
Abstract
Background
Inactive protein inclusion bodies occur commonly in Escherichia coli (E. coli) cells expressing heterologous proteins. Previously several independent groups have found that active protein aggregates or pseudo inclusion bodies can be induced by a fusion partner such as a cellulose binding domain from Clostridium cellulovorans (CBDclos) when expressed in E. coli. More recently we further showed that a short amphipathic helical octadecapeptide 18A (EWLKAFYEKVLEKLKELF) and a short beta structure peptide ELK16 (LELELKLKLELELKLK) have a similar property.
Results
In this work, we explored a third type of peptides, surfactant-like peptides, for performing such a "pulling-down" function. One or more of three such peptides (L6KD, L6K2, DKL6) were fused to the carboxyl termini of model proteins including Aspergillus fumigatus amadoriase II (AMA, all three peptides were used), Bacillus subtilis lipase A (LipA, only L6KD was used, hereinafter the same), Bacillus pumilus xylosidase (XynB), and green fluorescent protein (GFP), and expressed in E. coli. All fusions were found to predominantly accumulate in the insoluble fractions, with specific activities ranging from 25% to 92% of the native counterparts. Transmission electron microscopic (TEM) and confocal fluorescence microscopic analyses confirmed the formation of protein aggregates in the cell. Furthermore, binding assays with amyloid-specific dyes (thioflavin T and Cong red) to the AMA-L6KD aggregate and the TEM analysis of the aggregate following digestion with protease K suggested that the AMA-L6KD aggregate may contain structures reminiscent of amyloids, including a fibril-like structure core.
Conclusions
This study shows that the surfactant-like peptides L6KD and it derivatives can act as a pull-down handler for converting soluble proteins into active aggregates, much like 18A and ELK16. These peptide-mediated protein aggregations might have important implications for protein aggregation in vivo, and can be explored for production of functional biopolymers with detergent or other interfacial activities.
Similar content being viewed by others
Background
Inactive inclusion bodies are commonly formed during the overexpression of heterologous proteins in recombinant hosts such as E. coli [1]. Only a limited number of them, often small proteins with no or few cysteine residues, can be recovered through refolding [2]. It has been generally accepted that these nonfunctional inclusion bodies are noncrystalline, amorphous structures [3]. One notable exception was the inclusion bodies of beta-galactosidase obtained from overexpression in E. coli, which were found to be biologically active [4]. In recent years, however, several groups have strikingly observed the spontaneous formation of pseudo inclusion bodies which are active, when the target proteins are fused to an aggregation-prone domain or peptide [5–7]. For example, D-amino acid oxidase from Trigonopsis variabilis (TvDAO) fused with a cellulose binding domain from Clostridium cellulovorans (CBDclos) yielded an enzyme aggregate retaining high specific activity [5]. Similarly, MalE31, an aggregation-prone variant of the maltose-binding protein, and a β-amyloid peptide variant Aβ(F19D) have also been used as fusion partners for inducing active protein aggregates [6, 7]. These domains and peptide presumbly provide the specific self-associating modules for the fusion protiens and thus drive the target proteins into aggregates [5].
In our previous studies [8, 9], we unexpectedly found that a short amphipathic α helical octadecapeptide 18A (EWLKAFYEKVLEKLKELF) was able to induce several normally soluble proteins into active protein aggregates when expressed in E. coli. The fourier transform infrared (FTIR) spectra of the 18A peptide induced protein aggregates revealed enhanced α helical secondary structures, suggesting it was the assoication of the 18A peptide that led to the formation of pseudo inclusion bodies. Since amphipathic alpha peptides are abundent in protein strucutres [10], this observation might have implications for protein aggregation in general for biological systems. We subsequently considered whether there were peptides of a different structure other than alpha that could act as a pulling down handler, and a second peptide, ELK16 (LELELKLKLELELKLK) which is beta sturcure in nature was found to have a similar property [9].
To study the generality of this short-peptide induced protein aggregation, in this work we set out to test a third type of small peptides, i.e., surfactant-like peptides that were designed to mimic surfactants and normally do not resemble either α-helical or β-sheet structure [11, 12]. These peptides consist of a hydrophobic tail and a hydrophilic head similar to surfactant molecules, and can spontaneously form nanostructures in aqueous solution. To this end, we attached three of such surfactant-like peptides (L6KD, L6K2, DKL6) to the carboxyl termini of model proteins, and expressed the fusions in E. coli. Most of the fusion proteins were indeed found to be largely insoluble but retained high biological activity, showing that terminally attached surfactant-like peptides can also drive proteins into biologically active pseudo inclusion boides in E. coil.
Results
Surfactant-like peptides induced proteins to form active protein aggregates
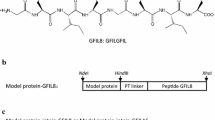
The surfactant-like peptides normally contain a hydrophobic tail and a hydrophilic head. We selected or re-designed three short peptides L6K2, L6KD, and DKL6 based on the literature (Figure 1) [11, 12]. The hydrophobic moiety of all three peptides is six leucines. Leucine has a large hydrophobic side chain, and can pack with each other more easily than other hydrophobic peptides [11]. The hydrophilic moiety of the molecule is two charged amino acids, the positively charged lysine and/or the negatively charged aspartic acid [11], with a total length of 2-3 nm in the extended conformation [11]. Starting from L6K2, to assess the charge effect, we replaced one of the lysines with one aspartic acid to yield L6KD to neutralize the charge, and to assess the positional effect of the hydrophilic head in relation to the model protein, we reversed the sequence of L6KD to yield DKL6. L6D2 was not tested as it was reported that L6K2 and L6D2 showed little difference in terms of self-assembly property [11, 12].

Schematics for surfactant-like peptide and fusion protein constructs. (A) The sequences of the surfactant-like peptides used in this study. (B) Genetic constructs of the surfactant-like peptide fusion proteins. Four model proteins: Aspergillus fumigatus amadoriase II (AMA), Bacillus subtilis lipase A (LipA), Bacillus pumilus xylosidase (XynB), and green fluorescent protein (GFP); linker, PTPPTTPTPPTTPTPTP.
These three peptides were first fused to the C-terminus of the model protein Aspergillus fumigatus amadoriase II (AMA) via a PT linker PTPPTTPTPPTTPTPTP (Figure 1), and expressed in E. coli at 30°C. The expression of these fusion proteins had little effect on the cell growth as judged by OD600 measurements. Cells were harvested and lysed, and lysates were separated into soluble and insoluble fractions by centrifugation, and analyzed by SDS-PAGE analyses. Most of the fusion proteins were found in the insoluble fractions, in the range of 64%~77% in terms of mass, markedly different from the native protein (~6%). All three fusion proteins in the insoluble fractions were found to be active. For L6K2 fusion, the activity of the insoluble fraction accounted for about 36.8% of the total activity, and for L6KD and DKL6 fusions, it was 60.5% (Figure 2A) and 56.1%, respectively. The differences between the mass-based percentages and the activity-based percentages resulted from the different specific activities of the fusion proteins in the soluble and insoluble fractions.

Distributions of enzyme activities in the soluble and insoluble fractions of cell lysates. (B) AMA-native and AMA-L6KD. (B) LipA-native and LipA-L6KD. (C) XynB-native and XynB-L6KD. The activities were determined using three independent experiments and normalized to the total activities of the respective native enzyme extracted from a same amount of cells (OD600). Standard deviations are also shown.
Since AMA-L6KD fusion protein produced the highest percentage of active aggregate, this suggested that this peptide was more efficient as a "pulling-down" handler, and thus was chosen for further testing using two additional model proteins, Bacillus subtilis lipase A (LipA), and Bacillus pumilus xylosidase (XynB) (Figure 2B and 2C). In agreement with the above results, both the LipA-L6KD and XynB-L6KD fusion proteins formed obvious active aggregates. For LipA-L6KD, the insoluble fraction accounted for about 80.2% of the total activity, and for XynB-L6KD, it was 83.7%. The relative specific activities of the insoluble fractions for three fusions were showed in Table 1. For AMA-L6KD, it was 92.6% of the native counterpart, and for LipA-L6KD and XynB-L6KD, it was 30.2% and 25.6%, respectively.
Microscopic analyses of active protein aggregates
To study the intracellular locations of these pseudo inclusion bodies, transmission electron microscopic (TEM) analyses were performed for recombinant cells expressing the fusion protein AMA-L6KD. From the TEM images (Figure 3A and 3B), cytoplasmic inclusion bodies were clearly observed for AMA-L6KD, with the diameters of about several micrometers. Further studies showed that the fusion proteins LipA-L6KD and XynB-L6KD had a similar pattern of aggregation (data not shown).

Intracellular localization of fusion proteins in E. coli. (A) and (B), TEM microscopic images for AMA-native and AMA-L6KD, respectively. (C) Confocal fluorescent micrograph for GFP-L6KD. Size bars are also shown.
To further confirm the cellular locations of the active protein aggregates, we similarly constructed GFP-L6KD fusion, and subjected the E. coli cells expressing GFP-L6KD to confocal fluorescence microscopic analyses. As shown in Figure 3C, clearly a localized pattern of fluorescence distribution was observed in the recombinant cells, different from the cells expressing native GFP in which a uniformed distribution of fluorescence was observed [9].
Analyses of amyloid-like properties
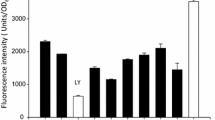
Recently, several research groups reported that inclusion bodies formed in E. coli may contain amyloidal structures [13–16] commonly found in protein deposits associated with diseases such as Alzheimer's disease [17–19]. Amyloids characteristically contain fibril-like cores and bind to specific dyes such as thioflavin T (ThT) and Congo red (CR) [18, 20, 21]. To gain an insight into the molecular mechanism in the surfactant-like peptide-mediated protein aggregation observed in our study, we thus explored the structural property of AMA-L6KD aggregate by using thioflavin T and Congo red. Thioflavin T will exhibit a significantly enhanced fluorescence at 480 nm relative to free dye upon binding to amyloid fibrils [21]. As shown in Figure 4, the ThT binding assay for AMA-L6KD clearly resulted in a 24-fold increase in the fluorescence at 480 nm (Figure 4). The CR binding assay also showed a shift of absorbance maximum to about 508 nm, and a band at about 541 nm in the differential spectrum, characteristic of binding of Congo red to amyloids (data not shown) [20]. We further used TEM coupled with protease K digestion to explore the morphology of the AMA-L6KD aggregate [14, 15]. As shown in Figure 5, irregularly organized fibrils can be clearly observed, suggesting an amyloid-like structure for the aggregate.

Binding of AMA-L 6 KD aggregate to thioflavin T (ThT). The histograms show the fluorescence intensity of ThT at 480 nm (excited at 445 nm) in the presence and in the absence of AMA-L6KD aggregate, in arbitrary units (a. u.).

Fibrillar structure of AMA-L 6 KD protein aggregate. The micrograph shows the fibers of AMA-L6KD aggregate after proteolytic treatment by protease K. Size bar is also shown.
Discussion
This study demonstrates that terminally attached surfactant-like peptides like L6KD can convert soluble proteins into active aggregates, with specific activities relative to the native counterparts ranging from 25% to 92%. As aggregation inducers, L6K2, L6KD, DKL6 are only eight residues in length, even shorter than the peptides 18A (18 residues in length) or ELK16 (16 residues in length). Judging from the activities of the insoluble fractions for their fusions with AMA as the model protein, it seems that a hydrophilic head with a neutral charge (as in the case of AMA-L6KD or AMA-DKL6) is better at promoting active protein aggregation than a hydrophilic head with a charge (as in the case of AMA-L6K2). Notably however, the position of the hydrophilic head in relation to the target protein in the fusion, i.e., between the target protein and the hydrophobic tail (as in the case of AMA-DKL6) or toward the C-terminal end of the hydrophobic tail (as in the case of AMA-L6KD), makes much less of a difference.
Judging from the microscopic analyses, the morphology of these fusion aggregates is similar to that of those induced by aggregation-prone domains and peptides as reported earlier [7–9]. While the exact structural detail by which these surfactant-like peptides drive the target proteins into active aggregates remain to be determined, our analysis of the AMA-L6KD aggregate following digestion of protease K revealed fibril-like structures (Figure 4), and the positive binding assay results with amyloid-specific dyes suggest that the AMA-L6KD aggregate possesses amyloid-like structures. This is consistent with the view that these pseudo inclusion bodies may contain amyloid-like structures [13–15, 17].
Recent studies have shown that, in addition to aggregation-prone domain such as the cellulose binding domain CBDclos [5], several peptides can also induce the formation of protein aggregates or inclusion bodies in vivo, such as the human β-amyloid peptide Aβ42(F19D) [7], a modified apolipoprotein A-I mimetic amphipathic peptide 18A [8], and the ELK16 peptide derived from EAK16 [9]. These peptides are alpha or beta structure in nature. Our surfactant-like peptides provide a third type of peptide structure beyond alpha or beta that can drive proteins into active aggregates. This peptide-mediated protein aggregation might have important implications for protein aggregation in vivo, and for protein aggregation-related diseases. The active protein aggregation induced by these peptides has potential biotechnological applications. For example, it can be used to design a facile expression and purification scheme for protein [22, 23], or to obtain active protein aggregates for direct use as biocatalysts [24, 25].
Along this line, it is interesting to compare the efficiency of these three short peptides (18A, ELK16 and L6KD) as aggregation tags, in terms of both pulling down efficiency (PDE) and specific enzyme activity relative to native enzyme (SArN). As shown in Table 2, ELK16 is a better tag for model enzymes AMA and XynB (LipA lost activity when attached to ELK16), compared with 18A and L6KD. L6KD is generally comparable with 18A both in terms of PDE and SArN, except that the SArN for LipA-18A fusion aggregate (84%) is much higher than that for LipA-L6KD (30.2%). It thus seems that L6KD has no superiority in terms of PDE and SArN compared with ELK16 or 18A, except for its smallest length which in some cases may confer advantage for fusion construction albeit in the cost of pulling down efficiency. Additionally, given its unique surfactant property, it is worthwhile to explore the possible use of this tag for design and mass-production of functional biopolymers with detergent or other interfacial activities via microorganism.
Conclusions
Our study reveals that the presence of surfactant-like peptides can convert fusion proteins into active aggregates in vivo which may contain amyloid-like structures. These peptide-mediated protein aggregates may be useful for protein purification, biocatalysis and biosurfactant design and production. Further exploration of this type of protein aggregates may provide new insights into protein aggregation and perhaps related cellular processes and diseases.
Materials and methods
Plasmid construction
To construct plasmid pET30a-AMA-L6KD, two primers (AMA-up: 5'-TTCTGGACATATG GCGGTAACCAAGTCATC-3', AMA-L6KD-down: 5'-ATGAACTCGAG TCAATCTTTCAGCAGCAGCAGCAGCAGCGGCGTCGGGGTTGGGGTG-3', the restriction sites Nde I and Xho I were underlined, respectively) were used to amplify the gene encoding AMA-L6KD from the previously constructed plasmid pET30a-AMA-C18 [8]. Then the amplified DNA fragment was digested with Nde I and Xho I, and inserted into similarly digested plasmid pET30a (+) (Novagen) to yield pET30a-AMA-L6KD. Plasmids pET30a-AMA-L6K2 and pET30a-AMA-DKL6 were similarly constructed using primers AMA-L6K2-down (5'-ATGAACTCGAG TCATTTTTTCAGCAGCAGCAGCAGCAGCGGCGTCGGGGTTGGGGT-3') and AMA-DKL6-down (5'-ATGAACTCGAG TCACAGCAGCAGCAGCAGCAGTTTATCCGGCGTCGGGGTTGGGGTG-3'). Plasmids pET30a-Lip A-L6KD, pET30a-XynB-L6KD and pET30a-GFP-L6KD were obtained by replacing the sequence encoding AMA in pET30a-AMA-L6KD with that of Lip A, XynB and GFP, respectively.
Expression and extraction of protein aggregates
E. coli BL21 (DE3) (Novagen) was used for all the experiments. The recombinant strains harboring the plasmids were cultured in Luria-Bertani (LB) medium supplemented with 50 mg/l kanamycin at 37°C. Isopropyl β-D-1-thiogalactopyranoside (IPTG, at a final concentration of 0.2 mM) was added to the culture medium at 30°C to induce fusion protein expression, when the cell optical density at 600 nm (OD600) reached 0.4-0.6. After 6 h, cells were harvested by centrifugation, and cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, 50 mM NaCl, 5% glycerol, pH 7.2) with a final concentration of 10 OD600 per ml. The resuspended cells were lysed by ultrasonication on ice, and the lysates separated by centrifugation. The insoluble fractions were washed twice with 1 ml of lysis buffer, and resuspended again in a same volume of lysis buffer. The amounts of target proteins in all samples were determined densitometrically by denaturing polyacrylamide gel electrophoresis (SDS PAGE, 12%) using bovine serum albumin (BSA) as standard, followed by staining with Coomassie Brilliant Blue G-250. The values of target protein amounts were calculated with Quantity One software (Bio-Rad Laboratories, Hercules, CA).
Determination of enzyme activities
The enzyme activities in both the soluble and insoluble fractions were assayed with a SPECTRAMAX M2 microplate reader (Molecular Device, CA). The amadoriase activity [26] was measured by monitoring the formation of a quinone dye in a peroxidase-coupling reaction at 555 nm (e = 39.2 cm2/mmol) at 37°C. 5 ml of enzyme was added to 175 ml of reaction mixture (100 mM potassium phosphate buffer (pH 8.0), 2.7 purpurogallin units of peroxidase, 0.45 mM 4-aminoantipyrine, 0.5 mM N-ethyl-N-(2-hydroxy-3-sulfopropyl)-m-toluidine (TOOS), and 5.0 mM D-fructosyl-glycine). One unit of amadoriase was defined as the amount of enzyme that produced 1 nmol H2O2 per min. LipA activity [27] and XynB activity [28] were measured by monitoring the formation of p-nitrophenol at 405 nm (e = 18.7 cm2/mmol) at 37°C. For lipase, 5 ml of diluted enzyme was added to 175 ml of reaction mixture (50 mM sodium phosphate buffer, pH 8.0; 0.4 mM p-nitrophenyl palmitate; 0.2% sodium deoxycholate, and 0.1% gum arabic). For β-xylosidase, 5 ml of enzyme was added to 175 ml of reaction mixture (50 mM phosphate buffer, pH 6.0, 2.5 mM p-nitrophenyl β-D-xylopyranoside). One unit of lipase was defined as the amount of enzyme producing 1 μmol of p-nitrophenol (pNP) per min while one unit of xylosidase was defined as the amount of enzyme producing 1 nmol of pNP per min.
Laser scanning confocal microscopic (LSCM) analyses
The cells expressing fusion protein GFP-L6KD were cultivated at 23°C for 22 h after induction with 0.2 mM IPTG. Cells were harvested and washed twice with phosphate buffered saline (PBS). The cell pellets were then fixed with 4% paraformaldehyde and photographed at 488 nm using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Germany).
Transmission electron microscopic (TEM) analyses
TEM was used to analyze the intracellular location and the morphology of protein aggregates. For the intracellular analyses, recombinant cells were harvested after expression 6 h at 30°C and fixed with fixing solution (2.5% glutaraldehyde and 2% osmium tetraoxide, dehydrated). The fixed cells were dehydrated through a graded-ethanol serial dehydration step, and embedded in epoxy resins. The embedded cells were then sectioned into ultrathin slices, stained by stain solution (containing uranyl acetate solution and lead citrate), and observed with a Hitachi H-7650B (Hitachi, Japan) transmission electron microscope at an accelerating voltage of 80 kV. For the morphology analyses [14], extracted protein aggregates were digested with DNAse and RNAse A (25 μg/ml) for 1 h at 37°C in PBS in the presence of 10 mM MgSO4. After that, the protein aggregates were washed with 0.5% Triton-X solution and then PBS. The protein aggregates (50 μg/ml) were then digested using protease K (20 μg/ml) at 37°C for 30 min in PBS, and then washed with PBS. The insoluble fractions were resuspended in a same volume of deionized distilled water, and spotted on copper grids for 5 min. The grids were washed with water, and stained with 1% (w/v) aqueous uranyl formate solution. The prepared samples were then analyzed with a Hitachi H-7650B transmission electron microscope at an accelerating voltage of 75 kV.
Binding assays of amyloid-specific dyes to AMA-L6KD
Thioflavin T (ThT) fluorescence assays were measured with a SPECTRAMAX M2 microplate reader (Molecular Device, CA) with an excitation wavelength of 445 nm and an emission range from 475 nm to 570 nm at 37°C [21, 29]. The protein aggregate (in a final concentration of 10 μM) was mixed with 10 μM ThT in PBS in a 96-well black plate. For the Congo red (CR) staining experiment [16, 30], 10 μM CR in PBS was incubated in the presence or absence of the protein aggregate (in a final concentration of 10 μM) for 20 min at room temperature and the absorbance spectrum form 360 nm to 700 nm was recorded with a Beckman UV/Vis spectrophotometer.
References
Marston FAO: The purification of eukaryotic polypeptides synthesized in Escherichia coli . Biochem J 1986,240(1):1-12.
Palmer I, Wingfield PT: Preparation and extraction of insoluble (inclusion-body) proteins from Escherichia coli . In Curr Protoc Protein Sci. Volume 6. Edited by: Coligan JE. Hoboken: John Wiley & Sons Inc; 2004:6.3.1-6.3.15.
Bowden GA, Paredes AM, Georgiou G: Structure and morphology of protein inclusion bodies in Escherichia coli . Nat Biotechnol 1991,9(8):725-730. 10.1038/nbt0891-725
Worrall DM, Goss NH: The formation of biologically active beta-galactosidase inclusion bodies in Escherichia coli . Aust J Biotechnol 1989,3(1):28-32.
Nahalka J, Nidetzky B: Fusion to a pull-down domain: A novel approach of producing Trigonopsis variabilis D-amino acid oxidase as insoluble enzyme aggregates. Biotechnol Bioeng 2007,97(3):454-461. 10.1002/bit.21244
Arie JP, Miot M, Sassoon N, Betton JM: Formation of active inclusion bodies in the periplasm of Escherichia coli . Mol Microbiol 2006,62(2):427-437. 10.1111/j.1365-2958.2006.05394.x
Garcia-Fruitos E, Gonzalez-Montalban N, Morell M, Vera A, Ferraz RM, Aris A, Ventura S, Villaverde A: Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb Cell Fact 2005, 4: 27. 10.1186/1475-2859-4-27
Wu W, Xing L, Zhou BH, Lin ZL: Assembly of active protein aggregates in vivo induced by terminally attached amphipathic peptide. Biotechnol Adv, in press.
Wu W, Xing L, Zhou BH, Lin ZL: Active protein aggregates induced by terminally attached self-assembling peptide ELK16 in Escherichia coli . Microb Cell Fact 2011,10(1):9. 10.1186/1475-2859-10-9
Sharadadevi A, Sivakamasundari C, Nagaraj R: Amphipathic alpha-helices in proteins: Results from analysis of protein structures. Proteins 2005,59(4):791-801. 10.1002/prot.20459
Vauthey S, Santoso S, Gong HY, Watson N, Zhang SG: Molecular self-assembly of surfactant-like peptides to form nanotubes and nanovesicles. Proc Natl Acad Sci USA 2002,99(8):5355-5360. 10.1073/pnas.072089599
von Maltzahn G, Vauthey S, Santoso S, Zhang SU: Positively charged surfactant-like peptides self-assemble into nanostructures. Langmuir 2003,19(10):4332-4337. 10.1021/la026526+
Carrio M, Gonzalez-Montalban N, Vera A, Villaverde A, Ventura S: Amyloid-like properties of bacterial inclusion bodies. J Mol Biol 2005,347(5):1025-1037. 10.1016/j.jmb.2005.02.030
Morell M, Bravo R, Espargaro A, Sisquella X, Aviles FX, Fernandez-Busquets X, Ventura S: Inclusion bodies: Specificity in their aggregation process and amyloid-like structure. Biochim Biophys Acta-Mol Cell Res 2008,1783(10):1815-1825. 10.1016/j.bbamcr.2008.06.007
Wang L, Maji SK, Sawaya MR, Eisenberg D, Riek R: Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol 2008,6(8):1791-1801.
Dasari M, Espargaro A, Sabate R, del Amo JML, Fink U, Grelle G, Bieschke J, Ventura S, Reif B: Bacterial inclusion bodies of Alzheimer's disease beta-Amyloid peptides can be employed to study native-like aggregation intermediate states. ChemBioChem 2011,12(3):407-423. 10.1002/cbic.201000602
Mitraki A: Protein aggregation: form inclusion bodies to amyloid and biomaterials. In Adv Protein Chem Struct Biol. Volume 79. Edited by: McPherson A. San Diego: Elsevier Academic Press Inc; 2010:89-125.
Reinke AA, Gestwicki JE: Insight into amyloid structure using chemical probes. Chem Biol Drug Des 2011,77(6):399-411. 10.1111/j.1747-0285.2011.01110.x
Sabate R, de Groot NS, Ventura S: Protein folding and aggregation in bacteria. Cell Mol Life Sci 2010,67(16):2695-2715. 10.1007/s00018-010-0344-4
Howie AJ, Brewer DB: Optical properties of amyloid stained by Congo red: History and mechanisms. Micron 2009,40(3):285-301. 10.1016/j.micron.2008.10.002
Naiki H, Higuchi K, Hosokawa M, Takeda T: Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Analytical Biochemistry 1989,177(2):244-249. 10.1016/0003-2697(89)90046-8
Ge X, Yang DSC, Trabbic-Carlson K, Kim B, Chilkoti A, Filipe CDM: Self-cleavable stimulus responsive tags for protein purification without chromatography. J Am Chem Soc 2005,127(32):11228-11229. 10.1021/ja0531125
Xing L, Wu W, Zhou BH, Lin ZL: Streamlined protein expression and purification using cleavable self-aggregating tags. Microb Cell Fact 2011, 10: 42. 10.1186/1475-2859-10-42
Roessl U, Nahalka J, Nidetzky B: Carrier-free immobilized enzymes for biocatalysis. Biotechnol Lett 2010,32(3):341-350. 10.1007/s10529-009-0173-4
Sheldon RA: Cross-linked enzyme aggregates as industrial biocatalysts. Org Process Res Dev 2011,15(1):213-223. 10.1021/op100289f
Zheng J, Guan H, Xu LH, Yang R, Lin ZL: Engineered amadoriase II exhibiting expanded substrate range. Appl Microbiol Biotechnol 2010,86(2):607-613. 10.1007/s00253-009-2319-7
Winkler UK, Stuckmann M: Glycogen, hyaluronate, and some other polysaccharides greatly enhance the formation of exolipase by Serratia-marcescens . J Bacteriol 1979,138(3):663-670.
Chen TJ, Zhang JQ, Liang L, Yang R, Lin ZL: An in vivo , label-free quick assay for xylose transport in Escherichia coli . Anal Biochem 2009,390(1):63-67. 10.1016/j.ab.2009.03.048
Levine H: Thioflavin T interaction with synthetic Alzheimer's disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci 1993,2(3):404-410.
Klunk WE, Jacob RF, Mason RP: Quantifying amyloid beta-peptide (A beta) aggregation using the Congo red A beta (CR-A beta) spectrophotometric assay. Anal Biochem 1999,266(1):66-76. 10.1006/abio.1998.2933
Acknowledgements
This work was supported by a grant from the National Basic Research Program of China (2009CB724704), and a grant for the National Engineering Lab for Industrial Enzymes from the National Development and Reform commission of China. The authors are grateful to the center of biomedical analysis at Tsinghua University for expert technical assistance with TEM and LSCM analyses.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
BZ designed part of the experiments, performed most of the experiments, and prepared the manuscript draft. LX and WW participated in the enzymatic assays and instrumental analyses. ZL and XZ conceived the study, designed and supervised the experiments, and revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhou, B., Xing, L., Wu, W. et al. Small surfactant-like peptides can drive soluble proteins into active aggregates. Microb Cell Fact 11, 10 (2012). https://doi.org/10.1186/1475-2859-11-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-11-10