness is also thought to involve chronic inflammation and the recruitment of myeloid suppressor cells and T-regulatory cells that effectively obstruct innate and adaptive anti-tumor immune responses [6, 12]. This antigenic tolerance is lacking in atopic diseases [13] which reportedly have an inverse association with glioma risk [14–23] (see Table 1). In the atopic immune response, aberrant recognition of antigen/allergen by antigen presentation cells (APCs: e.g. Dendritic cell, B cell) allows for the processing of antigen/allergen into a peptide for presentation via major histocompatibility complex (MHC) [24]. Full activation of APCs requires MHC plus peptide interaction with the T cell receptor (TCR) and CD40 ligation whereas full activation of T cells requires MHC:peptide:TCR interaction and CD28 ligation to CD80/86 [25]. Further interactions involving the B cell surface marker, CD21, with soluble CD23 and B cell cytokine stimulation (IL-4, IL-13) induces the generation of plasma cells specific for the immunoglobulin IgE, the most common immunoglobulin in allergy and asthma [26, 27]. High serum CD23 and IgE levels are associated with increased GBM patient survival [28, 29]. These molecules are also found to activate of mast cells and eosinophils [30]. Cytokines and chemokines produced by activated mast cells, T cells, and APCs increase vascular permeability for the enhanced recruitment of granulocytic immune cells (e.g., eosinophils, macrophages, and neutrophils) and the development of chronic inflammation [31]. In the tumor immunological response, chronic inflammation is also found to occur but the biological features distinctly differ [12]. Immuno-suppressive cytokines (IL-10, TGF-β) secreted by tumor cells, suppressor macrophages, and T regulatory (CD4+ Treg) cells that in association with additional mediators or cell:cell interactions inhibit the pro-inflammatory functions of dendritic cells, provoke chronic inflammation associated with tumorigenesis, and prevent a specific adaptive immune response required in tumor eradication [12, 32]. Thus, the distinct immune activation parameters in an allergic response may be imperative to immunotherapeutic treatments in cancer. In this regard, a comparison of the immune responses observed in cancer versus atopic diseases is summarized in Figure 1.

The immune response in cancer and atopic disease. (1) Full activation of antigen presentation cells (APCs: e.g. Dendritic cell, B cell) and T cells. (2) T cell cytokines (IL-4, IL-13) and soluble CD23 ligation to CD21, induces B cell differentiation, the generation of plasma cells, the production of IgE, and the subsequent IgE-dependent activation of mast cells. (3) Activated mast cells, APCs, and T cells produce chemokines and cytokines that recruit granulocytic cells (eosinophils, macrophages, neutrophils). (4) Immuno-suppressive cytokines (IL-10, TGF-β) are produced by tumor cells, suppressor macrophages, and T regulatory (CD4+ Treg) cells. These cytokines and additional mediators or cell:cell interactions prevent a specific adaptive immune response required in tumor eradication (see text for additional details).
Eosinophils
Eosinophils are myeloid cells known to accumulate at specific sites, such as the lung and gastrointestinal tract, in the pathobiology of atopic disease [33]. The functions of eosinophils are diverse and include organ development, tissue homeostasis, antigen presentation, wound repair, tissue remodeling, cytotoxic clearance of pathogens, nerve growth, and the production of various chemokines and cytokines known to influence both innate and adaptive immune responses [34–36]. In asthma, eosinophil recruitment has been characterized by early phase IgE-mediated activation of mast cells, the production of pro-inflammatory cytokines (e.g.: IL-2, IL-4, IL-5, GM-CSF) and the late phase recruitment of Th2 cells and eosinophils [37]. These events are preceded by the generation of IgE producing plasma cells (see Figure 1). Murine in vitro and in vivo studies suggest that eosinophils are required for the long-term maintenance of plasma cells [38, 39]. Cytokines produced by mast cells or CD4+ T cells (e.g.: IL-3, IL-5, GM-CSF) are known to induce the differentiation, activation, and survival of eosinophils [40, 41]. Activated eosinophils produce cytotoxic mediators, pro-inflammatory cytokines, pro-fibrotic and angiogenic factors that may alter innate (basophils, mast cells, neutrophils, dendritic cells) and adaptive (T cells) immune responses [35, 42]. The activation state of eosinophils may therefore also affect the tumor microenviroment and tumor development.
Eosinophils and cancer
Previous research has suggested that organs interfacing with the external environment (i.e., mouth, gastrointestinal tract, cervix) are more likely to exhibit inverse associations between allergy and cancer risk than non-interfacing organs (i.e., ovary, breast, prostate) [43]. The possible interaction of neural stem cells and glial progenitor cells with airborne pathogens via the olfactory bulb in the lateral ventricles of the brain suggests that glioma development may also be precluded as a result of an interface with the external environment [43, 44]. Eosinophils are an established effector cell in atopic disease [33] and may therefore participate in the reported inverse associations between atopic disease (allergy, asthma, eczema) and the risk of glioma (see Table 1), oral cancers [45], and gastrointestinal tract cancers [46, 47]. Despite differences in clinical protocols, organ microenvironment, and measurements to identify eosinophilia, a link between certain tumors (colon, stomach, brain, oral/mouth, penile, and uterine/cervix; see Table 2) and eosinophilia has been identified at various stages of disease progression and in association with enhanced patient survival [48–57]. Similar studies with respect to genitourinary cancers, however, are not uniform in outcome [43] (see Table 2). This observation may be a result of patient exposure to certain viruses [58], the organ interface with the external environment [43], hormonal influence [59, 60] or a lack of relevant investigational research. Of interest, eosinophilia in human cancers involving the immune system (Hodgkin's disease, cutaneous T cell lymphoma (CTCL)) has been associated with reduced patient survival (Table 2). The progression of these same diseases is positively influenced by immune factors (IgE, IL-5) known to promote the allergic response and induce the recruitment and activation of eosinophils [30, 37, 49, 61–63]. These human cancers also exhibit a strong Th2 (CD4+) response but lack specific Th1 cytotoxic T cell (CD8+) populations [64, 65]. Human atopic diseases are characterized not only by CD4+ T cell influx but also by CD8+ T cell effector functions [66, 67], suggesting that the eosinophilia associated with adaptive CD8+ T cell immune responses may be essential in the host defense to tumors and allergens.
Eosinophils and GBM
Eosinophils accumulate in various human central nervous system disorders (eosinophilic meningoencephalitis, idiopathic hypereosinophilic syndrome encephalopathy, eosinophilic meningitis, peripheral neuropathy), including tumors of the brain (neuroblastoma, leiomyoma, glioblastoma) [52, 68–75]. Interestingly, eosinophilic meningitis has been identified in a case of disseminated GBM [74]. Eosinophils have also been shown in an in vivo murine model to be recruited to necrotic tissue [76], which is also a primary determinant of human GBM [77]. Clinico-pathological assessment of human eosinophil migration to the brain has been indicated to occur in the development of subdural hematomas [78, 79], a condition that emerges in response to increased intracranial pressure in some human GBM case studies [80, 81]. Marked eosinophilia in sediments of spinal fluid has been identified in patients with intracerebral neoplasms, including one case of GBM [82]. In two separate clinical trials, enhanced GBM patient survival was associated with tissue eosinophilia found after postoperative treatments with interleukin-2 (IL-2) [52, 75]. Human eosinophils in an in vitro study have also been reported to be responsive to S100B [83], a possible blood marker in some GBM cases that is known to be released by CD8+ T cells, astrocytes, oligodendrocytes, and tumor cells [84–86]. However, the mechanisms by which eosinophils may function in tissue destruction or remodeling and repair are not clearly understood [35, 87]. Thus, the purpose of this review is to examine the potential roles of eosinophils in the stages of GBM development and the tumor immune response (see Figure 2).

The potential role(s) of tumor associated eosinophils. Tumor development has been characterized as proceeding through several stages (initiation, promotion, progression). The initiation stage is a period of mutagenesis where genetic and/or epigenetic alternations in stem cells or progenitor cells are established. The promotion stage invokes cellular growth (mitogenesis) that is induced by growth factors and altered apoptotic cell signal pathways. This increased cell division creates a microenvironment of metabolic stress, hypoxia and necrotic cell death that has also been associated with thrombosis. The progression stage involves additional genetic and epigenetic events that confer phenotypic changes necessary for tumor cell autonomous growth, invasiveness, and migration. Eosinophils are able to produce growth factors, cytokines, chemokines, blood coagulants, and cytotoxic mediators that may affect each stage of tumor development.
Initiation
Known factors in initiating GBM tumor formation
Malignant gliomas are thought to originate from neuroectodermal stem cells or tumor progenitor cells [88]. Genetic and/or epigenetic alterations in these cells promote the dysregulation of several signaling molecules/networks involving intracellular (MDM2, PTEN, TP53, annexin A7) and extracellular (platelet-derived growth factor, epidermal growth factor, vascular endothelial growth factor, fibroblast growth factor) protein function [88, 89]. While the development of this genomic instability is not clearly known, rare genetic disorders (Li-Fraumeni syndrome, neurofibromatosis, Turcot's syndrome), ionizing radiation, and oxidative stress from toxic chemical exposure or biological aging have been implicated in gliomagenesis [90–93]. Evidence supporting human cytomegalovirus (HCMV) infections and interleukin-4 receptor alpha (IL-4Rα) or IL-13 single nucleotide polymorphisms (SNPs) as GBM risk factors have also been found [94–98], suggesting that inflammation may play a role in GBM etiology.
IL-4 and cancer
The cytokines, IL-4 and IL-13, share a common receptor component, the IL-4Rα, and can initiate many similar immune responses [99]. These cytokines also play a pivotal role in the allergic response by stimulating B cell IgE synthesis and inducing epithelial production of pro-eosinophilic chemokines (eotaxin, monocyte chemotactic protein-1 (MCP-1)) [99]. As a result, clinical assessment of anti-IL-13 antibodies is being explored in the treatment of asthma with some improvement in lung function indicated [100]. However, in one asthmatic study involving 56 patients, the recruitment of eosinophils was not affected by anti-IL-13 therapy [101], perhaps due to alternative responses via IL-4 [99]. In a Phase I clinical trial of IL-4 involving cancer patients with non-hematological refractory malignancies, systemic eosinophil degranulation was identified in patient serum, urine, and skin biopsies of rashes via increased identification of the eosinophil granule protein, major basic protein (MBP) [102]. Patient sera were also examined for eosinophil viability factors where the cytokines IL-5, IL-3 and GM-CSF were identified in mediating the response [102]. Because of the identified activity of eosinophils, additional Phase I and II trials were explored but no significant tumor response was obtained in examinations of refractory malignancies [103, 104]. Further assessment involving the co-administration of IL-4 and IL-2 resulted in eosinophilia of unknown significance in all patients studied [53] (see Table 2). Efforts to understand eosinophilia and the tumor response to IL-4 in rodent models, including studies of GBM, revealed that the release of IL-4 at the tumor site induced significant eosinophil influx, tumor rejection, and the prolonged survival of nude mice [105–107]. Another in vivo murine study demonstrated that IL-4-mediated tumor suppression involved the production of the cytokine interferon-gamma (IFN-γ) [108], which supports subsequent findings indicating that IL-4-transfected tumor cell vaccines promoted Th1 immunity [109]. In asthmatic patients, production of IFN-γ by CD8+ T cells has been identified [110, 111]. In ovalbumin sensitized rats, production of IFN-γ by CD8+ T cells suppressed eosinophilia, and in human eosinophils, IFN-γ has also been found to enhance cytokine- (GM-CSF, IL-5) induced degranulation and superoxide anion production [112, 113]. Thus, in certain cases, effective GBM tumor eradication may occur in response to IL-4 and the concomitant recruitment of CD8+ T cells and eosinophils whereby the CD8+ T cells identify specific antigens and produce IFN-γ that enhances eosinophil activation and the release of cytotoxic granules. Modifications of these events via the aforesaid SNPs (IL-4Rα, IL-13) may allow for immune evasion and tumor formation.
Eosinophils and GBM initiation
Previous studies linking eosinophil function and tumor biology have indicated that eosinophil production of eosinophil peroxidase (EPO) and reactive oxygen species (ROS) may amplify oxidative damage and tumorigenesis in the lung [114], possibly via induced activation by the cytokine, GM-CSF, which has been shown in vitro to elicit these responses in human eosinophils [112]. Of note, human astrocytes and GBM tumor cells are also known to produce GM-CSF [115–117], which may enhance oxidative stress in a microenvironment involving eosinophils. Oxidative stress not only functions to induce DNA mutations but may also affect cell senescence and apoptosis in developing tumors [118]. Activated eosinophils are also known to produce eosinophil derived neurotoxin (EDN, RNase 2) and eosinophil cationic protein (ECP, RNase 3) [119]. EDN and ECP exhibit antiviral functions [120, 121] that may play role in preventing HCMV induced tumor formation. EDN has also been identified as a toll-like receptor-2 (TLR2) ligand that can promote the in vivo activation of murine dendritic cells (DCs) [122]. In experimental GBM models, TLR2-ligands have been indicated to induce an influx of tumor-infiltrating immune cells (DCs, CD8+ T effector cells) and significant tumor regression [123, 124], which raises the possibility that EDN may operate comparatively. In addition, ECP can alter cell membrane permeability and induce toxicity in cancer cell lines [121]. EDN and ECP may therefore be effectual eosinophilic components in preventing tumor formation (see Figure 2).
Promotion
Eosinophils and growth factors in GBM promotion
The promotion phase of carcinogenesis involves mitogenesis that is dependent upon apoptotic inhibition and growth stimulation [125]. In GBM, the cell signal cascades that regulate the activation of members of the NF-κB transcription factor family are altered, which in turn leads to enhanced expression of the anti-apoptotic molecules Bcl-2 and survivin [126, 127]. This pathway is known to be stimulated by various growth factors, ROS, and viruses such as HCMV [128]. Platelet-derived growth factor (PDGF) and PDGF receptors are expressed in GBM tumor cells and found to regulate NF-κB activation and cell proliferation [129–131]. Eosinophils likewise express PDGF receptors and PDGF has been reported to activate eosinophils [132]. Activated eosinophils may then release their cytotoxic granules and encourage anti-tumor and/or anti-viral responses during tumor promotion. Alternatively, activated eosinophils may enhance the promotion process through the production of tumor promoting growth factors (see Figure 3) [117].

Eosinophils in tumor promotion. A microenvironment involving rapidly dividing cells induces tumor necrosis and the production of damage associated molecular patterns (DAMPs) which include the RAGE ligands (e.g.: HMGB1 and S100 proteins). Eosinophils and additional innate immune cells (microglia, mast cells, neutrophils) are activated by GBM mediators (GM-CSF, PDGF, CXCL12, CXCL8) and DAMPs which may in turn induce the production of growth factors (amphiregullin, TGF-α, VEGF) and matrix metalloproteinases in promoting tumorigenesis. Alternatively, in certain cases, eosinophil release of cytotoxic granules (EDN, ECP, calprotectin) may function to prevent tumor promotion.
Eosinophils and additional innate immune cells (microglia, mast cells, neutrophils) are activated by GBM mediators (GM-CSF, PDGF, CXCL12, CXCL8) and damage associated molecular patterns (DAMPs: e.g.: S100 proteins, high mobility group box 1) which may in turn induce the production of growth factors and matrix metalloproteinases in promoting tumorigenesis [83, 84, 117, 132–141]. Human eosinophils stimulated in vitro with GM-CSF produce amphiregulin and transforming growth factor-alpha (TGF-α), which are ligands known to activate epidermal growth factor receptors (EGFR) [134, 135]. In primary GBM, amplification of the EGFR gene and subsequent over-expression of EGFR protein is the most common genetic alteration [142]. Increased expression of epidermal growth factor receptors, ligands and cell signals are highly implicated in the promotion of many tumors, including GBM [143]. Of interest, GBM cell lines cultured in eosinophil-conditioned media, generated in the presence or absence of GM-CSF, demonstrated increased cell growth compared to controls [117]. Thus, because GBM tumors are known to produce GM-CSF [115–117], a paracrine loop may develop where eosinophils promote GBM development by producing amphiregulin, TGF-α, or other growth factors in response to GBM-derived GM-CSF.
Eosinophils and RAGE in GBM promotion
Chronic inflammation associated with tumor promotion has also been linked to the activity of the receptor for advanced glycation end-products (RAGE) and RAGE ligands via an in vitro analysis of GBM cell lines and in an in vivo murine model of skin carcinogenesis [144, 145]. Eosinophils and GBM tumor cells each express RAGE and RAGE ligands where cell viability and migration are reported RAGE-mediated responses in these cells [83, 145]. The S100A8 and S100A9 proteins are RAGE ligands [146], DAMPs [147], markers of myeloid-derived suppressor cells [148], GM-CSF induced cytokines in eosinophils [83], and tumor promoting factors in experimental models [144, 149]. Interestingly, the S100A8 and S100A9 complex, calprotectin, is indicated to induce apoptosis in cancer cell lines [150]. Benign lesions associated with normal brain aging (corpora amylacea) also express both S100A8 and S100A9 [151]. In human GBM primary tumor parenchyma, S100A8 and S100A9 have been identified with higher levels of S100A9 noted in the tumor regrowth parenchyma of patients that received primary resection plus irradiation compared to primary resection alone [152]. Of interest, radiation treatment of pelvic cancers has been found to induce eosinophilia and increase ECP serum levels [153, 154]. Eosinophils cultured in GBM-cell line conditioned media in vitro have also been shown to release S100A9 [117]. Whether eosinophils are functional in promoting or preventing benign or cancerous lesions in the brain via S100 proteins and RAGE-mediated responses in these various disease states is not clear but the interactions may represent an important link between eosinophils and glioblastoma biology (see Figure 3).
Progression
GBM is a progressed disease
The progression stage in cancer biology arises from additional genetic and epigenetic events that confer phenotypic changes that are necessary for tumor cell autonomous growth, invasiveness, and migration [155]. At diagnosis, primary GBM (World Health Organization (WHO) Grade IV astrocytoma) presents at a progressed stage and is distinguished histopathologically from anaplastic astrocytoma (WHO Grade III astrocytoma) by the presence of necrosis, microvascular hyperplasia and possibly thrombosis [77]. Necrotic cell death in a developing tumor may occur in response to the increased metabolic demands of rapidly dividing cells, resulting in hypoxia, intravascular occlusion and thrombosis followed by the production of pro-angiogenic and pro-inflammatory mediators (see Figure 2) [77, 88, 156].
Eosinophils and dexamethasone in GBM
Recruitment of murine eosinophils to the tumor microenvironment is indicated to occur in response to necrotic cell death [76]. In rodent models, eosinophil numbers and recruitment to the lung are reduced by dexamethasone [157, 158], a common corticosteroid administered to GBM patients with peritumoral edema [159]. In some GBM cases, dexamethasone therapy (16 mg/day) has been observed to reduce the imaging of lesions on contrast-enhanced scans [160, 161]. A proposed mechanism underlying this phenomenon involves a reduction in capillary permeability at the brain-tumor barrier [162]. In vitro, dexamethasone has been found to inhibit the release of GM-CSF from human primary T cells [163] and GBM cell lines [117]. This steroid is also indicated to reduce GM-CSF-induced survival of human primary eosinophils in tissue culture experiments [164, 165]. Accordingly, the short-term response to dexamethasone therapy in a subset of GBM patients may partly reflect an increase in eosinophil death and the release of eosinophilic cytotoxic products in response to reduced tumor cell-derived GM-CSF. In addition, dexamethasone is also indicated to reduce the release of neurotrophins from human eosinophils in vitro [166]. Neurotrophin receptors have been identified in human GBM and are reportedly integral to disease progression [167, 168]. Human eosinophils activated with GM-CSF in tissue culture have also been shown to produce vascular endothelial growth factor (VEGF), a pro-angiogenic mediator [133]. Thus, dexamethasone-induced occlusion of capilliaries may lower the recruitment of eosinophils and the production of eosinophil-derived VEGF, implicating eosinophils as potential participants in disease progression.
Eosinophils, thrombosis, and neurotoxicity in GBM
The increased presence of eosinophils in the peripheral blood has been characterized as a pro-thrombotic condition and a potential premonitory sign of occult cancer [169–171]. Expression of tissue factor (TF) by immune cells or cancer cells is suspected to enhance the activation of the extrinsic blood coagulation pathway, resulting in the hypercoagulable state of advanced malignancy [172]. Human eosinophils in vitro are known to express cell membrane TF upon activation with GM-CSF [173]. Also, cell culture experiments have revealed that platelets are activated by incubation with two of the four eosinophil granule proteins (EPO, MBP), leading to the release of serotonin and β-thromboglobulin [174]. Of interest, EDN and ECP, the remaining granule proteins, have been found to induce the Gordon phenomenon, a neurotoxic event involving purkinje cell degeneration after intracerebral injection of human eosinophils into animals [175, 176]. These functions of eosinophils may aid in our understanding of GBM progression and the clinical observations (motor weakness/loss, lack of coordination, altered mental function [177]) associated with the disease (see Figure 2).
The immune response
The innate immune response is known as the first line of defense against tumors [178]. In GBM, this includes natural killer (NK) cells, microglia, granulocytes (e.g.: eosinophils, mast cells, neutrophils), the complement system, and various immune activators (e.g.: pathogen- or damage- associated molecular patterns (PAMPs or DAMPs) or the recognition of non-self/foreign peptides) [178, 179]. The adaptive immune response involves the specific identification and elimination of tumor antigens via the activation of CD8+ T cells and the generation of antibodies that target tumor-specific antigens [179, 180]. The combined innate and adaptive immune responses that effectively suppress tumor formation have been termed immunosurveillance [181]. Emerging information indicating that immune cells may not only be involved in tumor prevention but also tumor development has resulted in an additional term called immunoediting [181, 182]. This latter concept appears relevant to GBM in that various reports, particularly with respect to innate immune cells, indicate that the function of immune cells is altered by tumor cells to support rather than prevent tumorigenesis (see Figure 3) [117, 136, 137, 139, 183, 184]. Because atopic diseases reportedly have an inverse association with glioma risk [15, 18, 19, 185], immune activators in atopy may exhibit anti-tumor responses in GBM (see Figure 4). The cytokines, IL-4 and IL-13, are known to be up-regulated in allergy/asthma and have been characterized as integral proteins in GBM biology [95, 96, 98, 99]. These cytokines in association with CD23:CD21 ligation, drive the generation of IgE antibodies [26]. High serum CD23 and IgE levels are associated with increased GBM patient survival and the activation of mast cells and eosinophils [28–30]. TLR-ligands are components of the innate immune system, regulators of immune activation in allergy/asthma, and recently examined adjuvants in a GBM clinical trial involving dendritic cells where improved survival in certain patient subsets was identified [178, 179, 186, 187]. In vitro and in vivo experiments indicate that TLR-ligand activated dendritic cells or mast cells encourage CD8+ T cell recruitment [186, 188, 189]. Of interest, in vivo tumor models involving IL-4 also exhibited Th1 cell immunity as well as significant eosinophil influx, tumor rejection/suppression, and prolonged survival of the host [105–109]. Additional in vivo research revealed that IL-4-producing Th2 cells were critical for natural killer cell activation (perforin, granzyme-B) and tumor rejection [190]. These data concur with in vitro evidence indicating a function of IL-4 in suppressing the induction of tumor growth factor (TGF)-β-induced T regulatory cells [191–193]. Thus, the immune parameters in atopic disease (e.g.: IgE, TLR ligands, IL-4) may propel innate and adaptive immune responses toward tumor eradication.

GBM innate and adaptive immunity. Cytokines and chemokines produced by tumor cells are indicated to alter the tumor suppressive functions of innate immune cells (natural killer cell, microglia, neutrophil, mast cell) thereby creating a microenvironment that is conducive to tumor development. Immune activators associated with allergy/asthma (IL-4, IL-13, CD23:CD21 ligation, IgE, TLR ligands) induce the recruitment and activation of immune cells (mast cells, eosinophils, natural killer cells, CD8+ T cells), the suppression of CD4+ Treg development, tumor rejection, and enhanced host survival (see text for additional details).
Conclusions
The mechanisms and immunobiology of GBM tumor development are not clearly known and represent areas of active investigation. In this regard, emerging evidence reveals that eosinophils may hold a functional role in the initiation, promotion and progression of developing GBM tumors. Understanding the complex nature of the innate and adaptive immune responses may foster more effective immunotherapeutic approaches in treating GBM. Because of the multiple associations of eosinophils in tumorigenesis, further study of this diverse immune cell with respect to cancer appears warranted.
Abbreviations
- APCs:
-
antigen presentation cells
- DAMP:
-
damage associated molecular pattern
- ECP:
-
eosinophil cationic protein
- EDN:
-
eosinophil derived neurotoxin
- EGFR:
-
epidermal growth factor receptor
- EPO:
-
eosinophil peroxidase
- GBM:
-
glioblastoma
- GM-CSF:
-
granulocyte macrophage colony-stimulating factor
- HCMV:
-
human cytomegalovirus
- MHC:
-
major histocompatibility complex
- MBP:
-
major basic protein
- PAMPs:
-
pathogen associated molecular pattern
- PDGF:
-
platelet derived growth factor
- ROS:
-
reactive oxygen species
- TCR:
-
T cell receptor
- TF:
-
tissue factor
- TGF:
-
tumor growth factor
- TLR:
-
toll-like receptor
- VEGF:
-
vascular endothelial growth factor
- RAGE:
-
receptor for advanced glycation end-products
References
Ito N, Hasegawa R, Imaida K, Hirose M, Asamoto M, Shirai T: Concepts in multistage carcinogenesis. Crit Rev Oncol Hematol 1995,21(1–3):105–133.
Mantovani A, Allavena P, Sica A, Balkwill F: Cancer-related inflammation. Nature 2008,454(7203):436–444.
Galea I, Bechmann I, Perry VH: What is immune privilege (not)? Trends Immunol 2007,28(1):12–18.
Mitchell DA, Fecci PE, Sampson JH: Immunotherapy of malignant brain tumors. Immunol Rev 2008, 222:70–100.
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H: The brain tumor microenvironment. Glia 2011,59(8):1169–1180.
Albesiano E, Han JE, Lim M: Mechanisms of local immunoresistance in glioma. Neurosurg Clin N Am 2010,21(1):17–29.
Cancer facts and figures 2011 [http://www.cancer.org]
Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ, Uhm JH: Central nervous system tumors. Mayo Clin Proc 2007,82(10):1271–1286.
2011 CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007 [http://www.cbtrus.org/reports/2009]
Reardon DA, Wen PY: Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist 2006,11(2):152–164.
Gomez GG, Kruse CA: Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther Mol Biol 2006,10(A):133–146.
Whiteside TL: The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008,27(45):5904–5912.
Allan SE, Broady R, Gregori S, Himmel ME, Locke N, Roncarolo MG, Bacchetta R, Levings MK: CD4+ T-regulatory cells: toward therapy for human diseases. Immunol Rev 2008, 223:391–421.
Schlehofer B, Blettner M, Preston-Martin S, Niehoff D, Wahrendorf J, Arslan A, Ahlbom A, Choi WN, Giles GG, Howe GR, et al.: Role of medical history in brain tumour development. Results from the international adult brain tumour study. Int J Cancer 1999,82(2):155–160.
Wiemels JL, Wiencke JK, Sison JD, Miike R, McMillan A, Wrensch M: History of allergies among adults with glioma and controls. Int J Cancer 2002,98(4):609–615.
Brenner AV, Linet MS, Fine HA, Shapiro WR, Selker RG, Black PM, Inskip PD: History of allergies and autoimmune diseases and risk of brain tumors in adults. Int J Cancer 2002,99(2):252–259.
Schoemaker MJ, Swerdlow AJ, Hepworth SJ, McKinney PA, van Tongeren M, Muir KR: History of allergies and risk of glioma in adults. Int J Cancer 2006,119(9):2165–2172.
Wigertz A, Lonn S, Schwartzbaum J, Hall P, Auvinen A, Christensen HC, Johansen C, Klaeboe L, Salminen T, Schoemaker MJ, et al.: Allergic conditions and brain tumor risk. Am J Epidemiol 2007,166(8):941–950.
Linos E, Raine T, Alonso A, Michaud D: Atopy and risk of brain tumors: a meta-analysis. J Natl Cancer Inst 2007,99(20):1544–1550.
Wiemels JL, Wilson D, Patil C, Patoka J, McCoy L, Rice T, Schwartzbaum J, Heimberger A, Sampson JH, Chang S, et al.: IgE, allergy, and risk of glioma: update from the San Francisco Bay Area Adult Glioma Study in the temozolomide era. Int J Cancer 2009,125(3):680–687.
Berg-Beckhoff G, Schuz J, Blettner M, Munster E, Schlaefer K, Wahrendorf J, Schlehofer B: History of allergic disease and epilepsy and risk of glioma and meningioma (INTERPHONE study group, Germany). Eur J Epidemiol 2009,24(8):433–440.
Il'yasova D, McCarthy B, Marcello J, Schildkraut JM, Moorman PG, Krishnamachari B, Ali-Osman F, Bigner DD, Davis F: Association between glioma and history of allergies, asthma, and eczema: a case-control study with three groups of controls. Cancer Epidemiol Biomarkers Prev 2009,18(4):1232–1238.
Lachance DH, Yang P, Johnson DR, Decker PA, Kollmeyer TM, McCoy LS, Rice T, Xiao Y, Ali-Osman F, Wang F, et al.: Associations of high-grade glioma with glioma risk alleles and histories of allergy and smoking. Am J Epidemiol 2011,174(5):574–581.
Germain RN: MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 1994,76(2):287–299.
van Kooten C, Banchereau J: CD40-CD40 ligand. J Leukoc Biol 2000,67(1):2–17.
Bonnefoy JY, Gauchat JF, Life P, Graber P, Aubry JP, Lecoanet-Henchoz S: Regulation of IgE synthesis by CD23/CD21 interaction. Int Arch Allergy Immunol 1995,107(1–3):40–42.
Gould HJ, Sutton BJ: IgE in allergy and asthma today. Nat Rev Immunol 2008,8(3):205–217.
Zhou M, Wiemels JL, Bracci PM, Wrensch MR, McCoy LS, Rice T, Sison JD, Patoka JS, Wiencke JK: Circulating levels of the innate and humoral immune regulators CD14 and CD23 are associated with adult glioma. Cancer Res 2010,70(19):7534–7542.
Wrensch M, Wiencke JK, Wiemels J, Miike R, Patoka J, Moghadassi M, McMillan A, Kelsey KT, Aldape K, Lamborn KR, et al.: Serum IgE, tumor epidermal growth factor receptor expression, and inherited polymorphisms associated with glioma survival. Cancer Res 2006,66(8):4531–4541.
Stone KD, Prussin C, Metcalfe DD: IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol 2010,125(2 Suppl 2):S73–80.
Rothenberg ME, Zimmermann N, Mishra A, Brandt E, Birkenberger LA, Hogan SP, Foster PS: Chemokines and chemokine receptors: their role in allergic airway disease. J Clin Immunol 1999,19(5):250–265.
Akdis CA, Akdis M: Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J Allergy Clin Immunol 2009,123(4):735–746. quiz 747–738
Gleich GJ, Adolphson CR, Leiferman KM: The biology of the eosinophilic leukocyte. Annu Rev Med 1993, 44:85–101.
Foster EL, Simpson EL, Fredrikson LJ, Lee JJ, Lee NA, Fryer AD, Jacoby DB: Eosinophils increase neuron branching in human and murine skin and in vitro. PLoS One 2011,6(7):e22029.
Kita H: Eosinophils: multifaceted biological properties and roles in health and disease. Immunol Rev 2011,242(1):161–177.
Rothenberg ME, Hogan SP: The eosinophil. Annu Rev Immunol 2006, 24:147–174.
Trivedi SG, Lloyd CM: Eosinophils in the pathogenesis of allergic airways disease. Cell Mol Life Sci 2007,64(10):1269–1289.
Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, Lee JJ, Lohning M, Berek C: Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol 2011,12(2):151–159.
Chu VT, Berek C: Immunization induces activation of bone marrow eosinophils required for plasma cell survival. Eur J Immunol 2011, in press.
Minai-Fleminger Y, Levi-Schaffer F: Mast cells and eosinophils: the two key effector cells in allergic inflammation. Inflamm Res 2009,58(10):631–638.
Asquith KL, Ramshaw HS, Hansbro PM, Beagley KW, Lopez AF, Foster PS: The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J Immunol 2008,180(2):1199–1206.
Akuthota P, Wang HB, Spencer LA, Weller PF: Immunoregulatory roles of eosinophils: a new look at a familiar cell. Clin Exp Allergy 2008,38(8):1254–1263.
Sherman PW, Holland E, Sherman JS: Allergies: their role in cancer prevention. Q Rev Biol 2008,83(4):339–362.
Sanai N, Alvarez-Buylla A, Berger MS: Neural stem cells and the origin of gliomas. N Engl J Med 2005,353(8):811–822.
Vena JE, Bona JR, Byers TE, Middleton E Jr, Swanson MK, Graham S: Allergy-related diseases and cancer: an inverse association. Am J Epidemiol 1985,122(1):66–74.
El-Zein M, Parent ME, Ka K, Siemiatycki J, St-Pierre Y, Rousseau MC: History of asthma or eczema and cancer risk among men: a population-based case-control study in Montreal, Quebec, Canada. Ann Allergy Asthma Immunol 2010,104(5):378–384.
Negri E, Bosetti C, La Vecchia C, Levi F, Tomei F, Franceschi S: Allergy and other selected diseases and risk of colorectal cancer. Eur J Cancer 1999,35(13):1838–1841.
Moezzi J, Gopalswamy N, Haas RJ Jr, Markert RJ, Suryaprasad S, Bhutani MS: Stromal eosinophilia in colonic epithelial neoplasms. Am J Gastroenterol 2000,95(2):520–523.
Kural YB, Su O, Onsun N, Uras AR: Atopy, IgE and eosinophilic cationic protein concentration, specific IgE positivity, eosinophil count in cutaneous T Cell lymphoma. Int J Dermatol 2010,49(4):390–395.
Iwasaki K, Torisu M, Fujimura T: Malignant tumor and eosinophils. I. Prognostic significance in gastric cancer. Cancer 1986,58(6):1321–1327.
von Wasielewski R, Seth S, Franklin J, Fischer R, Hubner K, Hansmann ML, Diehl V, Georgii A: Tissue eosinophilia correlates strongly with poor prognosis in nodular sclerosing Hodgkin's disease, allowing for known prognostic factors. Blood 2000,95(4):1207–1213.
Hayes RL, Arbit E, Odaimi M, Pannullo S, Scheff R, Kravchinskiy D, Zaroulis C: Adoptive cellular immunotherapy for the treatment of malignant gliomas. Crit Rev Oncol Hematol 2001,39(1–2):31–42.
Whitehead RP, Friedman KD, Clark DA, Pagani K, Rapp L: Phase I trial of simultaneous administration of interleukin 2 and interleukin 4 subcutaneously. Clin Cancer Res 1995,1(10):1145–1152.
Dorta RG, Landman G, Kowalski LP, Lauris JR, Latorre MR, Oliveira DT: Tumour-associated tissue eosinophilia as a prognostic factor in oral squamous cell carcinomas. Histopathology 2002,41(2):152–157.
Ono Y, Ozawa M, Tamura Y, Suzuki T, Suzuki K, Kurokawa K, Fukabori Y, Yamanaka H: Tumor-associated tissue eosinophilia of penile cancer. Int J Urol 2002,9(2):82–87.
Moroni M, Porta C, De Amici M, Quaglini S, Cattabiani MA, Buzio C: Eosinophils and C4 predict clinical failure of combination immunotherapy with very low dose subcutaneous interleukin-2 and interferon in renal cell carcinoma patients. Haematologica 2000,85(3):298–303.
Bethwaite PB, Holloway LJ, Yeong ML, Thornton A: Effect of tumour associated tissue eosinophilia on survival of women with stage IB carcinoma of the uterine cervix. J Clin Pathol 1993,46(11):1016–1020.
Montgomery SM, Ehlin AG, Sparen P, Bjorksten B, Ekbom A: Childhood indicators of susceptibility to subsequent cervical cancer. Br J Cancer 2002,87(9):989–993.
Grossmann ME, Huang H, Tindall DJ: Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst 2001,93(22):1687–1697.
Karr JP, Pontes JE, Schneider S, Sandberg AA, Murphy GP: Clinical aspects of steroid hormone receptors in human renal cell carcinoma. J Surg Oncol 1983,23(2):117–124.
Samoszuk M, Ramzi E: IgE, Reed-Sternberg cells, and eosinophilia in Hodgkin's disease. Leuk Lymphoma 1993,9(4–5):315–319.
Di Biagio E, Sanchez-Borges M, Desenne JJ, Suarez-Chacon R, Somoza R, Acquatella G: Eosinophilia in Hodgkin's disease: a role for interleukin 5. Int Arch Allergy Immunol 1996,110(3):244–251.
Chong BF, Wilson AJ, Gibson HM, Hafner MS, Luo Y, Hedgcock CJ, Wong HK: Immune function abnormalities in peripheral blood mononuclear cell cytokine expression differentiates stages of cutaneous T-cell lymphoma/mycosis fungoides. Clin Cancer Res 2008,14(3):646–653.
Poppema S, Potters M, Visser L, van den Berg AM: Immune escape mechanisms in Hodgkin's disease. Ann Oncol 1998,9(Suppl 5):S21–24.
Miyagaki T, Sugaya M: Erythrodermic cutaneous T-cell lymphoma: how to differentiate this rare disease from atopic dermatitis. J Dermatol Sci 2011,64(1):1–6.
Robinson DS: The role of the T cell in asthma. J Allergy Clin Immunol 2010,126(6):1081–1091. quiz 1092–1083
Hennino A, Jean-Decoster C, Giordano-Labadie F, Debeer S, Vanbervliet B, Rozieres A, Schmitt AM, Nicolas JF: CD8+ T cells are recruited early to allergen exposure sites in atopy patch test reactions in human atopic dermatitis. J Allergy Clin Immunol 2011,127(4):1064–1067.
Graeff-Teixeira C, da Silva AC, Yoshimura K: Update on eosinophilic meningoencephalitis and its clinical relevance. Clin Microbiol Rev 2009,22(2):322–348. Table of Contents
Kono Y, Itoh Y: Diffusion-weighted imaging of encephalopathy related to idiopathic hypereosinophilic syndrome. Clin Neurol Neurosurg 2009,111(6):551–553.
Lo Re V, Gluckman SJ: Eosinophilic meningitis. Am J Med 2003,114(3):217–223.
Chao CC, Hsieh ST, Shun CT, Hsieh SC: Skin denervation and cutaneous vasculitis in eosinophilia-associated neuropathy. Arch Neurol 2007,64(7):959–965.
Russell HV, Strother D, Mei Z, Rill D, Popek E, Biagi E, Yvon E, Brenner M, Rousseau R: A phase 1/2 study of autologous neuroblastoma tumor cells genetically modified to secrete IL-2 in patients with high-risk neuroblastoma. J Immunother 2008,31(9):812–819.
Hua W, Xu F, Mao Y, Zhang J, Wang Y, Mao R, Zhou L: Primary intracranial leiomyomas: Report of two cases and review of the literature. Clin Neurol Neurosurg 2009,111(10):907–912.
Defendini R, Hunter SB, Schlesinger EB, Leifer E, Rowland LP: Eosinophilic meningitis in a case of disseminated glioblastoma. Arch Neurol 1981,38(1):52–53.
Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ, Pierz DM, Chen DK, Budzilovich GN, Ransohoff J: Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer 1995,76(5):840–852.
Cormier SA, Taranova AG, Bedient C, Nguyen T, Protheroe C, Pero R, Dimina D, Ochkur SI, O'Neill K, Colbert D, et al.: Pivotal Advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J Leukoc Biol 2006,79(6):1131–1139.
Tehrani M, Friedman TM, Olson JJ, Brat DJ: Intravascular thrombosis in central nervous system malignancies: a potential role in astrocytoma progression to glioblastoma. Brain Pathol 2008,18(2):164–171.
Golden J, Frim DM, Chapman PH, Vonsattel JP: Marked tissue eosinophilia within organizing chronic subdural hematoma membranes. Clin Neuropathol 1994,13(1):12–16.
Muller W, Firsching R: Significance of eosinophilic granulocytes in chronic subdural hematomas. Neurosurg Rev 1990,13(4):305–308.
Lee EL, Armstrong TS: Increased intracranial pressure. Clin J Oncol Nurs 2008,12(1):37–41.
Wang AM, Chinwuba CE, O'Reilly GV, Kleefield J: Subdural hematoma in patients with brain tumor: CT evaluation. J Comput Assist Tomogr 1985,9(3):511–513.
Ciembroniewicz J, Kolar O: Eosinophilic response in glioblastoma tissue culture after addition of autologous lymphocytes. Science 1967,157(792):1054–1055.
Curran CS, Bertics PJ: Human eosinophils express RAGE, produce RAGE ligands, exhibit PKC-delta phosphorylation and enhanced viability in response to the RAGE ligand, S100B. Int Immunol 2011, in press.
Steiner J, Marquardt N, Pauls I, Schiltz K, Rahmoune H, Bahn S, Bogerts B, Schmidt RE, Jacobs R: Human CD8(+) T cells and NK cells express and secrete S100B upon stimulation. Brain Behav Immun 2011,25(6):1233–1241.
Steiner J, Bogerts B, Schroeter ML, Bernstein HG: S100B protein in neurodegenerative disorders. Clin Chem Lab Med 2011,49(3):409–424.
Gartner W, Ilhan A, Neziri D, Base W, Weissel M, Wohrer A, Heinzl H, Waldhor T, Wagner L, Preusser M: Elevated blood markers 1 year before manifestation of malignant glioma. Neuro Oncol 2010,12(9):1004–1008.
Jacobsen EA, Taranova AG, Lee NA, Lee JJ: Eosinophils: singularly destructive effector cells or purveyors of immunoregulation? J Allergy Clin Immunol 2007,119(6):1313–1320.
Louis DN: Molecular pathology of malignant gliomas. Annu Rev Pathol 2006, 1:97–117.
Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, Yadav AK, Vogel H, Scheck AC, Tibshirani R, et al.: A network model of a cooperative genetic landscape in brain tumors. Jama 2009,302(3):261–275.
Gu J, Liu Y, Kyritsis AP, Bondy ML: Molecular epidemiology of primary brain tumors. Neurotherapeutics 2009,6(3):427–435.
Bhatti P, Stewart PA, Hutchinson A, Rothman N, Linet MS, Inskip PD, Rajaraman P: Lead exposure, polymorphisms in genes related to oxidative stress, and risk of adult brain tumors. Cancer Epidemiol Biomarkers Prev 2009,18(6):1841–1848.
Squier TC: Oxidative stress and protein aggregation during biological aging. Exp Gerontol 2001,36(9):1539–1550.
Kumar P, Devi U, Ali S, Upadhya R, Pillai S, Raja A, Rao S, Rao A: Plasma protein oxidation in patients with brain tumors. Neurol Res 2009,31(3):270–273.
Miller G: Brain cancer. A viral link to glioblastoma? Science 2009,323(5910):30–31.
Schwartzbaum J, Ahlbom A, Malmer B, Lonn S, Brookes AJ, Doss H, Debinski W, Henriksson R, Feychting M: Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res 2005,65(14):6459–6465.
Schwartzbaum JA, Ahlbom A, Lonn S, Malmer B, Wigertz A, Auvinen A, Brookes AJ, Collatz Christensen H, Henriksson R, Johansen C, et al.: An international case-control study of interleukin-4Ralpha, interleukin-13, and cyclooxygenase-2 polymorphisms and glioblastoma risk. Cancer Epidemiol Biomarkers Prev 2007,16(11):2448–2454.
Soroceanu L, Matlaf L, Bezrookove V, Harkins L, Martinez R, Greene M, Soteropoulos P, Cobbs CS: Human Cytomegalovirus US28 Found in Glioblastoma Promotes an Invasive and Angiogenic Phenotype. Cancer Res 2011.
Ruan Z, Zhao Y, Yan L, Chen H, Fan W, Chen J, Wu Q, Qian J, Zhang T, Zhou K, et al.: Single nucleotide polymorphisms in IL-4Ra, IL-13 and STAT6 genes occurs in brain glioma. Front Biosci (Elite Ed) 2011, 3:33–45.
Kuperman DA, Schleimer RP: Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med 2008,8(5):384–392.
Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, et al.: Lebrikizumab treatment in adults with asthma. N Engl J Med 2011,365(12):1088–1098.
Gauvreau GM, Boulet LP, Cockcroft DW, Fitzgerald JM, Carlsten C, Davis BE, Deschesnes F, Duong M, Durn BL, Howie KJ, et al.: Effects of interleukin-13 blockade on allergen-induced airway responses in mild atopic asthma. Am J Respir Crit Care Med 2011,183(8):1007–1014.
Sosman JA, Bartemes K, Offord KP, Kita H, Fisher SG, Kefer C, Ellis TA, Fisher RI, Higgins TJ, Gleich GJ: Evidence for eosinophil activation in cancer patients receiving recombinant interleukin-4: effects of interleukin-4 alone and following interleukin-2 administration. Clin Cancer Res 1995,1(8):805–812.
Atkins MB, Vachino G, Tilg HJ, Karp DD, Robert NJ, Kappler K, Mier JW: Phase I evaluation of thrice-daily intravenous bolus interleukin-4 in patients with refractory malignancy. J Clin Oncol 1992,10(11):1802–1809.
Margolin K, Aronson FR, Sznol M, Atkins MB, Gucalp R, Fisher RI, Sunderland M, Doroshow JH, Ernest ML, Mier JW, et al.: Phase II studies of recombinant human interleukin-4 in advanced renal cancer and malignant melanoma. J Immunother Emphasis Tumor Immunol 1994,15(2):147–153.
Yu JS, Wei MX, Chiocca EA, Martuza RL, Tepper RI: Treatment of glioma by engineered interleukin 4-secreting cells. Cancer Res 1993,53(13):3125–3128.
Tepper RI, Coffman RL, Leder P: An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 1992,257(5069):548–551.
Tepper RI, Pattengale PK, Leder P: Murine interleukin-4 displays potent anti-tumor activity in vivo. Cell 1989,57(3):503–512.
Platzer C, Richter G, Uberla K, Hock H, Diamantstein T, Blankenstein T: Interleukin-4-mediated tumor suppression in nude mice involves interferon-gamma. Eur J Immunol 1992,22(7):1729–1733.
Eguchi J, Kuwashima N, Hatano M, Nishimura F, Dusak JE, Storkus WJ, Okada H: IL-4-transfected tumor cell vaccines activate tumor-infiltrating dendritic cells and promote type-1 immunity. J Immunol 2005,174(11):7194–7201.
Cho SH, Stanciu LA, Holgate ST, Johnston SL: Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am J Respir Crit Care Med 2005,171(3):224–230.
Biller H, Bade B, Matthys H, Luttmann W, Virchow JC: Interferon-gamma secretion of peripheral blood CD8+ T lymphocytes in patients with bronchial asthma: in vitro stimulus determines cytokine production. Clin Exp Immunol 2001,126(2):199–205.
Yamaguchi T, Kimura H, Kurabayashi M, Kozawa K, Kato M: Interferon-gamma enhances human eosinophil effector functions induced by granulocyte-macrophage colony-stimulating factor or interleukin-5. Immunol Lett 2008,118(1):88–95.
Isogai S, Athiviraham A, Fraser RS, Taha R, Hamid Q, Martin JG: Interferon-gamma-dependent inhibition of late allergic airway responses and eosinophilia by CD8+ gammadelta T cells. Immunology 2007,122(2):230–238.
Mossman BT: Introduction to serial reviews on the role of reactive oxygen and nitrogen species (ROS/RNS) in lung injury and diseases. Free Radic Biol Med 2003,34(9):1115–1116.
Li JJ, Dickson D, Hof PR, Vlassara H: Receptors for advanced glycosylation endproducts in human brain: role in brain homeostasis. Mol Med 1998,4(1):46–60.
Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE: Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol 1999,155(5):1557–1567.
Curran CS, Evans MD, Bertics PJ: GM-CSF production by glioblastoma cells has a functional role in eosinophil survival, activation, and growth factor production for enhanced tumor cell proliferation. J Immunol 2011,187(3):1254–1263.
Visconti R, Grieco D: New insights on oxidative stress in cancer. Curr Opin Drug Discov Devel 2009,12(2):240–245.
Giembycz MA, Lindsay MA: Pharmacology of the eosinophil. Pharmacol Rev 1999,51(2):213–340.
Rosenberg HF: Eosinophil-derived neurotoxin/RNase 2: connecting the past, the present and the future. Curr Pharm Biotechnol 2008,9(3):135–140.
Boix E, Torrent M, Sanchez D, Nogues MV: The antipathogen activities of eosinophil cationic protein. Curr Pharm Biotechnol 2008,9(3):141–152.
Yang D, Chen Q, Su SB, Zhang P, Kurosaka K, Caspi RR, Michalek SM, Rosenberg HF, Zhang N, Oppenheim JJ: Eosinophil-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J Exp Med 2008,205(1):79–90.
Grauer OM, Molling JW, Bennink E, Toonen LW, Sutmuller RP, Nierkens S, Adema GJ: TLR ligands in the local treatment of established intracerebral murine gliomas. J Immunol 2008,181(10):6720–6729.
Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C, et al.: HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 2009,6(1):e10.
Trosko JE, Ruch RJ: Cell-cell communication in carcinogenesis. Front Biosci 1998, 3:d208–236.
Park S, Zhao D, Hatanpaa KJ, Mickey BE, Saha D, Boothman DA, Story MD, Wong ET, Burma S, Georgescu MM, et al.: RIP1 activates PI3K-Akt via a dual mechanism involving NF-kappaB-mediated inhibition of the mTOR-S6K-IRS1 negative feedback loop and down-regulation of PTEN. Cancer Res 2009,69(10):4107–4111.
Angileri FF, Aguennouz M, Conti A, La Torre D, Cardali S, Crupi R, Tomasello C, Germano A, Vita G, Tomasello F: Nuclear factor-kappaB activation and differential expression of survivin and Bcl-2 in human grade 2–4 astrocytomas. Cancer 2008,112(10):2258–2266.
Siebenlist U, Franzoso G, Brown K: Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol 1994, 10:405–455.
Nakamura T, Takeshita I, Fukui M: Glioma-derived PDGF-related protein presents as 17 kd intracellularly and assembled form induces actin reorganization. J Neurooncol 1991,11(3):215–224.
Smith D, Shimamura T, Barbera S, Bejcek BE: NF-kappaB controls growth of glioblastomas/astrocytomas. Mol Cell Biochem 2008,307(1–2):141–147.
Ranza E, Facoetti A, Morbini P, Benericetti E, Nano R: Exogenous platelet-derived growth factor (PDGF) induces human astrocytoma cell line proliferation. Anticancer Res 2007,27(4B):2161–2166.
Bach MK, Brashler JR, Stout BK, Johnson HG, Sanders ME, Lin AH, Gorman RR, Bienkowski MJ, Ishizaka T: Activation of human eosinophils by platelet-derived growth factor. Int Arch Allergy Immunol 1992,97(2):121–129.
Horiuchi T, Weller PF: Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol 1997,17(1):70–77.
Walz TM, Nishikawa BK, Malm C, Briheim K, Wasteson A: Transforming growth factor alpha expression in normal human blood eosinophils: differential regulation by granulocyte-macrophage colony-stimulating factor and interleukin-3. Leukemia 1994,8(4):612–619.
Matsumoto K, Fukuda S, Nakamura Y, Saito H: Amphiregulin production by human eosinophils. Int Arch Allergy Immunol 2009,149(Suppl 1):39–44.
Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B: Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS One 2011,6(8):e23902.
Polajeva J, Sjosten AM, Lager N, Kastemar M, Waern I, Alafuzoff I, Smits A, Westermark B, Pejler G, Uhrbom L, et al.: Mast Cell Accumulation in Glioblastoma with a Potential Role for Stem Cell Factor and Chemokine CXCL12. PLoS One 2011,6(9):e25222.
Yao XH, Ping YF, Chen JH, Chen DL, Xu CP, Zheng J, Wang JM, Bian XW: Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. J Neurooncol 2008,86(1):47–53.
Kast RE, Scheuerle A, Wirtz CR, Karpel-Massler G, Halatsch ME: The Rationale of Targeting Neutrophils with Dapsone during Glioblastoma Treatment. Anticancer Agents Med Chem 2011.
Zhang L, Liu W, Alizadeh D, Zhao D, Farrukh O, Lin J, Badie SA, Badie B: S100B attenuates microglia activation in gliomas: possible role of STAT3 pathway. Glia 2011,59(3):486–498.
Sick E, Brehin S, Andre P, Coupin G, Landry Y, Takeda K, Gies JP: Advanced glycation end products (AGEs) activate mast cells. Br J Pharmacol 2010,161(2):442–455.
Gan HK, Kaye AH, Luwor RB: The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci 2009,16(6):748–754.
Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS: Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007,19(10):2013–2023.
Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, Muller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, et al.: RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 2008,205(2):275–285.
Bassi R, Giussani P, Anelli V, Colleoni T, Pedrazzi M, Patrone M, Viani P, Sparatore B, Melloni E, Riboni L: HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: role in cell growth and migration. J Neurooncol 2008,87(1):23–33.
Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C, Los M: S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol 2008,83(6):1484–1492.
Srikrishna G: S100A8 and S100A9: New Insights into Their Roles in Malignancy. J Innate Immun 2011.
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G: Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 2008,181(7):4666–4675.
Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D: S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 2006,312(2):184–197.
Yui S, Nakatani Y, Mikami M: Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull 2003,26(6):753–760.
Hoyaux D, Decaestecker C, Heizmann CW, Vogl T, Schafer BW, Salmon I, Kiss R, Pochet R: S100 proteins in Corpora amylacea from normal human brain. Brain Res 2000,867(1–2):280–288.
Deininger MH, Pater S, Strik H, Meyermann R: Macrophage/microglial cell subpopulations in glioblastoma multiforme relapses are differentially altered by radiochemotherapy. J Neurooncol 2001,55(3):141–147.
Muggia FM, Ghossein NA, Wohl H: Eosinophilia following radiation therapy. Oncology 1973,27(2):118–127.
Bowen JM, Newbold K, Blake P, Wild G, Egner W, Norman AR, Andreyev HJ: Do serum levels of eosinophil granule-derived protein change in patients undergoing pelvic radiotherapy? Clin Oncol (R Coll Radiol) 2005,17(5):382–384.
Trosko JE, Ruch RJ: Gap junctions as targets for cancer chemoprevention and chemotherapy. Curr Drug Targets 2002,3(6):465–482.
Rong Y, Durden DL, Van Meir EG, Brat DJ: 'Pseudopalisading' necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 2006,65(6):529–539.
Ito A, Miyake M, Morishita M, Ito K, Torii S, Sakamoto T: Dexamethasone reduces lung eosinophilia, and VCAM-1 and ICAM-1 expression induced by Sephadex beads in rats. Eur J Pharmacol 2003,468(1):59–66.
Leick-Maldonado EA, Kay FU, Leonhardt MC, Kasahara DI, Prado CM, Fernandes FT, Martins MA, Tiberio IF: Comparison of glucocorticoid and cysteinyl leukotriene receptor antagonist treatments in an experimental model of chronic airway inflammation in guinea-pigs. Clin Exp Allergy 2004,34(1):145–152.
McClelland S, Long DM: Genesis of the use of corticosteroids in the treatment and prevention of brain edema. Neurosurgery 2008,62(4):965–967. discussion 967–968
Goh JJ, See SJ, Ang E, Ng WH: Vanishing glioblastoma after corticosteroid therapy. J Clin Neurosci 2009,16(9):1226–1228.
Hasegawa H, Pal D, Ramirez R, Ismail A, Marks P: Glioblastoma multiforme fades on CT imaging after dexamethasone therapy. J Clin Neurosci 2009.
Gu YT, Qin LJ, Qin X, Xu F: The molecular mechanism of dexamethasone-mediated effect on the blood-brain tumor barrier permeability in a rat brain tumor model. Neurosci Lett 2009,452(2):114–118.
Bergmann MW, Staples KJ, Smith SJ, Barnes PJ, Newton R: Glucocorticoid inhibition of granulocyte macrophage-colony-stimulating factor from T cells is independent of control by nuclear factor-kappaB and conserved lymphokine element 0. Am J Respir Cell Mol Biol 2004,30(4):555–563.
Meagher LC, Cousin JM, Seckl JR, Haslett C: Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J Immunol 1996,156(11):4422–4428.
Walsh GM, Wardlaw AJ: Dexamethasone inhibits prolonged survival and autocrine granulocyte-macrophage colony-stimulating factor production by human eosinophils cultured on laminin or tissue fibronectin. J Allergy Clin Immunol 1997,100(2):208–215.
Noga O, Hanf G, Gorges D, Dinh QT, Groneberg DA, Suttorp N, Kunkel G: Regulation of NGF and BDNF by dexamethasone and theophylline in human peripheral eosinophils in allergics and non-allergics. Regul Pept 2005,132(1–3):74–79.
Wadhwa S, Nag TC, Jindal A, Kushwaha R, Mahapatra AK, Sarkar C: Expression of the neurotrophin receptors Trk A and Trk B in adult human astrocytoma and glioblastoma. J Biosci 2003,28(2):181–188.
Johnston AL, Lun X, Rahn JJ, Liacini A, Wang L, Hamilton MG, Parney IF, Hempstead BL, Robbins SM, Forsyth PA, et al.: The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol 2007,5(8):e212.
Liao YH, Su YW, Tsay W, Chiu HC: Association of cutaneous necrotizing eosinophilic vasculitis and deep vein thrombosis in hypereosinophilic syndrome. Arch Dermatol 2005,141(8):1051–1053.
Liapis H, Ho AK, Brown D, Mindel G, Gleich G: Thrombotic microangiopathy associated with the hypereosinophilic syndrome. Kidney Int 2005,67(5):1806–1811.
Aderka D, Brown A, Zelikovski A, Pinkhas J: Idiopathic deep vein thrombosis in an apparently healthy patient as a premonitory sign of occult cancer. Cancer 1986,57(9):1846–1849.
Milsom C, Yu J, May L, Magnus N, Rak J: Diverse roles of tissue factor-expressing cell subsets in tumor progression. Semin Thromb Hemost 2008,34(2):170–181.
Moosbauer C, Morgenstern E, Cuvelier SL, Manukyan D, Bidzhekov K, Albrecht S, Lohse P, Patel KD, Engelmann B: Eosinophils are a major intravascular location for tissue factor storage and exposure. Blood 2007,109(3):995–1002.
Rohrbach MS, Wheatley CL, Slifman NR, Gleich GJ: Activation of platelets by eosinophil granule proteins. J Exp Med 1990,172(4):1271–1274.
Fredens K, Dahl R, Venge P: The Gordon phenomenon induced by the eosinophil cationic protein and eosinophil protein X. J Allergy Clin Immunol 1982,70(5):361–366.
Durack DT, Sumi SM, Klebanoff SJ: Neurotoxicity of human eosinophils. Proc Natl Acad Sci USA 1979,76(3):1443–1447.
Chandana SR, Movva S, Arora M, Singh T: Primary brain tumors in adults. Am Fam Physician 2008,77(10):1423–1430.
Lotfi R, Schrezenmeier H, Lotze MT: Immunotherapy for cancer: promoting innate immunity. Front Biosci 2009, 14:818–832.
Friese MA, Steinle A, Weller M: The innate immune response in the central nervous system and its role in glioma immune surveillance. Onkologie 2004,27(5):487–491.
de Visser KE, Eichten A, Coussens LM: Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 2006,6(1):24–37.
Dunn GP, Old LJ, Schreiber RD: The three Es of cancer immunoediting. Annu Rev Immunol 2004, 22:329–360.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011, 29:235–271.
Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT: TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol 2010,12(1):7–13.
Subleski JJ, Jiang Q, Weiss JM, Wiltrout RH: The split personality of NKT cells in malignancy, autoimmune and allergic disorders. Immunotherapy 2011,3(10):1167–1184.
Wiemels JL, Wiencke JK, Patoka J, Moghadassi M, Chew T, McMillan A, Miike R, Barger G, Wrensch M: Reduced immunoglobulin E and allergy among adults with glioma compared with controls. Cancer Res 2004,64(22):8468–8473.
Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, Nelson SF, Liau LM: Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res 2011,17(6):1603–1615.
Chen K, Xiang Y, Yao X, Liu Y, Gong W, Yoshimura T, Wang JM: The active contribution of Toll-like receptors to allergic airway inflammation. Int Immunopharmacol 2011,11(10):1391–1398.
Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ: Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol 2003,4(10):974–981.
Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S: TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood 2005,106(3):978–987.
Kitajima M, Ito T, Tumes DJ, Endo Y, Onodera A, Hashimoto K, Motohashi S, Yamashita M, Nishimura T, Ziegler SF, et al.: Memory type 2 helper T cells induce long-lasting antitumor immunity by activating natural killer cells. Cancer Res 71(14):4790–4798.
Mantel PY, Kuipers H, Boyman O, Rhyner C, Ouaked N, Ruckert B, Karagiannidis C, Lambrecht BN, Hendriks RW, Crameri R, et al.: GATA3-driven Th2 responses inhibit TGF-beta1-induced FOXP3 expression and the formation of regulatory T cells. PLoS Biol 2007,5(12):e329.
Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, Kato S, Yoshimura A, Kobayashi T: STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem 2008,283(22):14955–14962.
Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, et al.: IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat Immunol 2008,9(12):1347–1355.
Acknowledgements
This work was supported by NIH grants AI070503, HL0885940, and HL069116 (to P.J.B.). In Memoriam: This manuscript is dedicated to the memory of my mentor, Paul J. Bertics, Ph.D. (1956-2011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
This article was written principally by CSC, with conceptual and editorial contributions from PJB. Both authors have read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Curran, C.S., Bertics, P.J. Eosinophils in glioblastoma biology. J Neuroinflammation 9, 11 (2012). https://doi.org/10.1186/1742-2094-9-11
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-9-11