
Abstract
Background
Plasmodium vivax is one of the five species causing malaria in human beings, affecting around 391 million people annually. The development of an anti-malarial vaccine has been proposed as an alternative for controlling this disease. However, its development has been hampered by allele-specific responses produced by the high genetic diversity shown by some parasite antigens. Evaluating these antigens’ genetic diversity is thus essential when designing a completely effective vaccine.
Methods
The gene sequences of Plasmodium vivax p12 (pv12) and p38 (pv38), obtained from field isolates in Colombia, were used for evaluating haplotype polymorphism and distribution by population genetics analysis. The evolutionary forces generating the variation pattern so observed were also determined.
Results
Both pv12 and pv38 were shown to have low genetic diversity. The neutral model for pv12 could not be discarded, whilst polymorphism in pv38 was maintained by balanced selection restricted to the gene’s 5′ region. Both encoded proteins seemed to have functional/structural constraints due to the presence of s48/45 domains, which were seen to be highly conserved.
Conclusions
Due to the role that malaria parasite P12 and P38 proteins seem to play during invasion in Plasmodium species, added to the Pv12 and Pv38 antigenic characteristics and the low genetic diversity observed, these proteins might be good candidates to be evaluated in the design of a multistage/multi-antigen vaccine.
Similar content being viewed by others


Background
Malaria is a disease caused by protozoan parasites from the Plasmodium genus, five of which cause the disease in human beings (Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae and Plasmodium knowlesi) [1, 2]. This parasite is transmitted by the bite of an infected Anopheles female mosquito. Around 3.3 billon people are at risk of malaria annually, mainly in tropical and subtropical areas of the world, children aged less than five years and pregnant women being the most vulnerable [3]. Plasmodium falciparum is responsible for the disease’s most lethal form, being predominantly found on the African continent whilst P. vivax is widely distributed around the world. Even though it has been thought that infection caused by the latter species was benign, recent studies have shown that P. vivax can cause clinical complications [4]. It has been found that 2,488 million people are at risk of becoming infected by P. vivax on the continents of Asia and America, 132 to 391 million cases occurring annually [5].
In spite of control strategies having been introduced in different countries, malaria continues to be a public health problem due to the parasite’s resistance to anti-malarial treatments [6] and the vector’s resistance to insecticides [7], among other causes. More effective measures have thus to be implemented for controlling such disease, including the development of an anti-malarial vaccine.
Several antigens have been characterized as promising candidates for inclusion in a vaccine [8, 9], however, the genetic diversity of some of them [10–18] has hampered the development of such vaccine [19, 20] as these genetic variations provoke allele-specific responses [21, 22] making them become a mechanism for evading the immune system [23]. It has been necessary to focus vaccine development on conserved domains or antigens to avoid such responses [24], since these regions could have functional constraint and have had slower evolution [25].
Developing a multi-antigen vaccine against the parasite’s blood stage has been focused on blocking all host-pathogen interactions to stop merozoite entry to red blood cells (RBC) [26]. A group of proteins anchored to the membrane via glycosylphosphatidylinositol (GPI) has been identified in P. falciparum, predominantly located in detergent-resistant membrane (DRM) domains [27, 28]; they have been implicated in the parasite’s initial interaction with RBC [29–33] and some have been considered as being candidates for being included in a vaccine [34, 35]. One group of proteins belonging to the 6-cystein (6-Cys) family is particularly noteworthy among these DRMs (i.e., Pf12, Pf38, Pf41 and Pf92) as they have been characterized by having s48/45 domains (ID in PFAM: PF07422). Members of this family are expressed during different parasite stages [28, 36] and some of them (e.g., Pf48/45, Pf230) have been considered as vaccine candidates for the sexual stage [36, 37].
Pf12 and Pf38 are expressed during late stages of the intra-erythrocyte cycle, each having two high binding peptides, suggesting an active role during invasion of RBC [30]. Orthologous genes encoding these proteins have been characterized recently in P. vivax[38, 39]. Both proteins have a signal peptide, a GPI anchor sequence and have been associated with DRMs [38, 39]. Pv12 has two s48/45 domains [39] whilst Pv38 has a single domain located towards the C-terminal end [38]. These proteins have been shown to be antigenic [38–40], suggesting that they are exposed to the immune system, probably during P. vivax invasion of RBC.
The present study involved a population genetics analysis for evaluating the genetic diversity of pv12 and pv38 loci and the evolutionary processes generating this variation pattern; the results revealed these antigens’ low genetic diversity in the Colombian population, possibly due to functional/structural constraints in s48/45 domains. Since the proteins encoded by these genes share structural characteristics with other vaccine candidates, added to the fact that Pv12 and Pv38 are targets for the immune response [38–40] and have conserved domains, they should be considered when designing a multistage/multi-antigen anti-malarial vaccine.
Methods
Ethics statement
The parasitized DNA used in this study was extracted from total blood collected from different Colombian areas (Antioquia, Atlántico, Bogotá, Caquetá, Cordoba, Chocó, Guainía, Guaviare, Magdalena, Meta, Nariño, and Tolima) from 2007 to 2010. All P. vivax-infected patients who provided blood samples were notified about the object of the study and signed an informed consent form if they agreed to participate. All procedures involved in taking blood samples were approved by Fundación Instituto de Inmunología de Colombia (FIDIC) ethics committee.
Parasitized DNA presence and integrity
Parasitized DNA presence and integrity in 100 samples stored at -20°C (2007-2010) at FIDIC (from different areas of Colombia) were evaluated by 18S ribosomal RNA gene amplification using specific primers for P. vivax (SSU-F 5′-ATGAACGAGATCTTAACCTGC-3′ and SSU-R 5′-CATCACGATATGTA5TGATAAAGATTACC-3′) in a touchdown PCR [41]. The reaction contained: 1x Mango Taq reaction buffer (Bioline), 2.5 mM MgCl2, 0.25 mM dNTPs, 0.5 mM of each primer, 0.1 U Mango Taq DNA polymerase (Bioline) and 10-40 ng gDNA in 10 mL final volume. The PCR thermal profile was: one initial denaturing cycle at 95°C (5 min), followed by ten cycles at 95°C (20 sec), annealing at 65°C (30 sec) and an extension step at 72°C (45 sec). Annealing temperature was reduced by 1°C in each cycle until reaching 55°C; 35 additional cycles were run at this temperature followed by a final extension cycle at 72°C (10 min). PCR products were visualized by electrophoresis on 1.5% agarose gel in 1× TAE, using 1 μL SYBR-Safe (Invitrogen).
Identifying infection caused by single Plasmodium vivax strain
Infection by the single P. vivax strain was identified by PCR-RFLP of the pvmsp-1 polymorphic marker. The pvmsp-1 gene fragment 2 (blocks 6, 7 and 8) was amplified using direct 5′-AAAATCGAGAGCATGATCGCCACTGAGAAG-3′ and reverse 5′-AGCTTGTACTTTCCATAGTGGTCCAG-3′ primers [42]. The amplified fragments were digested with Alu I and Mnl I restriction enzymes, as described elsewhere [42]. The products were visualized by electrophoresis on 3% agarose gel in 1× TAE, using 1 μL SYBR-Safe (Invitrogen).
PCR amplification of pv12 and pv38 genes
A set of primers was designed for amplifying each of the genes based on Sal-I reference strain sequences (accession numbers in PlasmoDB: PVX_113775 for pv12 and PVX_097960 for pv38). The following primers were used: for pv12, pv12- direct 5′-GTACCGCTTAACACCGC-3′ and pv12- reverse 5′-GCACTACATTATAAAGAAAAGGACC-3′ and for pv38, pv38- direct 5′-CGCTTCTTTCACCGCTTC-3′ and pv38- reverse 5′-CACACATTAACGCTGCTTCG-3′. The PCR reaction mixture contained 10 mM Tris HCL, 50 mM KCl (GeneAmp 10× PCR Buffer II [Applied Biosystems]), 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.5 μM of each primer, 0.76 U Amplitaq Gold DNA polymerase (Applied Biosystems) and 10-40 ng gDNA in a 50 μL final volume. The PCR thermal profile was as follows: one cycle at 95°C (7 min), 40 cycles at 95°C (20 sec), 56°C (30 sec), 72°C (1 min) and a final extension cycle at 72°C (10 min). PCR products were purified using a commercial UltraClean PCR Clean-up kit (MO BIO). The purified PCR products were sequenced in both directions with the amplification primers using the BigDye method with capillary electrophoresis, using ABI-3730 XL (MACROGEN, Seoul, South Korea). Two independent PCR products were sequenced to ensure that errors were ruled out.
Analysing genetic diversity
The electropherograms obtained by sequencing were analysed and forward and reverse sequences were assembled using CLC Main workbench software v.5 (CLC bio, Cambridge, MA, USA). The pv12 and pv38 genes were analysed and compared to reference sequences obtained from several sequencing projects [43, 44] (accession numbers, pv12: XM_001616094.1, AFBK01001496.1, AFNI01000939.1, AFMK01001167.1 and AFNJ01001458.1; pv38: XM_001613202.1, AFNI01000834.1, AFNJ01000090.1, AFMK01001057.1 and AFBK01001340.1) or those reported in the GenBank database (accession numbers for pv12: GU476521.1; and for pv38: JF427569.1 and JF427570.1). Gene Runner software was used for translating the sequences for deducing the amino acid sequences. These sequences were then aligned using the MUSCLE algorithm [45], and manually edited. Amino acid alignment was then used for inferring DNA using PAL2NAL software [46].
DnaSP software (v.5) [47] was used for evaluating intrapopulation genetic polymorphism by calculating: the number of polymorphic segregating sites (Ss), the number of singleton sites (s), the number of parsimony-informative sites (Ps), the number of haplotypes (H), haplotype diversity (Hd, which was multiplied by (n-1)/n according to Depaulis and Veuille [47, 48]), the Watterson estimator (θw) and nucleotide diversity per site (π). DNA sequence variation was calculated using the sequences obtained from the aforementioned databases, plus the Colombian ones (worldwide isolates, global diversity) and just those obtained for the Colombian population (local diversity). The frequency for each Colombian haplotype was also estimated by count and year.
Two test families were used for evaluating the neutral molecular evolution model for the Colombian population: (1) frequency spectrum test, and (2) haplotype test. The former involved calculating Tajima’s D statistics [49], Fu and Li’s D* and F* [50] and Fay and Wu’s H statistic [51]. Tajima’s D statistic compares the difference between segregating sites and the average of nucleotide differences between two randomly taken sequences. Fu and Li’s D* statistic takes the difference between the number of singleton sites and the total of mutations, whilst F* takes the difference between the number of singleton sites and the average of nucleotide differences between two randomly taken sequences. Fay and Wu’s H statistic is based on the difference of the average number of nucleotide differences between pairs of sequences and the frequency of the derived variants. Fu’s Fs statistic [52], K-test and H-test [48] are tests for calculating haplotype distribution. The Fs statistic compares the number of haplotypes observed to the expected number of haplotypes in a random sample. K-test and H-test [48] are based on haplotype number and haplotype diversity, respectively; these statistics are conditioned by sample size (n) and the number of segregating sites (Ss). Test significance was determined by coalescence simulations using DnaSP (v.5) [47] and ALLELIX software (kindly supplied by Dr Sylvain Mousset). Sites having gaps were not taken into account in any of the tests performed.
The effect of natural selection was evaluated regarding intra and interspecies; the average number of non-synonymous substitutions per non-synonymous site (dN) and the average number of synonymous substitutions per synonymous site (dS) were calculated for the former by using the modified Nei-Gojobori method [53]. The significant differences between the above were determined by using Fisher’s exact test (suitable for dN and dS < 10) and codon-based Z-test incorporated in MEGA software (v.5) [54]. Differences between dN and dS per site were calculated by using SLAC, FEL, REL [55], IFEL [56], MEME [57], and FUBAR [58] methods. The average number of non-synonymous divergence substitutions per non-synonymous site (KN) and the average number of synonymous divergence substitutions per synonymous site (KS) were calculated using the modified Nei-Gojobori method [53], with Jukes-Cantor correction [59], to infer natural selection signals which may have prevailed during malarial parasite evolutionary history (interspecies; using Plasmodium cynomolgi (accession number BAEJ01001076.1) and P. knowlesi (accession number NC_011912.1) orthologous sequences). The significant differences between KN and KS were determined by using a codon-based Z-test incorporated in MEGA software (v.5) [54]. The McDonald-Kreitman test [60] was also calculated; this is based on a comparison of intraspecific polymorphism to interspecific divergence (using Plasmodium cynomolgi (accession number BAEJ01001076.1) and P. knowlesi (accession number NC_011912.1) orthologous sequences). This test involved using a web server [61], which takes Jukes-Cantor divergence correction into account [59]. All the above tests were calculated using the sequences obtained from the databases plus the Colombian ones and just those obtained for the Colombian population.
ZnS[62] and ZZ [63] statistics were calculated for evaluating the influence of linkage disequilibrium (LD) and intragenic recombination, respectively. The minimum number of recombination (Rm) events was also calculated; this included calculating effective population size and the probability of recombination between adjacent nucleotides per generation [64]. Additionally, the GARD method [65] available at the Datamonkey web server [66] was performed. These tests were performed using the sequences obtained from the Colombian population.
Results and discussion
The presence of genomic DNA (gDNA) and identification of single Plasmodium vivax strain infection
An 18S subunit rRNA gene fragment was amplified from 100 samples of P. vivax collected from different areas of Colombia and stored from 2007 to 2010. Seventy-seven samples revealed an amplicon at the expected size, indicating the presence of P. vivax gDNA. A region of the pvmsp-1 gene was then amplified and digested with restriction enzymes, showing that seven of the 77 samples proving positive for P. vivax had multiple infections. Only 70 samples were thus considered for later analysis. Due to the low number of samples collected from some areas, they were grouped according to geographical localisation and epidemiological conditions (South-west: Chocó, Nariño; South-east: Caquetá, Guainía, Guaviare, Meta; Midwest: Bogota, Tolima; North-west: Atlántico, Antioquia Cordoba, Magdalena).
Genetic diversity in pv12
Seventy samples amplified a 1,200 base pair (bp) fragment corresponding to the pv12 gene (South-west n = 6; South-east: n = 20; Midwest: n = 8; North-west: n =36). These amplicons were purified and sequenced; the sequences were then analysed, compared to different reference sequences obtained from various sequencing projects [43, 44] and those having a different haplotype were deposited in the GenBank database (accession numbers KF667328 and KF667329).
Four single nucleotide polymorphisms (SNP) were observed throughout the pv12 gene sequence (Figure 1A) located in positions 375 (N125K), 379 (T127A), 539 (L180W) and 662 (N221S). Only one SNP (nucleotide 375) was found in the Colombian population. A repeat region was observed; it was formed by previously reported amino acids N[A/V][H/Q] [39], in which an insertion was observed in the North Korean sequence (Figure 1A, haplotype 1) and deletions in the Colombian sequences (Figure 1A, haplotypes 2 and 3).

pv12 and pv38 haplotype alignment. A. Alignment of the six haplotypes found in the pv12 gene. Haplotypes 2, 3, 5 and 6 were found in the Colombian population. B. Alignment of the 17 haplotypes found in pv38, 14 of them found in the Colombian population (haplotypes 1, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, and 17). The dots indicate nucleotide identity and dashes indicate nucleotide absence. Numbering is based on the Sal-I reference sequence.
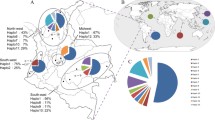
Six haplotypes were found in pv12 (Figure 1A and Table 1) around the world, four of which are present in Colombia at 8.7, 5.8, 10.1, and 75.4% frequency for haplotypes 2, 3, 5 and 6, respectively. Haplotypes 2, 5 and 6 were present in the different Colombian locations (Additional file 1), haplotype 6 being the most predominant per year (2007 n = 9; 2008 n = 17; 2009 n = 15; 2010 n = 29) and per location, having higher than 70% frequency (Figure 2A and Additional file 1). The remaining haplotypes were absent or had low frequency (Figure 2A and Additional file 1). Interestingly, haplotype 3 was present in Colombia during 2009 but absent in the other years studied (Figure 2A). The percentage of samples from the South-east area (some of them presenting haplotype 3) was greater than for other years, suggesting that haplotype 3 was restricted to a particular geographical area (Additional file 1) and/or that this had very low frequency in different Colombian subpopulations. Haplotype 2 was absent from 2007 to 2008 but present between 2009 and 2010 (Figure 2A); differently to haplotype 3, this haplotype was present everywhere, except in the South-west location (Additional file 1). This appeared to be consistent with previous studies which have reported numerous private haplotypes in American Plasmodium vivax populations [67]. These results suggested that the Colombian population had one predominant pv12 haplotype and several low frequency alleles, which are geographically isolated or were not detected during some periods of time. Since P. vivax populations within countries seem to be strongly structured [67], new pv12 haplotypes could appear in other parasite populations.

Frequency of the pv12 and pv38 haplotypes present in the Colombian population. A. Frequency per year for the four pv12 haplotypes found in the Colombian population: n = 9 in 2007, n = 17 in 2008, n = 15 in 2009 and n = 29 in 2010. B. Frequency per year for the 14 pv38 haplotypes found in the Colombian population. n = 6 in 2007, n = 6 in 2008, n = 8 in 2009 and n = 26 in 2010.
This gene had 0.0004 ± 0.0001 global nucleotide diversity (π) and 0.0003 ± 0.0001 for the Colombian population (Table 1). This value was about 2.5 times less than that reported for its orthologue in P. falciparum (π = 0.001) [68]; however, both values were low when compared to other membrane proteins [10–14, 17], suggesting that this gene is highly conserved in different Plasmodium species. This value places pv12 among the most conserved antigen-encoding genes characterized to date in P. vivax.
Mutations in pv12 appear to be selectively neutral
Several tests for evaluating the hypothesis that mutations in pv12 are neutral were performed. No significant values were found for the Tajima, Fu and Li, Fay and Wu or Fu tests (Table 2); likewise, the Colombian population’s number of haplotypes (4) and haplotype diversity (0.406 ± 0.07) (Table 2) were as expected under neutrality according to the K-test and H-test. Since neutrality could not be ruled out, the mutations or haplotypes found in pv12 could have been randomly fixed; this might explain the possible geographical isolation of haplotype 3, since different alleles could have become fixed in different populations according to the neutral hypothesis. Alternatively, the geographical isolation of haplotype 3 could have resulted from the structured P. vivax population in America, where haplotypes may have diversified in situ.
Natural selection in pv12
The gene was split into two regions: region A, nucleotides 1-546 (amino acids 1-182 including one s48/45 domain) and region B, nucleotides 547-1,095 (amino acids 183-365 including the other s48/45 domain). Synonymous substitution per synonymous site (dS) and non-synonymous substitution per non-synonymous site rates (dN) were calculated using the gene’s total length to evaluate whether natural selection had any effect on pv12 evolution. Full length gene and split regions had non-significant values (Table 3); likewise, when dN and dS were estimated for s48/45 domains, no significant values were observed (Additional file 2), contrary to that suggested for pf12, where purifying selection action has been reported [69]. The Datamonkey server was used for calculating dN and dS rates for each codon; no selected sites were found, indicating (once more) that the gene did not appear to deviate from neutrality.
However, assessing how natural selection acts on low genetic diversity antigens is not easy [70]; the fact that Plasmodium vivax shares its most recent common ancestor with parasites infecting primates (e.g. P. cynomolgi and P. knowlesi) led to inferring patterns which may have prevailed during their evolutionary history [70, 71]. When synonymous divergence substitution per synonymous site (KS) and non-synonymous divergence substitution per non-synonymous site (KN) rates were calculated, a significantly higher KS than KN was found (Table 4). Moreover, a sliding window for ω (dN/dS and/or KN/KS) revealed < 1 values throughout the gene (Figure 3), which could have been a consequence of negative selection. Moreover, significant values were observed when the McDonald-Kreitman (MK) test was used for comparing intraspecific polymorphism and interspecific divergence (using all the haplotypes found for this gene): PN/PS > DN/DS (Table 5), revealing (similar to KS rates) a large accumulation of synonymous substitutions between species, which could be interpreted as negative selection. Such accumulation of interspecies synonymous substitutions suggested that evolution tried to maintain protein structure by eliminating all deleterious mutations. However, when the MK test was done with haplotypes found in Colombia (and in spite of the accumulation of synonymous substitutions between species), no significant values were observed in this population (Table 5). Although Pv12 is exposed to the immune system [39, 40], it had a high level of conservation. This pattern could have been because pv12 had diverged by negative selection, due to a possible functional/structural constraint imposed by the presence of s48/45 domains [72] which seem to play an important role during host cell recognition [30, 69, 72].

Sliding window analysis for ω rates. The ω (dN/dS) values for Plasmodium vivax p12 and p38 are shown in blue, whereas the divergence (ω: KN/KS) between Plasmodium vivax and Plasmodium cynomolgi (Pcyn) and Plasmodium vivax and Plasmodium knowlesi (Pkno) is displayed in magenta and purple, respectively. A gene diagram is shown below the sliding window. Regions encoding signal peptides (brown), GPI anchors (green), s48/45 domains (dark cyan) as well as the N[A/V][H/Q] repeat (black) are indicated. Non-synonymous (red) and synonymous (orange) substitutions are shown with vertical lines above each gene.
Genetic diversity in pv38
Only 46 out of 70 samples could be amplified for the pv38 gene, giving a 1,121 bp fragment (South-west n = 6; South-east: n = 13; Midwest: n = 4; North-west: n = 23). The 46 sequences obtained from Colombian isolates were compared to and analysed regarding reference sequences obtained from different regions of the world [43, 44]. Colombian sequences that have a different haplotype to that of previously reported ones can be found in GenBank (accession numbers KF667330-KF667340).
Nine SNPs were observed in the pv38 gene (Figure 1B), most of which were no-synonymous (nucleotides: 88 (R30S), 206/207 (A69V), 209 (R70L), 524/525 (T175N), 880 (M294L), and 998 (S333N)), similar to that found in Pf38[73]. Positions 525 and 969 produced synonymous substitutions (a change in protein sequence was generated when the substitution in position 525 was accompanied with another one in position 524). The parasite population in Colombia has eight of these nine SNPs, all being informative-parsimonious sites. Similar to that reported for its orthologue in P. falciparum[73], most substitutions were found in the gene’s 5′ region.
Seventeen haplotypes were identified from alignment (including sequences from different regions of the world) (Figure 1B), 14 of which were found in Colombia’s parasite population at different frequencies: 11% haplotype 1, 4% haplotype 5, 2% haplotype 6, 20% haplotype 7, 15% haplotype 8, 7% haplotype 9, 2% haplotype 10, 4% haplotype 11, 13% haplotype 12, 4% haplotype 13, 2% haplotype 14, 4% haplotype 15, 7% haplotype 16, and 4% haplotype 17. Most haplotypes were found in intermediate frequencies per year (2007 n = 6; 2008 n = 6; 2009 n = 8; 2010 n = 26) and none exceeded 50% (Figure 2B). The absence of some haplotypes in determined years, or in some locations, could not just have been due to the low frequency which they might have had but also to the difference in the number of samples for each year (n = 6 in 2007, n = 6 in 2008, n = 8 in 2009 and n = 26 in 2010) or because American P. vivax populations appear to be structured and therefore several privative haplotypes might be found [67].
π in this gene was 0.0026 ± 0.0002 worldwide and 0.0024 ± 0.0002 in the Colombian population (Table 1), this being 1.3 times lower than that for its orthologue in P. falciparum (π = 0.0034) [68, 73] showing that the pv38 gene had low diversity, at least in the two main species affecting human beings.
Deviation from the neutral model of molecular evolution in pv38
Tajima’s D, Fu and Li’s D* and F*, Fay and Wu’s H and Fu’s Fs neutrality tests did not reveal statistically significant values (Table 2), suggesting that the gene might follow the neutral evolution model. However, the presence of 14 haplotypes and 0.890 ± 0.02 haplotype diversity in the Colombian population was greater than that expected under neutrality according to K-test and H-test results (Table 2). This suggested balanced ancestral polymorphism [48], being similar to that reported for the P. falciparum p38 gene which showed evidence of balanced selection in 5’ region [73].
Natural selection in pv38
A modified Nei Gojobori method was used for calculating dN and dS rates for showing some type of selection in the pv38 gene. Similar to that used regarding pv12, the pv38 gene was divided into two regions: region A, covering position 1-459 (amino acids 1-153) and region B, nucleotides 460-1,065 (amino acids 154-355 including the s48/45 domain). There were more dN substitutions in region A than dS substitutions, whilst there were more dS substitutions in region B than dN ones, even though no significant values were observed (Table 4 and Additional file 2). Selection tests by codon revealed positive selection in codon 70 and negative selection in codons 175 and 323, suggesting that the gene was influenced by selection. When the long-term effect of natural selection was explored by comparing divergence rates (KS and KN), pv38 had a higher statistically significant KS rate than KN (Table 4), revealing ω values below 1 throughout the gene (Figure 3), suggesting divergence by negative selection.
The McDonald-Kreitman test revealed statistically significant values (Table 4), when intraspecific polymorphism and interspecific divergence was compared, showing PN/PS > DN/DS (p < 0.02). This result could have been the result of either a negative selection or a balanced selection [61, 74]. K-test and H-test results (Table 2) and the presence of different haplotypes at intermediate frequencies (Figure 2B) suggested that it is most probable that pv38 was influenced by balanced selection, similar to that reported for P. falciparum[73]. Such selection seemed to be domain specific. Significant values were observed for region A (p = 0.014) when intraspecific polymorphism and interspecific divergence was calculated in each region (Additional file 3), this being where most of the substitutions found became accumulated, whilst neutrality could not be ruled out for region B (p = 0.1). Functional/structural constraint due to the presence of an s48/45 domain was also probable for pv38, given this region’s low diversity, two negatively selected sites and a statistically significant KS > KN.
Linkage disequilibrium (LD) and recombination
Several statistics were calculated for determining possible associations between polymorphisms and/or the presence of recombination in pv38. ZnS did not reveal statistically significant values, indicating that pv38 polymorphisms were not associated. Lineal regression between linkage disequilibrium (LD) and nucleotide distance revealed a reduction in LD as nucleotide distance increased, indicating that intragenic recombination might have led to new variations being produced.
The ZZ statistic was calculated to confirm whether recombination affected pv38 evolution, showing no significant values (Table 2); however, 2 RM (minimum recombination events) were found. The GARD method (in Datamonkey web server) gave a recombination breakpoint in position 524. Prior studies have suggested that new haplotypes could be produced through recombination in spite of functional constraints [73]. Intragenic recombination could thus be one of the factors promoting diversity in the pv38 gene. Crosslinking during recombination could produce new combinations between the gene’s 5′ (region A) and 3′ region (region B) as the breakpoint found in this gene was located upstream of the region encoding the s48/45 domain (region B). As only one polymorphic site was found in pv12, the aforementioned tests were not carried for this gene.
pv12 and pv38 should be considered for an antimalarial vaccine
The lack of a totally effective vaccine against human malarial parasites is at least partly due to high genetic diversity found in proteins involved in red blood cell invasion. These molecules’ constant exposure to the host’s immune system allows the fixation of mutations generating an adaptive advantage preventing their recognition. Antigens such as pvmsp-1, pvdbp, pvmsp-3 α, pvmsp-5, pvmsp-7C, pvmsp-7H, pvmsp-7I and pvama-1 have shown high genetic diversity which appears to be maintained by positive-balancing selection [10–15, 75–78]; however, other antigens are highly conserved despite being exposed to the host’s immune system. Surface antigens such as pvmsp-4, pvmsp-7A, pvmsp-7 K, pvmsp-8, pvmsp-10, pv230 or others in the rhoptries (pvrap-1 and pvrap-2) appear to evolve more slowly due to a possible functional constraint in their encoded proteins [70, 71, 79–82]. Thus, most mutations have become eliminated from the population, maintaining a conserved protein structure, even throughout these parasites’ evolutionary history [70, 71]. The latter behaviour seems to have been directing pv12 and pv38 evolution, highlighting high conservation at both intra- and inter-species level due to the influence of negative selection exerted on s48/45 domains which are important for red blood cell recognition [30]. Although antigens having low genetic diversity are usually not immunogenic [83] nor do they induce protection-inducing responses [84], some limited polymorphism antigens have been shown to be able to induce immunogenicity and protection [85]. Therefore, pv12 and pv38 (or their s48/45 domains) should be evaluated regarding vaccine development because immune responses against 6-Cys family antigens appear to be directed against structural epitopes in s48/45 domains [86–88], blocking such domains should prevent invasion [30, 88] and being highly conserved and having a functional constraint, allele-specific immune responses are thus avoided.
Conclusions
The p12 and p38 genes in P. vivax were seen to have low genetic diversity; the regions encoding the s48/45 domains seemed to be functionally or structurally constrained. Several members of the 6-Cys family are found on the surface of malaria parasites in every stage [28, 36–39, 69] and some of them (e g, P48/45, P230) are considered to be promising (transmission-blocking) vaccine candidates [36, 37, 87]. Epitopes identified by monoclonal antibodies against this type of protein are structural and have been localized within s48/45 domains [86, 87] which seem to be involved in host-pathogen interaction [30, 72]. Since pv12 and pv38 share structural characteristics with members of the 6-Cys family, added to their antigenic characteristics [38–40] and the low genetic diversity found in this study, the proteins encoded by these genes or their functionally/structurally constrained (conserved) regions could be born in mind when designing a multistage, multi-antigen subunit-based anti-malarial vaccine.
References
Rich SM, Ayala FJ: Progress in malaria research: the case for phylogenetics. Adv Parasitol. 2003, 54: 255-280.
White NJ: Plasmodium knowlesi: the fifth human malaria parasite. Clin Infect Dis. 2008, 46: 172-173. 10.1086/524889.
WHO: World Malaria Report 2012. 2012, Geneva: World Health Organization,http://www.who.int/malaria/publications/world_malaria_report_2012/wmr2012_no_profiles.pdf,
Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM: Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007, 77: 79-87.
Gething PW, Elyazar IR, Moyes CL, Smith DL, Battle KE, Guerra CA, Patil AP, Tatem AJ, Howes RE, Myers MF, George DB, Horby P, Wertheim HF, Price RN, Müeller I, Baird JK, Hay SI: A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl Trop Dis. 2012, 6: e1814-10.1371/journal.pntd.0001814.
Drug resistance in malaria.http://www.who.int/csr/resources/publications/drugresist/malaria.pdf,
Ranson H, Rossiter L, Ortelli F, Jensen B, Wang X, Roth CW, Collins FH, Hemingway J: Identification of a novel class of insect glutathione S-transferases involved in resistance to DDT in the malaria vector Anopheles gambiae. Biochem J. 2001, 359: 295-304. 10.1042/0264-6021:3590295.
Carvalho LJ, Daniel-Ribeiro CT, Goto H: Malaria vaccine: candidate antigens, mechanisms, constraints and prospects. Scand J Immunol. 2002, 56: 327-343. 10.1046/j.1365-3083.2002.01160.x.
Jones TR, Hoffman SL: Malaria vaccine development. Clin Microbiol Rev. 1994, 7: 303-310.
Gomez A, Suarez CF, Martinez P, Saravia C, Patarroyo MA: High polymorphism in Plasmodium vivax merozoite surface protein-5 (MSP5). Parasitology. 2006, 133: 661-672. 10.1017/S0031182006001168.
Martinez P, Suarez CF, Cardenas PP, Patarroyo MA: Plasmodium vivax Duffy binding protein: a modular evolutionary proposal. Parasitology. 2004, 128: 353-366. 10.1017/S0031182003004773.
Putaporntip C, Jongwutiwes S, Seethamchai S, Kanbara H, Tanabe K: Intragenic recombination in the 3′ portion of the merozoite surface protein 1 gene of Plasmodium vivax. Mol Biochem Parasitol. 2000, 109: 111-119. 10.1016/S0166-6851(00)00238-3.
Putaporntip C, Udomsangpetch R, Pattanawong U, Cui L, Jongwutiwes S: Genetic diversity of the Plasmodium vivax merozoite surface protein-5 locus from diverse geographic origins. Gene. 2010, 456: 24-35. 10.1016/j.gene.2010.02.007.
Garzon-Ospina D, Lopez C, Forero-Rodriguez J, Patarroyo MA: Genetic diversity and selection in three Plasmodium vivax merozoite surface protein 7 (Pvmsp-7) genes in a Colombian population. PLoS One. 2012, 7: e45962-10.1371/journal.pone.0045962.
Figtree M, Pasay CJ, Slade R, Cheng Q, Cloonan N, Walker J, Saul A: Plasmodium vivax synonymous substitution frequencies, evolution and population structure deduced from diversity in AMA 1 and MSP 1 genes. Mol Biochem Parasitol. 2000, 108: 53-66. 10.1016/S0166-6851(00)00204-8.
Mascorro CN, Zhao K, Khuntirat B, Sattabongkot J, Yan G, Escalante AA, Cui L: Molecular evolution and intragenic recombination of the merozoite surface protein MSP-3alpha from the malaria parasite Plasmodium vivax in Thailand. Parasitology. 2005, 131: 25-35. 10.1017/S0031182005007547.
Escalante AA, Lal AA, Ayala FJ: Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. 1998, 149: 189-202.
Chenet SM, Tapia LL, Escalante AA, Durand S, Lucas C, Bacon DJ: Genetic diversity and population structure of genes encoding vaccine candidate antigens of Plasmodium vivax. Malar J. 2012, 11: 68-10.1186/1475-2875-11-68.
Takala SL, Plowe CV: Genetic diversity and malaria vaccine design, testing and efficacy: preventing and overcoming ‘vaccine resistant malaria’. Parasite Immunol. 2009, 31: 560-573. 10.1111/j.1365-3024.2009.01138.x.
Genton B, Reed ZH: Asexual blood-stage malaria vaccine development: facing the challenges. Curr Opin Infect Dis. 2007, 20: 467-475. 10.1097/QCO.0b013e3282dd7a29.
Fluck C, Smith T, Beck HP, Irion A, Betuela I, Alpers MP, Anders R, Saul A, Genton B, Felger I: Strain-specific humoral response to a polymorphic malaria vaccine. Infect Immun. 2004, 72: 6300-6305. 10.1128/IAI.72.11.6300-6305.2004.
Ouattara A, Takala-Harrison S, Thera MA, Coulibaly D, Niangaly A, Saye R, Tolo Y, Dutta S, Heppner DG, Soisson L, Diggs CL, Vekemans J, Cohen J, Blackwelder WC, Dube T, Laurens MB, Doumbo OK, Plowe CV: Molecular basis of allele-specific efficacy of a blood-stage malaria vaccine: vaccine development implications. J Infect Dis. 2013, 207: 511-519. 10.1093/infdis/jis709.
Zambrano-Villa S, Rosales-Borjas D, Carrero JC, Ortiz-Ortiz L: How protozoan parasites evade the immune response. Trends Parasitol. 2002, 18: 272-278. 10.1016/S1471-4922(02)02289-4.
Richie TL, Saul A: Progress and challenges for malaria vaccines. Nature. 2002, 415: 694-701. 10.1038/415694a.
Kimura M: The neutral theory of molecular evolution. Cambridge Cambridgeshire. 1983, New York: Cambridge University Press
O’Donnell RA, De Koning-Ward TF, Burt RA, Bockarie M, Reeder JC, Cowman AF, Crabb BS: Antibodies against merozoite surface protein (MSP)-1(19) are a major component of the invasion-inhibitory response in individuals immune to malaria. J Exp Med. 2001, 193: 1403-1412. 10.1084/jem.193.12.1403.
Nagao E, Seydel KB, Dvorak JA: Detergent-resistant erythrocyte membrane rafts are modified by a Plasmodium falciparum infection. Exp Parasitol. 2002, 102: 57-59. 10.1016/S0014-4894(02)00143-1.
Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ, McConville MJ, Schofield L, Hodder AN, Yates JR, Crabb BS: Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J Biol Chem. 2005, 280: 40169-40176. 10.1074/jbc.M509631200.
Arevalo-Pinzon G, Curtidor H, Vanegas M, Vizcaino C, Patarroyo MA, Patarroyo ME: Conserved high activity binding peptides from the Plasmodium falciparum Pf34 rhoptry protein inhibit merozoites in vitro invasion of red blood cells. Peptides. 2010, 31: 1987-1994. 10.1016/j.peptides.2010.07.009.
Garcia J, Curtidor H, Pinzon CG, Vanegas M, Moreno A, Patarroyo ME: Identification of conserved erythrocyte binding regions in members of the Plasmodium falciparum Cys6 lipid raft-associated protein family. Vaccine. 2009, 27: 3953-3962. 10.1016/j.vaccine.2009.04.039.
Garcia Y, Puentes A, Curtidor H, Cifuentes G, Reyes C, Barreto J, Moreno A, Patarroyo ME: Identifying merozoite surface protein 4 and merozoite surface protein 7 Plasmodium falciparum protein family members specifically binding to human erythrocytes suggests a new malarial parasite-redundant survival mechanism. J Med Chem. 2007, 50: 5665-5675. 10.1021/jm070773z.
Urquiza M, Rodriguez LE, Suarez JE, Guzman F, Ocampo M, Curtidor H, Segura C, Trujillo E, Patarroyo ME: Identification of Plasmodium falciparum MSP-1 peptides able to bind to human red blood cells. Parasite Immunol. 1996, 18: 515-526. 10.1046/j.1365-3024.1996.d01-15.x.
Rodriguez LE, Curtidor H, Urquiza M, Cifuentes G, Reyes C, Patarroyo ME: Intimate molecular interactions of P. falciparum merozoite proteins involved in invasion of red blood cells and their implications for vaccine design. Chem Rev. 2008, 108: 3656-3705. 10.1021/cr068407v.
Barrero CA, Delgado G, Sierra AY, Silva Y, Parra-Lopez C, Patarroyo MA: Gamma interferon levels and antibody production induced by two PvMSP-1 recombinant polypeptides are associated with protective immunity against P. vivax in Aotus monkeys. Vaccine. 2005, 23: 4048-4053. 10.1016/j.vaccine.2005.02.012.
Richards JS, Beeson JG: The future for blood-stage vaccines against malaria. Immunol Cell Biol. 2009, 87: 377-390. 10.1038/icb.2009.27.
Van Dijk MR, Van Schaijk BC, Khan SM, Van Dooren MW, Ramesar J, Kaczanowski S, Van Gemert GJ, Kroeze H, Stunnenberg HG, Eling WM: Three members of the 6-cys protein family of Plasmodium play a role in gamete fertility. PLoS Pathog. 2010, 6: e1000853-10.1371/journal.ppat.1000853.
Williamson KC: Pfs230: from malaria transmission-blocking vaccine candidate toward function. Parasite Immunol. 2003, 25: 351-359. 10.1046/j.1365-3024.2003.00643.x.
Mongui A, Angel DI, Guzman C, Vanegas M, Patarroyo MA: Characterisation of the Plasmodium vivax Pv38 antigen. Biochem Biophys Res Commun. 2008, 376: 326-330. 10.1016/j.bbrc.2008.08.163.
Moreno-Perez DA, Areiza-Rojas R, Florez-Buitrago X, Silva Y, Patarroyo ME, Patarroyo MA: The GPI-anchored 6-Cys protein Pv12 is present in detergent-resistant microdomains of Plasmodium vivax blood stage schizonts. Protist. 2013, 164: 37-48. 10.1016/j.protis.2012.03.001.
Chen JH, Jung JW, Wang Y, Ha KS, Lu F, Lim CS, Takeo S, Tsuboi T, Han ET: Immunoproteomics profiling of blood stage Plasmodium vivax infection by high-throughput screening assays. J Proteome Res. 2010, 9: 6479-6489. 10.1021/pr100705g.
Prajapati SK, Joshi H, Shalini S, Patarroyo MA, Suwanarusk R, Kumar A, Sharma SK, Eapen A, Dev V, Bhatt RM, Valecha N, Nosten F, Rizvi MA, Dash AP: Plasmodium vivax lineages: geographical distribution, tandem repeat polymorphism, and phylogenetic relationship. Malar J. 2011, 10: 374-10.1186/1475-2875-10-374.
Imwong M, Pukrittayakamee S, Gruner AC, Renia L, Letourneur F, Looareesuwan S, White NJ, Snounou G: Practical PCR genotyping protocols for Plasmodium vivax using Pvcs and Pvmsp1. Malar J. 2005, 4: 20-10.1186/1475-2875-4-20.
Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, Chapman SB, Dash AP, Anvikar AR, Sutton PL, Birren BW, Escalante AA, Barnwell JW, Carlton JM: The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012, 44: 1046-1050. 10.1038/ng.2373.
Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P, Cheng Q, Coulson RM, Crabb BS, Del Portillo HA, Essien K, Feldblyum TV, Fernandez-Becerra C, Gilson PR, Gueye AH, Guo X, Kang’a S, Kooij TW, Korsinczky M, Meyer EV, Nene V, Paulsen I, White O, Ralph SA, Ren Q, Sargeant TJ: Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008, 455: 757-763. 10.1038/nature07327.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Suyama M, Torrents D, Bork P: PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34: W609-W612. 10.1093/nar/gkl315.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Depaulis F, Veuille M: Neutrality tests based on the distribution of haplotypes under an infinite-site model. Mol Biol Evol. 1998, 15: 1788-1790. 10.1093/oxfordjournals.molbev.a025905.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Fay JC, Wu CI: Hitchhiking under positive Darwinian selection. Genetics. 2000, 155: 1405-1413.
Fu YX: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-925.
Zhang J, Rosenberg HF, Nei M: Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci U S A. 1998, 95: 3708-3713. 10.1073/pnas.95.7.3708.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28: 2731-2739. 10.1093/molbev/msr121.
Kosakovsky Pond SL, Frost SD: Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005, 22: 1208-1222. 10.1093/molbev/msi105.
Pond SL, Frost SD, Grossman Z, Gravenor MB, Richman DD, Brown AJ: Adaptation to different human populations by HIV-1 revealed by codon-based analyses. PLoS Comput Biol. 2006, 2: e62-10.1371/journal.pcbi.0020062.
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL: Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8: e1002764-10.1371/journal.pgen.1002764.
Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K: FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 2013, 30: 1196-1205. 10.1093/molbev/mst030.
Jukes TH, Cantor CR: Evolution of protein molecules. Mammalian Protein Metabolism. Edited by: Munro HN. 1969, New York: Academic Press
McDonald JH, Kreitman M: Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991, 351: 652-654. 10.1038/351652a0.
Egea R, Casillas S, Barbadilla A: Standard and generalized McDonald-Kreitman test: a website to detect selection by comparing different classes of DNA sites. Nucleic Acids Res. 2008, 36: W157-W162. 10.1093/nar/gkn337.
Kelly JK: A test of neutrality based on interlocus associations. Genetics. 1997, 146: 1197-1206.
Rozas J, Gullaud M, Blandin G, Aguade M: DNA variation at the rp49 gene region of Drosophila simulans: evolutionary inferences from an unusual haplotype structure. Genetics. 2001, 158: 1147-1155.
Hudson RR, Kaplan NL: Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985, 111: 147-164.
Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD: Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006, 23: 1891-1901. 10.1093/molbev/msl051.
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL: Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010, 26: 2455-2457. 10.1093/bioinformatics/btq429.
Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, Herrera S, Kim JY, Menard D, Póvoa MM, Villegas L, Mulyanto , Snounou G, Cui L, Zeyrek FY, Escalante AA: The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013, 30: 2050-2064. 10.1093/molbev/mst104.
Tetteh KK, Stewart LB, Ochola LI, Amambua-Ngwa A, Thomas AW, Marsh K, Weedall GD, Conway DJ: Prospective identification of malaria parasite genes under balancing selection. PLoS One. 2009, 4: e5568-10.1371/journal.pone.0005568.
Tonkin ML, Arredondo SA, Loveless BC, Serpa JJ, Makepeace KA, Sundar N, Petrotchenko EV, Miller LH, Grigg ME, Boulanger MJ: Structural and biochemical characterization of Plasmodium falciparum 12 (Pf12) reveals a unique interdomain organization and the potential for an antiparallel arrangement with Pf41. J Biol Chem. 2013, 288: 12805-12817. 10.1074/jbc.M113.455667.
Pacheco MA, Ryan EM, Poe AC, Basco L, Udhayakumar V, Collins WE, Escalante AA: Evidence for negative selection on the gene encoding rhoptry-associated protein 1 (RAP-1) in Plasmodium spp. Infect Genet Evol. 2010, 10: 655-661. 10.1016/j.meegid.2010.03.013.
Pacheco MA, Elango AP, Rahman AA, Fisher D, Collins WE, Barnwell JW, Escalante AA: Evidence of purifying selection on merozoite surface protein 8 (MSP8) and 10 (MSP10) in Plasmodium spp. Infect Genet Evol. 2012, 12: 978-986. 10.1016/j.meegid.2012.02.009.
Arredondo SA, Cai M, Takayama Y, MacDonald NJ, Anderson DE, Aravind L, Clore GM, Miller LH: Structure of the Plasmodium 6-cysteine s48/45 domain. Proc Natl Acad Sci U S A. 2012, 109: 6692-6697. 10.1073/pnas.1204363109.
Reeder JC, Wapling J, Mueller I, Siba PM, Barry AE: Population genetic analysis of the Plasmodium falciparum 6-cys protein Pf38 in Papua New Guinea reveals domain-specific balancing selection. Malar J. 2011, 10: 126-10.1186/1475-2875-10-126.
Parsch J, Zhang Z, Baines JF: The influence of demography and weak selection on the McDonald-Kreitman test: an empirical study in Drosophila. Mol Biol Evol. 2009, 26: 691-698.
Ord R, Polley S, Tami A, Sutherland CJ: High sequence diversity and evidence of balancing selection in the Pvmsp3alpha gene of Plasmodium vivax in the Venezuelan Amazon. Mol Biochem Parasitol. 2005, 144: 86-93. 10.1016/j.molbiopara.2005.08.005.
Kang JM, Ju HL, Kang YM, Lee DH, Moon SU, Sohn WM, Park JW, Kim TS, Na BK: Genetic polymorphism and natural selection in the C-terminal 42 kDa region of merozoite surface protein-1 among Plasmodium vivax Korean isolates. Malar J. 2012, 11: 206-10.1186/1475-2875-11-206.
Zakeri S, Sadeghi H, Mehrizi AA, Djadid ND: Population genetic structure and polymorphism analysis of gene encoding apical membrane antigen-1 (AMA-1) of Iranian Plasmodium vivax wild isolates. Acta Trop. 2013, 126: 269-279. 10.1016/j.actatropica.2013.02.017.
Ju HL, Kang JM, Moon SU, Kim JY, Lee HW, Lin K, Sohn WM, Lee JS, Kim TS, Na BK: Genetic polymorphism and natural selection of Duffy binding protein of Plasmodium vivax Myanmar isolates. Malar J. 2012, 11: 60-10.1186/1475-2875-11-60.
Garzon-Ospina D, Romero-Murillo L, Tobon LF, Patarroyo MA: Low genetic polymorphism of merozoite surface proteins 7 and 10 in Colombian Plasmodium vivax isolates. Infect Genet Evol. 2011, 11: 528-531. 10.1016/j.meegid.2010.12.002.
Garzon-Ospina D, Romero-Murillo L, Patarroyo MA: Limited genetic polymorphism of the Plasmodium vivax low molecular weight rhoptry protein complex in the Colombian population. Infect Genet Evol. 2010, 10: 261-267. 10.1016/j.meegid.2009.12.004.
Martinez P, Suarez CF, Gomez A, Cardenas PP, Guerrero JE, Patarroyo MA: High level of conservation in Plasmodium vivax merozoite surface protein 4 (PvMSP4). Infect Genet Evol. 2005, 5: 354-361. 10.1016/j.meegid.2004.12.001.
Doi M, Tanabe K, Tachibana S, Hamai M, Tachibana M, Mita T, Yagi M, Zeyrek FY, Ferreira MU, Ohmae H, Kaneko A, Randrianarivelojosia M, Sattabongkot J, Cao YM, Horii T, Torii M, Tsuboi T: Worldwide sequence conservation of transmission-blocking vaccine candidate Pvs230 in Plasmodium vivax. Vaccine. 2011, 29: 4308-4315. 10.1016/j.vaccine.2011.04.028.
Patarroyo MA, Calderon D, Moreno-Perez DA: Vaccines against Plasmodium vivax: a research challenge. Expert Rev Vaccines. 2012, 11: 1249-1260. 10.1586/erv.12.91.
Giraldo MA, Arevalo-Pinzon G, Rojas-Caraballo J, Mongui A, Rodriguez R, Patarroyo MA: Vaccination with recombinant Plasmodium vivax MSP-10 formulated in different adjuvants induces strong immunogenicity but no protection. Vaccine. 2009, 28: 7-13. 10.1016/j.vaccine.2009.09.046.
Rojas-Caraballo J, Mongui A, Giraldo MA, Delgado G, Granados D, Millan-Cortes D, Martinez P, Rodriguez R, Patarroyo MA: Immunogenicity and protection-inducing ability of recombinant Plasmodium vivax rhoptry-associated protein 2 in Aotus monkeys: a potential vaccine candidate. Vaccine. 2009, 27: 2870-2876. 10.1016/j.vaccine.2009.02.083.
Carter R, Coulson A, Bhatti S, Taylor BJ, Elliott JF: Predicted disulfide-bonded structures for three uniquely related proteins of Plasmodium falciparum, Pfs230, Pfs48/45 and Pf12. Mol Biochem Parasitol. 1995, 71: 203-210. 10.1016/0166-6851(94)00054-Q.
Tachibana M, Sato C, Otsuki H, Sattabongkot J, Kaneko O, Torii M, Tsuboi T: Plasmodium vivax gametocyte protein Pvs230 is a transmission-blocking vaccine candidate. Vaccine. 2012, 30: 1807-1812. 10.1016/j.vaccine.2012.01.003.
Feller T, Thom P, Koch N, Spiegel H, Addai-Mensah O, Fischer R, Reimann A, Pradel G, Fendel R, Schillberg S, Scheuermayer M, Schinkel H: Plant-based production of recombinant Plasmodium surface protein pf38 and evaluation of its potential as a vaccine candidate. PLoS One. 2013, 8: e79920-10.1371/journal.pone.0079920.
Acknowledgements
We would especially like to thank Dr Sylvain Mousset who provided the ALLELIX software for our analysis. We would also like to thank Jason Garry for translating the manuscript and Professor Manuel E. Patarroyo for his comments and suggestions. This work was financed by the “Departamento Administrativo de Ciencia, Tecnología e Innovación (COLCIENCIAS)” through contract RC # 0309-2013. JF-R received financing through COLCIENCIAS cooperation agreement # 0757-2012.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JF-R devised the study, participated in designing it, performed the experiments, made the population genetics analysis and wrote the manuscript. DG-O devised and designed the study, helped perform the experiments, carried out the population genetics analysis and wrote the manuscript. MAP devised and coordinated the study, and helped to write the manuscript. All the authors have read and approved the final manuscript.
Johanna Forero-Rodríguez, Diego Garzón-Ospina contributed equally to this work.
Electronic supplementary material
12936_2013_3119_MOESM1_ESM.doc
Additional file 1: pv12 and pv38 haplotypes distribution in the Colombian population. Haplotype distribution found in pv12 (A) and pv38 (B) from 2007 to 2010. (DOC 794 KB)
12936_2013_3119_MOESM2_ESM.docx
Additional file 2: Synonymous substitution per synonymous site rate (d S ) and non-synonymous substitution per non-synonymous site rate (d N ) in s48/45 domains from pv12 and pv38 genes. No statistically significant differences were found by codon-based Z-test or Fisher’s exact tests. se: Standard error. pv12 s48/45 domain in region A: nucleotides 82-471; pv12 s48/45 domain in region B: nucleotides 589-906; pv38 s48/45 domain in region B: nucleotides 481-852 -: There is no s48/45 domain in the pv38 region. Numbering is based on the Sal-I reference sequence. (DOCX 16 KB)
12936_2013_3119_MOESM3_ESM.docx
Additional file 3: McDonald-Kreitman test for evaluating the action of natural selection in pv12 and pv38 gene regions A and B. The McDonald-Kreitman test was done using sequences obtained from databases (worldwide isolates) together with Colombian ones, and just with those obtained in the Colombian population. The interspecies divergence data was obtained from comparing Plasmodium vivax sequences with two related species: Plasmodium cynomolgi and Plasmodium knowlesi. Significant values are underlined. pv12: region A, nucleotides 1-546 and region B, nucleotides 547-1,095. pv38: region A, nucleotides 1-459 and region B, nucleotides 460-1,065. (DOCX 21 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Forero-Rodríguez, J., Garzón-Ospina, D. & Patarroyo, M.A. Low genetic diversity and functional constraint in loci encoding Plasmodium vivax P12 and P38 proteins in the Colombian population. Malar J 13, 58 (2014). https://doi.org/10.1186/1475-2875-13-58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-13-58