
Abstract
Background
Plasmodium vivax is the second most prevalent malaria parasite affecting more than 75 million people each year, mostly in South America and Asia. In addition to major morbidity this parasite is associated with relapses and a reduction in birthweight. The emergence and spread of drug resistance in Plasmodium falciparum is a major factor in the resurgence of this parasite. P. vivax resistance to drugs has more recently emerged and monitoring the situation would be helped, as for P. falciparum, by molecular methods that can be used to characterize parasites in field studies and drug efficacy trials.
Methods
Practical PCR genotyping protocols based on polymorphic loci present in two P. vivax genetic markers, Pvcs and Pvmsp1, were developed. The methodology was evaluated using 100 P. vivax isolates collected in Thailand.
Results and Discussion
Analysis revealed that P. vivax populations in Thailand are highly diverse genetically, with mixed genotype infections found in 26 % of the samples (average multiplicity of infection = 1.29). A large number of distinguishable alleles were found for the two markers, 23 for Pvcs and 36 for Pvmsp1. These were generally randomly distributed amongst the isolates. A total of 68 distinct genotypes could be enumerated in the 74 isolates with a multiplicity of infection of 1.
Conclusion
These results indicate that the genotyping protocols presented can be useful in the assessment of in vivo drug efficacy clinical trials conducted in endemic areas and for epidemiological studies of P. vivax infections.
Similar content being viewed by others


Background
Plasmodium vivax is globally distributed and is the dominant species in many countries. Infection by this species is generally regarded as benign and mortality is a rare outcome. 1.2 billion inhabitants of the South East Asian and Pacific countries are at risk from malaria transmission, representing about a third of world population exposed to Plasmodium parasites [1, 2]. Most of the populations at risk in these countries (corresponding to the WHO regional groupings SEARO and WPRO) reside in hypoendemic and mesoendemic areas, where half of the recorded infections are due to P. vivax, suggesting that the global burden of vivax malaria morbidity must be close to that of falciparum malaria. It has recently been established that P. vivax infections during pregnancy are associated with reduced birthweight [3], and thereby, increase the risk of neonatal deaths. Thus, the urgency to develop and implement measures to control P. vivax should equal that attending P. falciparum.
The emergence and global spread of P. falciparum parasites resistant to chloroquine and pyrimethamine – sulfadoxine has led to an increase in morbidity and mortality [4], forcing many countries to abandon these relatively cheap drugs. Recent reports of P. vivax resistance to these drugs [5–13] are of concern and steps to restrict the emergence of drug resistance need to be taken. A critical component of malaria control is surveillance for resistance. Despite some disadvantages, in vitro assessment of the susceptibility of P. falciparum to antimalarial drugs provides a rapid means to monitor general levels of drug resistance. Although protocols to culture P. vivax parasites ex-vivo have been developed [14, 15], they are still unsuited for routine assays. Thus, well conducted clinical trials, an essential component of resistance monitoring, remain the only alternative for P. vivax. However, in vivo drug efficacy studies conducted in endemic areas are to some extent compromised by their inability to distinguish true recrudescences, i.e. treatment failures, from re-infections which become clinically or parasitologically manifest during the follow-up period, i.e. treatment successes. This has been largely overcome for P. falciparum by the application of genotyping based on well characterized polymorphic regions within the gene encoding msp1, msp2 and glurp [16].
A similar approach has not been adopted for P. vivax, a parasite species less well-studied at the molecular level than P. falciparum [17]. Four polymorphic P. vivax single copy genes have been independently used for molecular epidemiological studies: Pvgam1 coding for a gametocyte antigen, Pvcs coding for the circumsporozoite protein, Pvmsp1 and Pvmsp3α coding for the merozoite surface proteins 1 and 3 alpha, respectively. Amplification of Pvgam1 was found to be associated with artefacts [18] and should thus be excluded as a genetic marker. The suitability of Pvmsp3α as a marker has been recently validated [19]. The extent of diversity found for the remaining two genes was mainly established through sequencing of amplified fragments.
The objective of this work was to develop field applicable methods of genotyping which could also complement clinical trials in vivax malaria. The highly polymorphic genes Pvcs and Pvmsp1 were therefore selected to develop protocols for polymerase chain reaction (PCR) amplification and subsequent analysis of the polymorphic regions. The methodology was then assessed and validated with samples obtained from patients who acquired a P. vivax infection in Thailand.
Materials and Methods
Blood samples
Blood samples were collected from adult patients with symptomatic P. vivax malaria admitted to the Bangkok Hospital for Tropical Diseases, Thailand between 1995 to 1998 (n = 100). All patients gave fully informed consent to enrolment in these studies which were approved by the Ethics committee of the Faculty of Tropical Medicine, Mahidol university. Diagnosis was established by microscopy at the malaria laboratory through examination of thin and thick blood smears, stained with Field's stain. The blood samples were kept at -30°C until DNA extraction.
DNA template preparation
DNA was purified from blood sample using the commercially available DNA Blood Kit (QIAGEN, Germany). The final volume of the DNA solution used as a template for the amplification reactions was such that 1 μL corresponded to 5 μL of whole blood. Confirmation of the microscopic diagnosis as P. vivax and testing for the presence of other Plasmodium species were achieved by a previously described PCR-based protocol [20].
Amplification protocols
In order to increase the sensitivity of amplification, a nested or semi-nested PCR approach was adopted for all the fragments amplified except for one of the three Pvmsp1 regions. Oligonucleotide primers were designed using published sequences of Pvcs and of Pvmsp1 (their sequences are presented in Table 1). The optimal Mg2+ concentrations, annealing temperatures and numbers of cycles were individually determined for the different primer pairs and are presented in Table 1.
All amplification reactions were carried out in a total volume of 20 μL and in the presence of 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 250 nM of each oligonucleotide primers, 125 μM of each of the four dNTPs, and 0.4 units of AmpliTaq polymerase (Perkin Elmer Cetus, USA). Primary amplification reactions were initiated with one μL of the template genomic DNA prepared from the blood samples, and the one μL of the product of these reactions was used to initiate the secondary amplification reactions. The cycling parameters for PCR were as follows: an initial denaturation step at 95°C for five minutes preceded the cycles of annealing at a temperature defined for each primer pair (Table 1) for two minutes, extension at 72°C for two minutes, and denaturation at 94°C for one minute. After a final annealing step followed by five minutes of extension, the reaction was stopped. PCR products were stored at 4°C until analysis.
The lack of cross-contamination was monitored by the inclusion of multiple, randomly distributed, negative control samples (human DNA or no template) in each amplification run. A subset of the samples were analysed in triplicate in order to confirm the consistency of the results obtained.
Analysis of the amplification product
The DNA fragments obtained following PCR were analysed by electrophoresis in agarose gels. For direct analysis of the fragments, 10 μL of the amplified PCR product were mixed with two μL of loading buffer and applied to 2 % agarose gels. For restriction fragment length polymorphism analysis of the PCR products (PCR-RFLP), 10 μL of the amplified PCR product were first digested with a restriction enzyme (New England Biolabs Inc., UK) according to the suppliers specifications, for three hours in a total volume of 20 μL, before adding five μL of loading buffer and applying to 1.5 % or 1.8 % agarose gels. Electrophoresis was performed in TBE buffer, and the DNA was visualised on an ultraviolet transilluminator following ethidium bromide staining. The size of the amplified fragments was estimated by comparison to a 100 bp ladder marker set.
Selected amplified fragments were purified using QIAquick gel extraction kits (QIAGEN, Germany) and cloned using the TOPO -TA Cloning kit (Invitrogen, U.S.A.). Plasmid DNA containing the fragment was purified from positive bacterial colonies using the QIAquick Miniprep spin kit (QIAGEN, Germany) and sequenced using an ABI automated sequencer. Sequence alignments were performed using the Gene Jockey II program (Biosoft, United Kingdom).
Results
Specificity and sensitivity of the amplification reactions
Primers specific to Pvcs and Pvmsp1 were designed to hybridize to sequences conserved in all variants known at that time. The specificity of all the primer pairs was confirmed since amplification products were only observed when P. vivax DNA was included in the reaction, but not when genomic DNA from P. falciparum, Plasmodium malariae, Plasmodium ovale or humans were used as a template. A set of a 10-fold serial dilution of a template prepared from a blood sample containing P. vivax only, calibrated using a previously described protocol [20], was used to assess the sensitivity of the reactions. Consistent detection of 10 parasite genomes was achieved for Pvcs-specific primers, and of 50 parasite genomes for Pvmsp1-specific primers. These levels of sensitivity were in agreement with subsequent analyses of samples harbouring a known number of parasites.
Genotyping using Pvcs
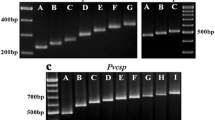
The circumsporozoite protein gene of P. vivax, as for other Plasmodium species, comprises a central repetitive domain flanked by two conserved domains. The majority of variations observed in Pvcs occur in the repeat region and the immediate pre- and post-repeat sequences. Thus the genotyping strategy was focused on an amplified fragment spanning these regions (Fig. 1A). In a given parasite line, the repetitive domain is composed of a 27 bp element repeated a variable number of times. Variations in the number of repeated elements result in size polymorphisms amenable to detection by electrophoresis. Seven allelic types distinguishable by size were observed in the Thai P. vivax isolates analysed (Fig. 1B), a number consistent with the variations in the number of repeat elements (15 to 21) previously observed for Pvcs [21, 22]. In P. vivax, two types of repeat elements are found, VK210 and VK247 [23], and a given Pvcs gene will exclusively bear either the VK210 type (type I repeats based on GDRADGQPA) or the VK247 type (type II repeats based on ANGAGNQPG). In order to differentiate between amplified fragments carrying the VK210 from those carrying the VK247 repeat element, the exclusive presence of Alu I recognition sites in the VK210-type and of Bst NI recognition sites in the VK247-type repeated sequences were exploited. Thus, following digestion with the appropriate enzyme, the amplified fragments are degraded into fragments of < 150 bp, easily distinguishable from the original fragments of > 700 bp (Fig. 1C). In this manner the PCR fragments obtained from an isolate can be grouped by size and by repeat type.

A. Schematic representation of the Pvcs gene, which consists of a conserved region (blank box) flanking a repeated region (grey box) which is itself flanked by pre- and post-repeat specific sequences (cross-hatched boxes). The bold horizontal line represents the PCR-amplified fragment used for further analysis. B. Example of the fragments (denoted A to G) of distinct size observed in different isolates, as single or as mixed infections (some lanes were not labelled because of lack of space). C. Fragments digested (D) or undigested (U) with Alu I or BstN I, two restriction enzymes with multiples sites in VK 210 type or VK247 type repeat sequences, respectively. Digestion cuts the fragment in small fragments of less than 150 bp in size. D. PCR-RFLP using specific restriction enzymes analysis for the presence/absence (yes/no) of specific pre- and post-repeat sequences in fragments carrying the VK210 or VK247 type repeats. Undigested fragments are denoted by (U) and Pr and Po denote digests to determine the presence of pre- and post-repeat sequence types, respectively. A 100 bp ladder, where the 600 bp band stains most intensely, was used a molecular weight marker (M) for all the gels.
In order to increase the genotyping resolution of Pvcs as a marker, so as to subdivide the P. vivax population into a larger number of Pvcs allelic types, sequence variations previously observed in the pre- and post-repeat regions [24] were used to develop an additional PCR-RFLP protocol (Fig. 1D). For some Pvcs genes, the pre-repeat consists of the Region I sequence (KLKQP) that directly abuts the repeated region, while for others amino acid residues are inserted between the two regions, namely a T, V or A residue for VK210-type Pvcs genes, or ED residues for VK247-type Pvcs genes. The Scr FI restriction endonuclease cuts VK210-type Pvcs fragments that do not have the T/A/V insertion following Region I, thus shortening them by 41 bp. VK247-type Pvcs bearing the ED insertion after Region I are cut by the Mbo II restriction endonuclease and the amplified fragment is thereby shortened by 55 bp. For the post-repeat region, a 36 bp insertion has been found for some VK210-type Pvcs. This insertion harbours a recognition site for the Bbs I enzyme, and digestion truncates the corresponding PCR fragments by 115 bp.
Genotyping of the 100 P. vivax isolates collected from Thailand was achieved successfully using the protocols described above. In total, parasites with a Pvcs bearing the VK210 type repeats only were found in 90 isolates (representing seven size polymorphisms). Parasites with a Pvcs bearing the VK247-type repeats only were found in nine isolates (representing only three size polymorphisms), and in one isolate parasites of both types were found. Thus, 10 different allelic forms of Pvcs were detected by simple analyses of the fragment size and repeat type. When associated with RFLP analysis of the pre- and post-repeat types, these could be divided into 23 different allelic types (Table 2), 18 for the VK210 type and five for the VK247 type. Mixed genotype infections were found in 20 of the isolates, for each of which the multiplicity of infection (MOI), i.e. the total number of different allelic types observed, was two. Thus, for the samples analysed, a total of 120 bands were observed, with an mean MOI of 1.2. The allelic variants were generally randomly distributed between the 120 bands; the highest frequencies, 0.2 and 0.18, were found for VK210k and VK210n, respectively (Fig 2).

Allele frequency of the distinct allelic variants of Pvcs observed in the 100 isolates from Thailand. Allelic types were defined according to repeat type (VK210 or VK247), fragment size and the presence or absence of defined pre- and post-repeat sequences.
Genotyping using Pvmsp1
The Pvmsp1 gene encodes a polypeptide of about 1,720 amino acids [25, 26], and sequence comparison revealed 13 regions of interallele conserved blocks and variable blocks [27]. Three main regions of sequence divergence were found through comparison of the full length Pvmsp1 sequences from two distinct P. vivax lines (Sal-1 and Belem). Three segments (labelled F1 to F3) corresponding to these regions were thus amplified for further analysis (Fig. 3A).

A. Schematic representation of the Pvmsp1 gene for the localization of interallele conserved blocks (blank boxes) and variable blocks (black boxes). The position of the three amplified segments (F1, F2 and F3) is indicated by the horizontal bold lines. B. Gel electrophoresis of PCR products of a selection of fragments, corresponding to the three segments, amplified from different isolates. The molecular size of the largest and smallest bands are indicated. C. Analysis of the diversity of the F2 segment by PCR-RFLP using Alu I or Mnl I. A selection of lanes is labelled to indicate the type of variant observed (corresponding to the nomenclature in Table 3). A 100 bp ladder was used a molecular weight marker (M) for all the gels.
For the 100 Thai P. vivax isolates, five distinguishable size variants were observed for F1 (ca. 350 bp – 450 bp), and four for F3 (ca. 250 bp – 350 bp), but only two were observed for the larger (ca. 1087 bp -1150 kb) F2 segment (Fig. 3B). One allelic variant dominated in frequency for each of the three amplified segments (Table 3): band C for F1 (42 % of the bands observed), band A for F2 (78 %) and band D for F3 (76 %). Mixed genotype infections were observed infrequently and only for the F1 (n = 3, 2 MOI of 2 and 1 MOI of 3) and for the F3 (n = 6, 4 MOI of 2 and 2 MOI of 3) segments.
A selection of amplified product from F1 (n = 18) and F3 (n = 8) representative of all the size variants were sequenced and compared to previously published sequences (Fig. 4). The number of distinct F1 variants was found to be higher than that revealed by electrophoretic separation, since 11 different allelic variants were observed for the 18 fragments derived from the Thai isolates. Sequence differences were observed within the variants assigned to the B, D or E size classes. Some of the bands within each class might be distinguished by the use of higher resolution agarose gels, though others had the same size and only exhibited subtle sequence differences. A similar pattern was observed for the F3 fragments, where sequencing distinguished six different variants.

Alignment of amino acid sequences of amplified F1 and F3 fragments of Pvmsp1. Sequences were aligned against the largest fragment obtained; dots represent identical residues and dashes represent gaps. Sequences obtained during this study are labelled by their GenBank accession numbers, while the Belem and Sal-1 sequences had been previously published (AF435594 and AF435593, respectively).
The F2 segment, which was comparatively poorly polymorphic in size in the Thai isolates, encompassed regions established as polymorphic in previous studies [27]. An RFLP strategy was thus adopted to distinguish between the different allelic variants. The restriction endonucleases Alu I and Mnl I which recognize multiple sites in F2, were used to reveal extensive polymorphism at the nucleotide level (Fig. 3C). Thus, nine different Alu I and eight different Mnl I RFLP patterns were observed in the 100 Thai isolates (Table 3). The occurrence of patterns indicative of partial digestion, or the presence of mixed F2 genotypes in individual samples were excluded since the sum of the RFLP fragments' sizes was not found to be greater than that of the uncut product for any isolate and the patterns observed were not unique to isolates where mixed genotypes were detected through size polymorphism of the F1 or F3 segments. When the data from both analyses (size and RFLP) were combined, 36 Pvmsp1 F2 allelic variants could be differentiated (Table 4). When the frequency of the allelic variants was considered individually for the two RFLP patterns, 50 % and 38 % of the isolates were of a single Alu I- and Mnl I-classified allelic variant, respectively. The remaining isolates were randomly distributed between the different variants (Fig. 5A). However, by combining the results for both restriction enzymes, the most dominant allele was found in only 27 % of the isolates (Fig. 5B), while the remainder were found at lower frequency (5 % or less).

Allele frequency of the distinct allelic variants of the F2 segment of Pvmsp1 observed in the 100 isolates from Thailand. Allelic variants were defined according to the RFLP patterns observed. A. Variants were divided according to the digestions patterns obtained individually with each of the two restriction enzymes Alu I (a1, a2, etc...) or Mnl I (m1, m2, etc...) for A or B, the two different sized fragments amplified, as in Table 3. B) Variants were classed according to size and the combined RFLP patterns obtained for both restriction enzymes (Alu I and Mnl I), as in Table 4.
Two locus genotyping
When the genotype analyses from the four polymorphic regions of the two genes were combined, the 100 isolates collected from Thai patients proved to be highly genetically diverse. Mixed genotype infections were detected in 26 of the isolates, mainly through the Pvcs gene (n = 20). Only one of the isolates identified as mixed genotype infections by Pvmsp1 (n = 7) was classed as such by Pvcs analysis. The mean MOI for isolates with a mixed genotype was 2.11, and that for the complete set of isolates was 1.29.
There were 74 isolates where a single genotype was detected after full analysis of the Pvcs and Pvmsp1 markers. Comparison of the genotyping patterns obtained for these isolates allows to ascertain the relative contribution of the polymorphic loci to distinguish between P. vivax populations. When the complete genotyping analyses are taken into account, a total of 68 distinct genotypes are enumerated. Sixty-three genotypes were observed in only one isolate, with a maximum of three isolates sharing the same genotype (highest genotype frequency = 0.040). No evidence for linkage disequilibrium could be detected when Pvcs and Pvmsp1 were considered. If the results from the Pvmsp1 F1 segment are omitted from the analysis, 58 distinct genotypes would be found, with a maximum of four isolates sharing the same genotype (highest genotype frequency = 0.054). Incremental omission of the Pvmsp1 F3 segment followed by that of the Pvcs pre- and post-repeat PCR-RFLP results would reduce the total number of distinct genotypes to 55 with four isolates sharing the same genotype (highest genotype frequency = 0.054), and to 49 with seven isolates sharing the same genotype (highest genotype frequency = 0.095), respectively.
Discussion
Populations of P. vivax, like those of other Plasmodium species, comprise genetically distinct lines that exhibit diversity in a number of factors of epidemiological, biological and clinical relevance. For example, the proportion of hypnozoites and the timing and frequency of their activation, differ between temperate and tropical strains of this parasite [28]. Resistance to drugs is linked to genetic mutations of defined genes [29] and recently the Pvcs repeat type was found to be associated with transmissibility by defined species of anophelines [30]. The ability to differentiate genetically different parasite populations would enhance a wide spectrum of investigations of the biology and epidemiology of P. vivax.
In this article, the potential of two P. vivax genes, Pvcs and Pvmsp1, as genetic markers, were assessed and practical protocols for their use in genotyping parasites collected from the field were described. PCR-based protocols targeted four polymorphic regions from two genetic markers, one from Pvcs and three from Pvmsp1, and were further associated to RFLP analyses of two of these regions, the repeat region of Pvcs and the F2 fragment of Pvmsp1, that allow the division of parasite populations into an incremental number of genetically distinct sub-groups.
For Pvmsp1, three polymorphic regions were independently assessed. The fragments F1, located at variable block 2 and F3 located at variable block 10, displayed moderate allelic size variation, five and four types respectively. The frequency of the size variants, in particular those of F3, displayed a biased frequency distribution among the 100 Thai isolates considered in this study, thus undermining their usefulness as genetic markers. Sequencing was carried out for a subset of F1 and F3 fragments amplified from the Thai isolates. This revealed that 13 F1 and 10 F2 allelic variants could actually be distinguished. These results confirm the high degree of complex polymorphisms observed for Pvmsp1 block 2 and block 10 observed in 31 isolates collected in Thailand, Brazil, Oceania and India, where 19 and 13 distinct allelic types were observed for block 2 and block 10 respectively [27]. Thus it is likely that the F1 and F3 segments could be exploited as useful genetic markers through the development of type-specific oligonucleotides, that would then be used in a series of nested PCR reactions. The amplified large central F2 fragment, located between variable block 6 and 8, showed two size variants only. However, when combined to RFLP analyses, using two restriction endonucleases, the variants were subdivided into a total of 36 different allelic types. For Pvcs, populations were divided into 10 subgroups through size and repeat type determination, and into 23 subgroups when sequence variations in the pre- and post-repeat sequences were assessed by RFLP.
The conclusion of this work is that the polymorphic repeat region of Pvcs and that of the Pvmsp1 F2 region can be considered suitable genetic markers of P. vivax populations, alone or in combination. This is supported by the fact that practical PCR-RFLP genotyping protocols revealed the presence of numerous distinct allelic variants in a sample of 100 isolates, among which they were randomly distributed without evidence of linkage disequilibrium between the two loci.
Interesting epidemiological indications could be derived from genotyping of the P. vivax isolates. Malaria is considered to be hypoendemic in Thailand where transmission, confined to regions bordering Cambodia, Lao PDR and Myanmar, rarely exceeds a few infective bites per person per year. A study of P. falciparum diversity, based on three genetic markers, in a refugee camp located in Thailand close to the Myanmar border, revealed levels of diversity and MOI (1.6, with mixed genotype infections observed in 60 % of the isolates) that were higher than expected from the low effective inoculation rates [31]. A recent study of P. vivax parasites collected in the same region and based on two genetic markers, showed that populations of this parasite species were also highly diverse and mixed genotype infections were observed in 35 % of the isolates [32]. The results of the present study indicated that 26 % of the isolates were of mixed genotype, with an overall MOI for all 100 isolates of 1.29. It should be stated that the three studies are not strictly comparable since the number and type of genetic markers, the genotyping protocols and the collection strategies that were employed differed. Furthermore, in the current study the origin of the P. vivax isolates was not confined to a single area, since the patients from whom blood was collected acquired their infection in diverse endemic areas of Thailand. Nonetheless, these studies indicated that the two biologically distinct parasite species have similar population characteristics. The production of infectious gametocytes early during the primary infection in P. vivax versus their late appearance, following the acute phase, in P. falciparum, and the existence of hypnozoites solely in P. vivax, might not be sufficient to explain the maintenance of diversity in areas of low transmission. Substantially prolonged duration of the infections, probably as a result of low level drug resistance leading to parasitological (though not clinical) failures, as suggested for P. falciparum [31], and/or an underestimation of the transmission intensity, might underlie the high genetic diversity and MOI observed for the two parasite species. These two non-mutually exclusive scenarios are consistent with the detection of a high level of mixed species infections observed in Thailand [33–36].
It is hoped that the methodologies presented here can be adopted as standard protocols for the genotyping of P. vivax parasites, not only for in vivo drug efficacy trials, but also in field-based investigations aimed at elucidating the biology, pathology and epidemiology of this important parasite species.
Conclusion
These results indicate that the genotyping protocols presented can be useful in the assessment of in vivo drug efficacy clinical trials conducted in endemic areas and for epidemiological studies of P. vivax infections.
Abbreviations
- Pvcs :
-
Circumsporozoite surface protein gene of Plasmodium vivax
- Pvmsp1 :
-
Merozoite surface protein 1 gene of Plasmodium vivax
- Pvmsp3α :
-
Merozoite surface protein 3 alpha gene of Plasmodium vivax PCR-Polymerase Chain Reaction
- RFLP:
-
Restriction Fragment Length Polymorphism
References
Mendis K, Sina BJ, Marchesini P, Carter R: The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001, 64 (1-2 Suppl): 97-106.
Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW: The global distribution and population at risk of malaria: past, present, and future. Lancet Infect Dis. 2004, 4 (6): 327-336. 10.1016/S1473-3099(04)01043-6.
Nosten F, McGready R, Simpson JA, Thwai KL, Balkan S, Cho T, Hkirijaroen L, Looareesuwan S, White NJ: Effects of Plasmodium vivax malaria in pregnancy. Lancet. 1999, 354 (9178): 546-549. 10.1016/S0140-6736(98)09247-2.
Trape JF: The public health impact of chloroquine resistance in Africa. Am J Trop Med Hyg. 2001, 64 (1-2 Suppl): 12-17.
Rieckmann KH, Davis DR, Hutton DC: Plasmodium vivax resistance to chloroquine?. Lancet. 1989, 2 (8673): 1183-1184. 10.1016/S0140-6736(89)91792-3.
Baird JK, Basri H, Purnomo, Bangs MJ, Subianto B, Patchen LC, Hoffman SL: Resistance to chloroquine by Plasmodium vivax in Irian Jaya, Indonesia. Am J Trop Med Hyg. 1991, 44 (5): 547-552.
Garavelli PL, Corti E: Chloroquine resistance in Plasmodium vivax: the first case in Brazil. Trans R Soc Trop Med Hyg. 1992, 86 (2): 128-10.1016/0035-9203(92)90535-K.
Thaithong S, Chan SW, Songsomboon S, Wilairat P, Seesod N, Sueblinwong T, Goman M, Ridley R, Beale G: Pyrimethamine resistant mutations in Plasmodium falciparum. Mol Biochem Parasitol. 1992, 52 (2): 149-157. 10.1016/0166-6851(92)90047-N.
Myat Phone K, Myint O, Myint L, Thaw Z, Kyin Hla A, Nwe Nwe Y: Emergence of chloroquine-resistant Plasmodium vivax in Myanmar (Burma). Trans R Soc Trop Med Hyg. 1993, 87 (6): 687-10.1016/0035-9203(93)90294-Z.
Murphy GS, Basri H, Purnomo, Andersen EM, Bangs MJ, Mount DL, Gorden J, Lal AA, Purwokusumo AR, Harjosuwarno S: Vivax malaria resistant to treatment and prophylaxis with chloroquine. Lancet. 1993, 341 (8837): 96-100. 10.1016/0140-6736(93)92568-E.
Garg M, Gopinathan N, Bodhe P, Kshirsagar NA: Vivax malaria resistant to chloroquine: case reports from Bombay. Trans R Soc Trop Med Hyg. 1995, 89 (6): 656-657. 10.1016/0035-9203(95)90432-8.
Baird JK, Leksana B, Masbar S, Fryauff DJ, Sutanihardja MA, Suradi, Wignall FS, Hoffman SL: Diagnosis of resistance to chloroquine by Plasmodium vivax: timing of recurrence and whole blood chloroquine levels. Am J Trop Med Hyg. 1997, 56 (6): 621-626.
Pukrittayakamee S, Chantra A, Simpson JA, Vanijanonta S, Clemens R, Looareesuwan S, White NJ: Therapeutic responses to different antimalarial drugs in vivax malaria. Antimicrob Agents Chemother. 2000, 44 (6): 1680-1685. 10.1128/AAC.44.6.1680-1685.2000.
Golenda CF, Li J, Rosenberg R: Continuous in vitro propagation of the malaria parasite Plasmodium vivax. Proc Natl Acad Sci U S A. 1997, 94 (13): 6786-6791. 10.1073/pnas.94.13.6786.
Chotivanich K, Silamut K, Udomsangpetch R, Stepniewska KA, Pukrittayakamee S, Looareesuwan S, White NJ: Ex-vivo short-term culture and developmental assessment of Plasmodium vivax. Trans R Soc Trop Med Hyg. 2001, 95 (6): 677-680. 10.1016/S0035-9203(01)90113-0.
Snounou G, Beck HP: The use of PCR-genotyping in the assessment of recrudescence or reinfection after antimalarial treatment. Parasitology today. 1998, 14 (11): 462-467. 10.1016/S0169-4758(98)01340-4.
Cui L, Escalante AA, Imwong M, Snounou G: The genetic diversity of Plasmodium vivax populations. Trends Parasitol. 2003, 19 (5): 220-226. 10.1016/S1471-4922(03)00085-0.
Imwong M, Pukrittakayamee S, Looareesuwan S, Poirriez J, Pasvol G, White NJ, Snounou G: Plasmodium vivax: polymerase chain reaction amplification artifacts limit the suitability of pvgam1 as a genetic marker. Exp Parasitol. 2001, 99 (3): 175-179. 10.1006/expr.2001.4646.
Bruce MC, Galinski MR, Barnwell JW, Snounou G, Day KP: Polymorphism at the merozoite surface protein-3alpha locus of Plasmodium vivax: global and local diversity. Am J Trop Med Hyg. 1999, 61 (4): 518-525.
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, Thaithong S, Brown KN: High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol. 1993, 61 (2): 315-320. 10.1016/0166-6851(93)90077-B.
Mann VH, Huang T, Cheng Q, Saul A: Sequence variation in the circumsporozoite protein gene of Plasmodium vivax appears to be regionally biased. Mol Biochem Parasitol. 1994, 68 (1): 45-52. 10.1016/0166-6851(94)00148-0.
Rongnoparut P, Supsamran N, Sattabongkot J, Suwanabun N, Rosenberg R: Phenotype and genotype diversity in the circumsporozoite proteins of Plasmodium vivax in Thailand. Mol Biochem Parasitol. 1995, 74 (2): 201-210. 10.1016/0166-6851(95)02504-9.
Rosenberg R, Wirtz RA, Lanar DE, Sattabongkot J, Hall T, Waters AP, Prasittisuk C: Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science. 1989, 245 (4921): 973-976.
Mann VH, Good MF, Saul AJ: Diversity in the circumsporozoite protein of Plasmodium vivax: Does it matter? . Parasitology today. 1995, 11: 33-36. 10.1016/0169-4758(95)80107-3.
del Portillo HA, Longacre S, Khouri E, David PH: Primary structure of the merozoite surface antigen 1 of Plasmodium vivax reveals sequences conserved between different Plasmodium species. Proc Natl Acad Sci U S A. 1991, 88 (9): 4030-4034.
Gibson HL, Tucker JE, Kaslow DC, Krettli AU, Collins WE, Kiefer MC, Bathurst IC, Barr PJ: Structure and expression of the gene for Pv200, a major blood-stage surface antigen of Plasmodium vivax. Mol Biochem Parasitol. 1992, 50 (2): 325-333. 10.1016/0166-6851(92)90230-H.
Putaporntip C, Jongwutiwes S, Sakihama N, Ferreira MU, Kho WG, Kaneko A, Kanbara H, Hattori T, Tanabe K: Mosaic organization and heterogeneity in frequency of allelic recombination of the Plasmodium vivax merozoite surface protein-1 locus. Proc Natl Acad Sci U S A. 2002, 99 (25): 16348-16353. 10.1073/pnas.252348999.
Garnham PC, Bray RS, Bruce-Chwatt LJ, Draper CC, Killick-Kendrick R, Sergiev PG, Tiburskaja NA, Shute PG, Maryon M: A strain of Plasmodium vivax characterized by prolonged incubation: morphological and biological characteristics. Bull World Health Organ. 1975, 52 (1): 21-32.
Imwong M, Pukrittayakamee S, Renia L, Letourneur F, Charlieu JP, Leartsakulpanich U, Looareesuwan S, White NJ, Snounou G: Novel point mutations in the dihydrofolate reductase gene of Plasmodium vivax: evidence for sequential selection by drug pressure. Antimicrob Agents Chemother. 2003, 47 (5): 1514-1521. 10.1128/AAC.47.5.1514-1521.2003.
Gonzalez-Ceron L, Rodriguez MH, Santillan F, Chavez B, Nettel JA, Hernandez-Avila JE, Kain KC: Plasmodium vivax: ookinete destruction and oocyst development arrest are responsible for Anopheles albimanus resistance to circumsporozoite phenotype VK247 parasites. Exp Parasitol. 2001, 98 (3): 152-161. 10.1006/expr.2001.4626.
Paul RE, Hackford I, Brockman A, Muller-Graf C, Price R, Luxemburger C, White NJ, Nosten F, Day KP: Transmission intensity and Plasmodium falciparum diversity on the northwestern border of Thailand. Am J Trop Med Hyg. 1998, 58 (2): 195-203.
Cui L, Mascorro CN, Fan Q, Rzomp KA, Khuntirat B, Zhou G, Chen H, Yan G, Sattabongkot J: Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. Am J Trop Med Hyg. 2003, 68 (5): 613-619.
Mayxay M, Pukritrayakamee S, Chotivanich K, Imwong M, Looareesuwan S, White NJ: Identification of cryptic coinfection with Plasmodium falciparum in patients presenting with vivax malaria. Am J Trop Med Hyg. 2001, 65 (5): 588-592.
Siripoon N, Snounou G, Yamogkul P, Na-Bangchang K, Thaithong S: Cryptic Plasmodium falciparum parasites in clinical P. vivax blood samples from Thailand. Trans R Soc Trop Med Hyg. 2002, 96 (1): 70-71. 10.1016/S0035-9203(02)90246-4.
Mayxay M, Pukrittayakamee S, Newton PN, White NJ: Mixed-species malaria infections in humans. Trends Parasitol. 2004, 20 (5): 233-240. 10.1016/j.pt.2004.03.006.
Snounou G, White NJ: The co-existence of Plasmodium: sidelights from falciparum and vivax malaria in Thailand. Trends Parasitol. 2004, 20 (7): 333-339. 10.1016/j.pt.2004.05.004.
Acknowledgements
This study was part of the Wellcome Trust-Mahidol University-Oxford Tropical Medicine Research Programme supported by the Wellcome Trust of Great Britain.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
GS, NJW and SP designed the study. GS and SP were responsible for the day-to-day supervision of the work. ACG, FL and LR and MI collaborated to obtain and analyze DNA sequences. SL and SP were responsible for patient recruitment and clinical management. MI developed the protocols with the help of GS, and carried out the vast majority of the laboratory work. GS and MI analyzed the data and composed the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Imwong, M., Pukrittayakamee, S., Grüner, A.C. et al. Practical PCR genotyping protocols for Plasmodium vivax using Pvcs and Pvmsp1. Malar J 4, 20 (2005). https://doi.org/10.1186/1475-2875-4-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-4-20