
Abstract
Background
The merozoite surface protein-3β of Plasmodium vivax (PvMSP-3β) is one of the candidate antigens for blood stage malaria vaccine development. The polymorphisms in PvMSP-3β have been reported in certain P. vivax isolates. However, the diversity of PvMSP-3β throughout its global distribution has not been well understood. In this study, the genetic diversity and the effects of natural selection in PvMSP-3β among P. vivax Korean isolates were analysed.
Methods
Blood samples were collected from 95 patients with vivax malaria in Korea. The region flanking full-length PvMSP-3β was amplified by polymerase chain reaction and cloned into a TA cloning vector. The PvMSP-3β sequence of each isolate was determined and the polymorphic characteristics and effects of natural selection were analysed using the DNASTAR, MEGA4, and DnaSP programs.
Results
Five different subtypes of PvMSP-3β were identified based on single nucleotide polymorphisms (SNPs), insertions, and deletions. Although a high level of sequence diversity was observed in the PvMSP-3β gene, the coiled-coil tertiary structure of the PvMSP-3β protein was well conserved in all of the sequences. The PvMSP-3β of Korean isolates is under natural selection. DNA polymerase slippage and intragenic recombination likely contributed to PvMSP-3β diversity in Korean P. vivax isolates.
Conclusions
The PvMSP-3β of Korean P. vivax isolates displayed polymorphisms, with SNPs, insertions and deletions scattered throughout of the gene. These results of parasite heterogeneity are relevant to the development of a PvMSP-3β based vaccine against P. vivax and the implementation of malaria control programmes in Korea.
Similar content being viewed by others

Background
Plasmodium vivax is the most widely distributed malaria parasite in worldwide, causing 70–80 million cases annually, and is a major public health problem contributing to the global burden of malaria morbidity in regions outside of Africa [1]. Plasmodium vivax is more difficult to control and eliminate than Plasmodium falciparum mainly because of its tendency to relapse. Moreover, vivax malaria has re-emerged in many temperate regions in recent years, including South Korea, where it had been largely eradicated during the global malaria control campaigns.
Understanding the population structures of the malaria parasites is an important factor in drug-resistance surveillance and estimating the performance of vaccines under development in a particular parasite population [2]. Although the population structures of P. falciparum isolates have been examined on a global scale, those of P. vivax remain largely less understood. With a global distribution that ranges from tropical to temperate regions, P. vivax isolates have exhibited variation in biological characteristics, such as relapse patterns and transmissibility, which can be used to distinguish geographical isolates and even subspecies [3]. Moreover, extensive genetic diversity has also been identified within a local P. vivax population. Therefore, understanding the nature of genetic polymorphisms of P. vivax populations in endemic areas might provide information regarding parasite heterogeneity that is relevant to malaria control efforts.
The P. vivax merozoite surface protein-3 (PvMSP-3) family consists of several related proteins, PvMSP-3α, PvMSP-3β and PvMSP-3γ. Although the level of shared amino acid sequence identity among the proteins is limited, all of them contain a central alanine-rich domain that forms a coiled-coil tertiary structure [4–6]. Limited sequence polymorphism has been observed in the MSP-3 of P. falciparum (PfMSP-3) [7, 8], but the PvMSP-3α is likely to be highly polymorphic and is reported to be a reliable genetic marker for population analysis [9–13]. PvMSP-3β also showed polymorphic patterns among several geographically different P. vivax isolates [14, 15], but its polymorphic pattern in worldwide isolates is poorly understood compared to other vaccine candidate antigens. As PvMSP-3β has been regarded as a potential vaccine target [16], a more thorough understanding of genetic diversity of the gene is required since genetic polymorphism in the candidate antigen can hamper the efficacy of a vaccine.
Following its re-emergence in Korea in 1993, vivax malaria has persisted, with varying numbers of indigenous cases annually in the country. Several recent studies have suggested that rapid genetic variation has occurred in the Korean P. vivax population in recent years [17–21], which supports settlement of local transmission of vivax malaria in Korea. In this study, the polymorphic nature of PvMSP-3β in Korean P. vivax isolates was analysed. The PvMSP-3β gene is radically polymorphic in Korean P. vivax population, with multiple gene sizes and single nucleotide polymorphisms (SNPs) which are scattered throughout the gene.
Methods
Blood samples
A total of 95 blood samples used in this study were collected from Korean patients infected with P. vivax in Korea between 2007 and 2012 (2007–2010, n = 15 for each year; 2011, n = 18; 2012, n = 17). All the patients inhabited in malaria endemic areas, Ilsan, Kimpo or Yonchon, and have not been abroad at least in recent 2 years when their blood samples were collected. The P. vivax infections were identified by microscopic examination of thin and thick blood smears, and confirmed by polymerase chain reaction (PCR) [10, 20]. About 5 ml of blood was collected from each individual. The blood was separated into packed cells and plasma and then stored at −80°C until use. Informed consent was obtained from all of the patients before blood collection. The study protocol was approved by the Ethics Committee of the Inha University School of Medicine.
Genomic DNA extraction and amplification of PvMSP-3β
Genomic DNA was extracted from 200 μl of whole blood sample using the QIAamp DNA Blood Kit (Qiagen, Hilden, Germany). The full-length region encoding PvMSP-3β was amplified using two rounds of PCR. The primers were designed based on PvMSP-3β sequences of Sal I (XM_001613146) and Belem (AF099662) strains deposited in GenBank. The primers used for the first round of PCR were 5′-TTCGCAACACTCGCCTTATTTCGCTCAACG-3′ and 5′-CCCCCAATTCGTCACCAATTTGTTTAGCAT-3′. The primers used for the nested PCR were 5′-TTTCGCTCAACGCGCGCATCTAAAATG-3′ and 5′-TTAGCATATTTTCTTCCGCCTCCTTTA-3′. The following thermal cycling conditions were used for both amplifications: 94°C for 5 min; 30 cycles of 94°C for 1 min, 52°C for 1 min and 72°C for 3 min, and a final extension at 72°C for 10 min. The PCR product was analysed on a 1.2% agarose gel, purified from the gel, and ligated into the T&A cloning vector (Real Biotech Corporation, Banqiao City, Taiwan). Each ligation mixture was transformed into Escherichia coli DH5α competent cells and positive clones with the appropriate insert were selected by colony PCR. The nucleotide sequences of the cloned insert were analysed by automatic DNA sequencing using the vector primers, M13 forward and M13 reverse primers. Sequencing analyses with two additional specific internal primers (5′-AGCAAAAACAGAAGCAGAAACAGCACA-3′ and 5′-GGAAATTTTCAGCTTCCGTTTTTGCTT-3′) were also performed to obtain the sequence of central region of the PvMSP-3β. At least two clones from each isolate were sequenced to ensure accuracy, and some isolates underwent three-fold sequence coverage to confirm the existence of rare polymorphisms. The nucleotide sequences reported in this study have been deposited in the GenBank database under the accession numbers JX667768-JX667772.
Sequence and phylogenetic analysis of PvMSP-3β
The nucleotide and deduced amino acid sequences of PvMSP-3β were analysed using EditSeq and SeqMan in the DNASTAR package (DNASTAR, Madison, WI, USA). The phylogenetic tree was constructed using the neighbour-joining method in MEGA4 computational program [22]. Bootstrap proportions were used to assess the robustness of the tree with 1,000 bootstrap replicates. The coiled-coil motifs in each of the sequences were predicted using the Paircoil2 structural analysis program [23].
Sequence polymorphism analysis
DNA sequence polymorphism analysis was performed on the 95 PvMSP-3β sequences. The number of segregating sites (S), haplotypes (H), haplotype diversity (Hd), nucleotide diversity (π), and the average number of pair-wise nucleotide differences within the population (K) were estimated using the DnaSP ver. 5.10.00 [24]. The π was calculated to estimate the step-wise diversity throughout the entire PvMSP-3β based on a sliding window of 100 bases with a step size of 25 bp. The rates of synonymous (dS) and non-synonymous (dN) substitutions were estimated and were compared using the Z-test (P <0.05) in MEGA4 program [22] using Nei and Gojobori’s method [25] with the Jukes and Cantor correction. To evaluate the neutral theory of evolution, the Tajima’s D test [26] was performed with the DnaSP ver. 5.10.00 [24]. Fu and Li’s D and F statistics [27] were also analysed using the DnaSP ver. 5.10.00 [24].
Recombination parameters and linkage disequilibrium
The recombination parameter (R), which included the effective population size and probability of recombination between adjacent nucleotides per generation, and the minimum number of recombination events (Rm) were determined using the DnaSP ver. 5.10.00 [24]. The linkage disequilibrium (LD) between the different polymorphic sites was computed based on the R2 index.
Results and discussion
The full-length region encoding to PvMSP-3β was amplified from the genomic DNA from each of 95 P. vivax Korean isolates. The approximate sizes of amplified products were varied ranging from 2.1 to 2.5 kb. The nucleotide sequence analysis of the sequences revealed SNPs scattered throughout of the gene, as well as small or large insertions and deletions that resulted in variations in the size between the sequences. Based on the results of the sequence analysis, the 95 PvMSP-3β sequences of Korean isolates were classified as five different subtypes (subtypes 1, 2, 3, 4, or 5) (Figure 1). The N-terminal and C-terminal domains of PvMSP-3β are fairly polymorphic, containing SNPs and small insertions. Regarding the two large segments of insert A and insert B, which were found in Sal I sequence but not in Belem sequence [14], only two subtypes (subtypes 4 and 5) had a 120 amino acid-length insert A, but not a 200 amino acid-length insert B. The remaining three subtypes (subtypes 1, 2 and 3) had neither insert A nor B. Subtypes 2, 3, 4, and 5 contained small inserts in the N-terminal portion, whereas subtype 1 had a 2 amino acid-length small insert in C-terminal region. Subtype 3 was the most prevalent (n = 40), followed by subtype 4 (n = 23). No significant difference was observed in the annual prevalence of the subtypes (Figure 2). The phylogenetic analysis revealed that the Korean PvMSP-3β subtypes were clustered into five different clades, which differ to presently reported PvMSP-3β sequences (Figure 3). Interestingly, the Korean subtypes were widely distributed among different isolates from distinct geographic regions. This result differs from the previously reported result, which was that PvMSP-3β alleles tended to cluster based on geographic location of the isolates [14]. But, considering that sequence data for PvMSP-3β from worldwide isolates are scant, further complicated studies of P. vivax isolates from different geographical areas are required to characterize patterns in PvMSP-3β polymorphisms on a global scale.

Sequence polymorphism of the PvMSP-3β in Korean Plasmodium vivax isolates. Sequence analysis of 95 P. vivax Korean isolates revealed five distinct subtypes of PvMSP-3β. The large-scale insert A and insert B are boxed with the blue line or dotted red line, respectively. The five small insertions (S1-S5) in the N-terminal and C-terminal domains are indicated with bold lines on the sequences. The total number of sequences for each subtype are listed in the right panel. The PvMSP-3β sequences of the Salvador I (Sal I), Belem and Chess were compared to those of Korean P. vivax isolates.

Annual distribution of PvMSP-3β subtypes during the study period. The 95 PvMSP-3β sequences from Korean isolates were analysed by year of collection.

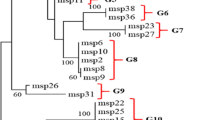
Phylogenetic analysis of the PvMSP-3β in Korean Plasmodium vivax isolates. The phylogenetic trees for the five PvMSP-3β Korean subtypes were constructed using a neighbour-joining method with the MEGA4 program. The numbers on the branches indicate the bootstrap proportions (1,000 replicates).
Although the biological function of PvMSP-3β is not fully understood, the alanine-rich central core, an important structural feature found in its amino acid sequence, suggests that the protein forms a coiled-coil tertiary structure [4, 5, 14]. Based on its expression on the surface of merozoites, PvMSP-3β is predicted to be associated with other merozoite surface molecules, possibly through protein-protein interactions involving the coiled-coil structure [4–6], in a similar manner to PfMSP-3 [7]. These overall coiled-coil structures were well conserved in all Korean PvMSP-3β subtypes, even though central coiled-coil domains were more polymorphic than the N- and C-terminal regions, which are relatively conserved (Figure 4). These results are consistent with those of previous studies that the overall tertiary structure of PvMSP-3β is well conserved in P. vivax isolates despite sequence variation among the isolates [14].

Conservation of the coiled-coil tertiary structure. The five subtypes of PvMSP-3β sequences of Korean P. vivax showed a high probability of forming coiled-coils. The portions corresponding to insert A and insert B are marked with the blue or dotted red lines, respectively.
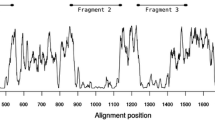
DNA sequence analyses were performed to examine the nucleotide diversity and genetic differentiation at PvMSP-3β among Korean P. vivax isolates. Nucleotide diversity was not consistently distributed within each domain or throughout the entire PvMSP-3β gene of Korean P. vivax isolates. The average number of pair-wise nucleotide differences (K) for the N-terminal domain of PvMSP-3β was 73.296 (Table 1). The overall haplotype diversity (Hd) and nucleotide diversity (π) for N-terminal domain was estimated to be 0.773 ± 0.028 and 0.07271 ± 0.00240, respectively (Table 1). The C-terminal domain of PvMSP-3β was more conserved than the N-terminal domain. The overall Hd and π for the C-terminal domain of PvMSP-3β was 0.733 ± 0.028 and 0.03040 ± 0.00179, respectively. Meanwhile, the insert A was highly conserved in subtypes 4 and 5, which represented a single haplotype. To examine whether natural selection had contributed to PvMSP-3β diversity within the Korean P. vivax population, the ratio of the average non-synonymous and synonymous mutation rates (dN/dS) in the PvMSP-3β sequences was analysed. The dN/dS ratio is widely used to evaluate the effects of natural selection on gene sequences. An excess of dN relative to dS (dN/dS ≥1) indicates positive selection, whereas a lack of dN relative to dS (dN/dS < 1) suggests negative or purifying selection imposed by functional constraints [25, 28]. The estimated dN/dS for the N-terminal domain was 1.286, indicating that positive natural selection may be occurring in the N-terminal domain of PvMSP-3β of Korean P. vivax isolates (Table 1). The dN/dS value of the C-terminal domain was less than 1 (0.455), which suggesting purifying selection pressure may act on the domain. In order to more closely explore the effects of natural selection on each domain of PvMSP-3β, Tajima’s D test was applied. The Tajima’s D values for N-terminal domain and C-terminal domain of PvMSP-3β were 3.15439 (P <0.01) and 2.27420 (P <0.05), respectively, indicating both domains had been influenced by positive natural selection (Table 1). Analysis of the sliding window plot (window length 100 bp, step size 25 bp) showed that most of the nucleotide diversities found in the N-terminal domain occurred primarily in the middle and C-terminal ends of the N-terminal domain (Figure 5A). Meanwhile, in the case of C-terminal domain, the diversities were concentrated in the N-terminal end, decreasing toward the C-terminal end of the domain (Figure 5A). These findings are consistent with those of a previous report of unequally distributed nucleotide diversity within PvMSP-3β [14].

Natural selection and recombination event in PvMSP-3β. (A) The sliding window plot of nucleotide diversity per site (π) was constructed to compare the level of genetic diversity in the N-terminal and C-terminal domains of PvMSP-3β. The π values were calculated using the DnaSP with a window length of 100 bp and step size of 25 bp. (B) The linkage disequilibrium (LD) plot showed non-random associations between the nucleotide variants in 95 Korean P. vivax isolates at different polymorphic sites. The R2 values were plotted against nucleotide distance using a two-tailed Fisher’s exact test for statistical significance.
Based on the occurrence of small indels in the sequence and the presence of degenerate repeat on either side of the indels, DNA polymerase slippage has been suggested as a possible mechanism generating diversity of PvMSP-3β [14]. Similar small indels and flanking degenerate repeats were also identified in PvMSP-3β sequences of Korean P. vivax isolates, which suggested slipped-strand mispairing during DNA replication may have contributed to PvMSP-3β diversity. The 11 amino acid-length repeated elements that were identified in the large-scale insertion [14] were also conserved in the end of the N-terminal domain and the end of insert A of PvMSP-3β of Korean P. vivax isolates. The diversity of Plasmodium antigens, including PvMSP-3α, is also known to be generated by intragenic recombination events, and is maintained by balancing selection [9, 10, 20, 28]. Similarly, it has been postulated that recombination is another major factor generating allelic diversity of PvMSP-3β [14]. To understand the role of the recombination event in PvMSP-3β in Korean P. vivax isolates, the recombination parameters and the linkage disequilibrium in the gene were analysed. For the N-terminal domain of PvMSP-3β, the minimum number of recombination events between adjacent polymorphic sites (Rm) was 13, whereas the R between adjacent sites (Ra) and per gene (Rb) was 0.0036 and 3.6, respectively. In the case of C-terminal domain of PvMSP-3β, the Rm was 4 with a Ra of 0.0029 and a Rb of 3.1. These high recombination parameter values suggested that recombination may have occurred between sites, contributing to genetic diversity in PvMSP-3β gene. The linkage disequilibrium index, R2, was also declined across the analysed region, which further indicates that intragenic recombination might have contributed to the diversity observed in both the N-terminal and C-terminal domains of PvMSP-3β in Korean P. vivax isolates (Figure 5B).
The genetic diversity of malaria parasites is closely associated with the levels of endemicity and intensity of transmission [29, 30]. The P. vivax population in hyperendemic areas, such as Papua New Guinea, is highly diverse and shows multiplicity of infections [31, 32]. However, complex genetic structures have also been identified in P. vivax populations in hypo-endemic areas, such as China, Myanmar and Thailand [10, 33–38]. Several recent studies have suggested that genetic diversity within the P. vivax population in Korea has increased in recent years [17–21]. Considering the low endemicity and transmission intensity in Korea, it remains unclear how genetic diversity of P. vivax in Korea is rapidly disseminated and maintained in recent years. An increased influx of international travellers and foreign workers might have contributed to the increasing allelic variation in the Korean P. vivax population through the introduction of malaria parasites from other endemic areas [17]. It is also possible that the Korean P. vivax population has evolved under evolutionary pressure, most probably related to host immune response. Further studies of genetic diversity in the Korean P. vivax population in Korea are warranted to clarify the patterns of genetic variation and the biological relevance of increasing P. vivax diversity.
Conclusions
Radical genetic polymorphism was identified in the PvMSP-3β gene of Korean P. vivax isolates. The size of the PvMSP-3β varied, as a result of large and small insertions/deletions, and numerous SNPs were observed throughout the gene. Despite the high level of sequence diversity, the coiled-coil tertiary structure of the protein was well conserved in all of the Korean P. vivax isolates, suggesting that the protein is under functional constraints. The genetic diversity of the PvMSP-3β of Korean P. vivax isolates has been influenced by natural selection, particularly in the N-terminal and C-terminal domains. Polymerase slippage during DNA replication and intragenic recombination might have contributed to the genetic diversity of PvMSP-3β in the Korean P. vivax population. These findings of parasite heterogeneity are relevant to the development of a PvMSP-3β-based vaccine against P. vivax and the implementation of malarial control programmes in Korea.
References
Mendis K, Sina BJ, Marchesini P, Carter R: The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001, 64: 97-106.
Cui L, Escalante AA, Imwong M, Snounou G: The genetic diversity of Plasmodium vivax populations. Trends Parasitol. 2003, 19: 220-226. 10.1016/S1471-4922(03)00085-0.
Li J, Collins WE, Wirtz RA, Rathore D, Lal A, McCutchan TF: Geographic subdivision of the range of the malaria parasite Plasmodium vivax. Emerg Infect Dis. 2001, 7: 35-42. 10.3201/eid0701.010105.
Galinski MR, Corredor-Medina C, Povoa M, Crosby J, Ingravallo P, Barnwell JW: Plasmodium vivax merozoite surface protein-3 contains coiled-coil motifs in an alanine-rich central domain. Mol Biochem Parasitol. 1999, 101: 131-147. 10.1016/S0166-6851(99)00063-8.
Galinski MR, Ingravallo P, Corredor-Medina C, Al-Khedery B, Povoa M, Barnwell JW: Plasmodium vivax merozoite surface proteins-3beta and-3gamma share structural similarities with P. vivax merozoite surface protein-3alpha and define a new gene family. Mol Biochem Parasitol. 2001, 115: 41-53. 10.1016/S0166-6851(01)00267-5.
Jiang J, Barnwell JW, Meyer EV, Galinski MR: Plasmodium vivax merozoite surface protein-3 (PvMSP3): expression of an 11 member multigene family in blood-stage parasites. PLoS One. 2013, 8: e63888-10.1371/journal.pone.0063888.
McColl DJ, Anders RF: Conservation of structural motifs and antigenic diversity in the Plasmodium falciparum merozoite surface protein-3 (MSP-3). Mol Biochem Parasitol. 1997, 90: 21-31. 10.1016/S0166-6851(97)00130-8.
Huber W, Felger I, Matile H, Lipps HJ, Steiger S, Beck HP: Limited sequence polymorphism in the Plasmodium falciparum merozoite surface protein 3. Mol Biochem Parasitol. 1997, 87: 231-234. 10.1016/S0166-6851(97)00067-4.
Rayner JC, Corredor V, Feldman D, Ingravallo P, Iderabdullah F, Galinski MR, Barnwell JW: Extensive polymorphism in the Plasmodium vivax merozoite surface coat protein MSP-3α is limited to specific domains. Parasitology. 2002, 125: 393-405.
Moon SU, Lee HW, Kim JY, Na BK, Cho SH, Lin K, Sohn WM, Kim TS: High frequency of genetic diversity of Plasmodium vivax field isolates in Myanmar. Acta Trop. 2009, 109: 30-36. 10.1016/j.actatropica.2008.09.006.
Véron V, Legrand E, Yrinesi J, Volney B, Simon S, Carme B: Genetic diversity of msp3alpha and msp1 b5 markers of Plasmodium vivax in French Guiana. Malar J. 2009, 8: 40-10.1186/1475-2875-8-40.
Schousboe ML, Rajakaruna RS, Amerasinghe PH, Konradsen F, Ord R, Pearce R, Bygbjerg IC, Roper C, Alifrangis M: Analysis of polymorphisms in the merozoite surface protein-3α gene and two microsatellite loci in Sri Lankan Plasmodium vivax: evidence of population substructure in Sri Lanka. Am J Trop Med Hyg. 2011, 85: 994-1001. 10.4269/ajtmh.2011.11-0338.
Rice BL, Acosta MM, Pacheco MA, Escalante AA: Merozoite surface protein-3 alpha as a genetic marker for epidemiologic studies in Plasmodium vivax: a cautionary note. Malar J. 2013, 12: 288-10.1186/1475-2875-12-288.
Rayner JC, Huber CS, Feldman D, Ingravallo P, Galinski MR, Barnwell JW: Plasmodium vivax merozoite surface protein PvMSP-2 beta is radically polymorphic through mutation and large insertions and deletions. Infect Genet Evol. 2004, 4: 309-319. 10.1016/j.meegid.2004.03.003.
Rungsihirunrat K, Chaijaroenkul W, Siripoon N, Seugorn A, Na-Bangchang K: Genotyping of polymorphic marker (MSP3α and MSP3β) genes of Plasmodium vivax field isolates from malaria endemic of Thailand. Trop Med Int Health. 2011, 16: 794-801. 10.1111/j.1365-3156.2011.02771.x.
Bitencourt AR, Vicentin EC, Jimenez MC, Ricci R, Leite JA, Costa FT, Ferreira LC, Russell B, Nosten F, Rénia L, Galinski MR, Barnwell JW, Rodrigues MM, Soares IS: Antigenicity and immunogenicity of Plasmodium vivax merozoite surface protein-3. PLoS One. 2013, 8: e56061-10.1371/journal.pone.0056061.
Choi YK, Choi KM, Park MH, Lee EG, Kim YJ, Lee BC, Cho SH, Rhie HG, Lee HS, Yu JR, Lee JS, Kim TS, Kim JY: Rapid dissemination of newly introduced Plasmodium vivax genotypes in South Korea. Am J Trop Med Hyg. 2010, 82: 426-432. 10.4269/ajtmh.2010.09-0245.
Han ET, Wang Y, Lim CS, Cho JH, Chai JY: Genetic diversity of the malaria vaccine candidate merozoite surface protein 1 gene of Plasmodium vivax field isolates in Republic of Korea. Parasitol Res. 2011, 109: 1571-1576. 10.1007/s00436-011-2413-5.
Honma H, Kim JY, Palacpac NM, Mita T, Lee W, Horii T, Tanabe K: Recent increase of genetic diversity in Plasmodium vivax population in the Republic of Korea. Malar J. 2011, 10: 257-10.1186/1475-2875-10-257.
Kang JM, Ju HL, Kang YM, Lee DH, Moon SU, Sohn WM, Park JW, Kim TS, Na BK: Genetic polymorphism and natural selection in the C-terminal 42 kDa region of merozoite surface protein-1 among Plasmodium vivax Korean isolates. Malar J. 2012, 11: 206-10.1186/1475-2875-11-206.
Ju HL, Kang JM, Moon SU, Bahk YY, Cho PY, Sohn WM, Park YK, Park JW, Kim TS, Na BK: Genetic diversity and natural selection of Duffy binding protein of Plasmodium vivax Korean isolates. Acta Trop. 2013, 125: 67-74. 10.1016/j.actatropica.2012.09.016.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
McDonnell AV, Jiang T, Keating AE, Berger B: Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006, 22: 356-358. 10.1093/bioinformatics/bti797.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451-1452. 10.1093/bioinformatics/btp187.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu YX, Li WH: Statistical tests of neutrality of mutations. Genetics. 1993, 133: 693-709.
Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA: Assessing the effect of natural selection in malaria parasites. Trends Parasitol. 2004, 20: 388-395. 10.1016/j.pt.2004.06.002.
Babiker HA, Lines J, Hill WG, Walliker D: Population structure of Plasmodium falciparum in villages with different malaria endemicity in east Africa. Am J Trop Med Hyg. 1997, 56: 141-147.
Anderson TJC, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bocharie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day K: Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000, 17: 1467-1482. 10.1093/oxfordjournals.molbev.a026247.
Kolakovich KA, Ssengoba A, Wojcik K, Tsuboi T, Al-Yaman F, Alpers M, Adams JH: Plasmodium vivax: favored gene frequencies of the merozoite surface protein-1 and the multiplicity of infection in a malaria endemic region. Exp Parasitol. 1996, 83: 11-19. 10.1006/expr.1996.0044.
Bruce MC, Galinski MR, Barnwell JW, Donnelly CA, Walmsley M, Alpers MP, Walliker D, Day K: Genetic diversity and dynamics of Plasmodium falciparum and P. vivax populations in multiply infected children with asymptomatic malaria infections in Papua New Guinea. Parasitology. 2000, 121: 257-272. 10.1017/S0031182099006356.
Cui L, Mascorro CN, Fan Q, Rzomp KA, Khuntirat B, Zhou G, Chen H, Yan G, Sattabongkot J: Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. Am J Trop Med Hyg. 2003, 68: 613-619.
Sheng HF, Zhou SS, Gu ZC, Zheng X: Malaria situation in the People’s Republic of China in 2002. Clin J Parasitol Parasit Dis. 2003, 21: 193-196.
Zakeri S, Djadid ND, Zeinali S: Sequence heterogeneity of the merozoite surface protein-1 gene (MSP-1) of Plasmodium vivax wild isolates in southeastern Iran. Acta Trop. 2003, 88: 91-97. 10.1016/S0001-706X(03)00192-X.
Leclerc MC, Menegon M, Cligny A, Noyer JL, Mammadov S, Aliyev N, Gasimov E, Majori G, Severini C: Genetic diversity of Plasmodium vivax isolates from Azerbaijan. Malar J. 2004, 3: 40-10.1186/1475-2875-3-40.
Moon SU, Na BK, Kang JM, Kim JY, Cho SH, Park YK, Sohn WM, Lin K, Kim TS: Genetic polymorphism and effect of natural selection at domain I of apical membrane antigen-1 (AMA-1) in Plasmodium vivax isolates from Myanmar. Acta Trop. 2010, 114: 7-15.
Ju HL, Kang JM, Moon SU, Kim JY, Lee HW, Lin K, Sohn WM, Lee JS, Kim TS, Na BK: Genetic polymorphism and natural selection of Duffy binding protein of Plasmodium vivax Myanmar isolates. Malar J. 2012, 11: 60-10.1186/1475-2875-11-60.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2011–0028135).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JMK and HLJ performed all the experiments and analysed the sequence data. PYC, SKA and SUM collected blood samples or performed sequence and phylogenetic analyses. BKN and TSK designed the study and supervised the study process. JMK and BKN wrote the paper. TSK, HWL and WMS assisted in writing and editing the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kang, JM., Ju, HL., Cho, P.Y. et al. Polymorphic patterns of the merozoite surface protein-3β in Korean isolates of Plasmodium vivax. Malar J 13, 104 (2014). https://doi.org/10.1186/1475-2875-13-104
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-13-104