
Abstract
Background
Idiopathic renal hypouricemia (iRHUC) is an autosomal recessive hereditary disorder, characterized by impaired tubular uric acid transport, re-absorption insufficiency and/or the acceleration of secretions. Some patients present with severe complications, such as exercise-induced acute kidney injury (EIAKI) and nephrolithiasis.
Case presentation
Herein, we report the case of a girl with severe iRHUC (serum urate 0.05 mg/dL, fractional excretion of uric acid 295.99%) associated with recurrent EIAKI, in whom the disease was caused by a homozygous mutation (g.68G > A in exon 3) in the SLC2A9 gene. Her family members (father, mother and brother) carried the same mutation but were heterozygous, without any signs of severe hypouricemia.
Conclusions
Our findings indicate that iRHUC is a rare disorder but that it should also be considered in patients with EIAKI, especially in those patients who manifest with moderately elevated or normal serum concentrations of uric acid during the acute phase of AKI. Mutational screening of the SLC2A9 gene is necessary for the diagnosis of iRHUC, and homozygous mutations of the SLC2A9 alleles can cause severe hypouricemia. Careful attention should be paid to any signs of hypouricemia during the recovery phase of AKI and long-term follow-up.
Similar content being viewed by others


Background
Idiopathic renal hypouricemia (iRHUC) is an autosomal recessive hereditary disorder characterized by impaired tubular uric acid (UA) transport, reabsorption insufficiency and/or the acceleration of secretion [1–4]. Currently, there are two subtypes of RHUC. Type 1 (RHUC1) is characterized by loss-of function mutations in the SLC22A12 gene, which encodes urate transporter 1 (URAT1). Mutations in the SLC22A12 gene are responsible for most cases of renal hypouricemia. In contrast, type 2 (RHUC2) was recently revealed to be caused by defects in the SLC2A9 gene [5]. Most of these patients are clinically asymptomatic and are detected incidentally, but some have nephrolithiasis or hematuria or are predisposed to exercise-induced acute kidney injury (EIAKI). Dinour et al. reported that homozygous mutations of SLC2A9 cause more severe hypouricemia than URAT1 mutations and are associated with a high incidence of renal calculus and EIAKI [6].
The diagnosis of iRHUC is based on biochemical markers: hypouricemia (<119 μmol/L or 2.0 mg/dL) and increased fractional excretion of uric acid (FE-UA) (>10%) [7]. Furthermore, the exclusion of secondary causes of hyperuricosuric hypouricemia (such as Wilson’s disease, Fanconi syndrome and drug-induced tubulopathy) is very important. Confirmation of the diagnosis is accomplished by molecular analysis of the SCL22A12 and/or SLC2A9 genes.
To present time [8], only more than 100 patients with SLC22A12 mutations, and a few patients with SLC2A9 defects have been characterized worldwidely. Here, we describe one case of idiopathic renal hypouricemia; the patient presented with recurrent EIAKI and had a novel homozygous nonsense mutation g.68G > A in exon 3 of the SLC2A9 gene.
Case presentation
First episode
An otherwise healthy 12.1-year-old girl presented to our clinical department on October 22, 2010, with nausea, vomiting and acute central abdominal pain for 4 days after an 800-meter run. Her parents denied any illness, viral prodrome or aminoglycoside or nonsteroidal anti-inflammatory drug ingestion prior to exercising. Her urea nitrogen is 32.62 mmol/L, creatinine 663.1 μmol/L (1 mg/dl = 88.4 μmol/L) and uric acid 201.8 μmol/L (1 mg = 59.48 μmol/L). Thus, she was admitted to our ward with acute kidney injury on October 25. Her past medical history was otherwise unremarkable. There was no family history of muscle disorders or renal diseases.
On physical examination, her blood pressure was 139/90 mmHg without orthostasis. Her weight and height were 40 kg and 146 cm, respectively. The examination was entirely unremarkable except for a mild lethargy. There was no muscle swelling or tenderness.
The laboratory evaluation revealed the following results: pH 7.443, potassium 4.7 mmol/L, bicarbonate 16.5 mmol/L, SBE -6.8 mmol/L, creatine kinase 43 U/L (normal range 30–200 U/L), serum myoglobin 25.1 μg/L (normal range 0.1-70 μg/L), and blood β2-microglobulin 4233 ng/mL. The urinalysis revealed a specific gravity of 1.020, a pH of 5.5-5.0, 0–4 red blood cells/μL, rare renal tubular epithelial cells and rare uric acid crystals. No glycosuria, aminoaciduria or crystallization was found. The 24-hour urinary protein and urine output were 604.3 mg and 1500 mL, respectively. Her urinary secretory IgA was 1.13 μg/mL (normal range 0.74-2.5 μg/mL), a1-microglobulin >32.8 mg/L (normal range 1.36-10.36 mg/L), microalbumin >50 μg/mL (normal range 3.03-16.81 μg/mL) and immunoglobulin G 30 μg/mL (normal range 2.29-5.45 μg/mL). The following parameters were within normal limits or negative: serum immunoglobulin; serum complement titer; antinuclear antibody (ANA); double-stranded DNA (dsDNA); myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA); perinuclear-antineutrophil cytoplasmic antibody (P-ANCA); cytosolic-antineutrophil cytoplasmic antibody (C-ANCA); PR3-ANCA; anti-streptolysin O antibody (ASO); HIV; EBV; TORCH; anti-glomerular basement membrane antibodies; HBsAg; and hepatitis C antibody. Tests with pyrazinamide or probenecid were not performed.
A renal biopsy was performed on day 9 after admission. The light microscope revealed normal glomeruli and arterioles, with vacuolar degeneration in the tubular epithelial cells, accompanied by interstitial edema, scattered lymphocytes and monocyte infiltration. Immunofluorescence stainings for IgG, IgA, IgM, C3, C4 and Fib were all negative. Electron microscopy showed focal effacement of foot processes and a normal basement membrane without any electron-dense deposits. These results revealed the kidney to be recovering from acute tubular necrosis.
With conservative therapy of fluid control, her renal function recovered gradually. After 15 days of admission, she was discharged with improved renal function (BUN 10.52 mmol/L and serum creatinine 106.9 μmol/L). During the 1-year follow-up, her eGFR calculated by the Schwartz formula [9] eventually recovered.
Second episode
Unfortunately, a relapse of EIAKI occurred nearly 1 year after the first episode. On December 23, 2011, the patient was admitted again to our hospital with nausea, vomiting, abdominal pain and loin pain after the same exercise. The physical examination did not reveal any abnormalities. Her blood pressure was 94/51 mmHg, and her 24-hr urinary output was 1400 mL and 24-hr urine protein excretion was 220 mg. In addition, Scr was 143 μmol/L, BUN was 12.59 mmol/L, and uric acid was 21 μmol/L. The serum copper ceruloplasmin was normal, and serum myoglobin was 21.3 μg/L. The 24-hour urinary excretion of calcium and copper was within the normal range. The renal ultrasound scan showed increased echogenicity in the cortex of both kidneys, but hydronephrosis was not observed. Her MRI was normal.
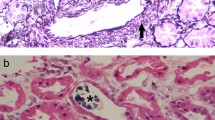
A second renal biopsy was conducted on December 28, 2011. Very slight mesangial cell proliferation and normal arterioles were observed by light microscope, with moderate vacuolar degeneration in the tubular epithelial cells, accompanied by interstitial edema and small amounts of lymphocytes and monocyte infiltration (Figure 1). Immunofluorescence stainings for IgG, IgA, IgM, C3, C4 and Fib were all negative. The present renal tubular-interstitial lesions appeared more moderate than previously.

Secondary renal biopsy revealed very slight mesangial cell proliferation and normal arterioles by light microscope, with moderate vacuolar degeneration in the tubular epithelial cells (↖), accompanied by interstitial edema and small amounts of lymphocytes and monocyte infiltration. A &B × 100, and C &D × 200.
This time, we noticed an extremely low concentration of uric acid in the serum. We performed a fractional excretion of urate (FEUA), and the result was 295.99%. All secondary causes of renal hypouricemia and EIAKI were excluded. A diagnosis of EIAKI by idiopathic renal hypouricemia was made retrospectively. After symptomatic treatment, the patient was discharged on day 13. During the follow-up, she was revealed to have normal Scr and eGFR, but severe hypouricemia (3.1–8.7 μmol/L) remained. Anaerobic activity was strictly prohibited, and no further episode of EIAKI occurred despite persistent hypouricemia at the 1.5-year follow-up. The laboratory findings of Scr and serum uric acid from each hospitalization are summarized in Figure 2.

Variation of eGFR (ml/ min) and serum uric acid (UA, μmol/ L) within 2.5- year follow- up in patient with idiopathic renal hypouricemia.
The serum UA level was also screened in other family members. Her parents’ serum urate levels were 220.6 μmol/L (mother) and 263.4 μmol/L (father). Her younger brother’s (8 years old) serum urate level was 115.6 μmol/L, and his fractional excretion of urate was 24.72%.
Mutational analysis
Secondary RHU, caused by isolated or generalized tubular defects, such as Fanconi syndrome, Wilson disease, cystinosis, heavy metal poisoning or drug-induced tubulopathy, was excluded before the molecular analysis of the SLC22A12 and SLC2A9 genes in the present study. Sequence analysis of the SLC22A12 and SLC2A9 genes was performed in the patient and her family members under the diagnosis of EIAKI associated with idiopathic renal hypouricemia. The clinical data collection and genomic analysis was approved by the institutional ethics committee, and all of the subjects provided their written informed consent. No significant sequence variants in SLC22A12 were found. The sequence analysis of the SLC2A9 gene revealed a novel homozygous nonsense mutation of g.68G > A in exon 3 (p.Trp23Stop), which resulted in prematurely truncated GLUT9 protein in the patient. No similar loss-of-function mutations have been previously reported. A heterozygous mutation of g.68G > A was also found in the parents and younger brother (Figure 3). The results of the analysis in the patient’s family members suggested an autosomal recessive mode of inheritance (Figure 4).

Electropherograms of partial sequences of exon 3 of SLC2A9 showing a novel homozygous mutation g.68G > A (p.Trp23Stop) in patient exhibiting clinical features compatible with idiopathic renal hypouricemia and same but heterozygous mutation in her father, mother and brother.

Pedigree of the family carrying novel homozygous or heterozygous mutation in SLC2A9 gene. The results of the analysis in the patient’s family members suggested an autosomal recessive mode of inheritance.
Discussion
Hypouricemia is arbitrarily defined as a lower serum uric acid (UA) concentration caused by decreased production or increased excretion, and idiopathic renal hypouricemia (IRHU) is a familial hereditary disease characterized by an increased renal urate clearance caused by an isolated inborn error of membrane transport for urate in the proximal renal tubules. Although most patients with IRHU have no clinical symptoms or complications, the major complications in this disease are urolithiasis and exercise-induced acute kidney injury (EIAKI).
EIAKI associated with IRHU was first reported in 1989 by Erley et al. [10], and a number of cases have been reported to date [8]. However, why renal hypouricemia causes exercise-induced AKI remains unclear. One possibility is that AKI might develop due to acute uric acid nephropathy, and tubular obstruction by uric acid has been suggested as causing AKI [11]. Circulatory disturbance of the kidney is an alternative mechanism, as most renal biopsies in cases of exercise-induced AKI show no uric acid crystallization. Because plasma uric acid is a powerful antioxidant, anaerobic exercise induces an accumulation of oxygen-free radicals, which are vasoconstrictive, and this accumulation can result in a reduced glomerular filtration rate. Uric acid seems to play a protective role in the kidney, and the decreased antioxidant potential in renal hypouricemia might lead to kidney injury caused by ROS [12]. Furthermore, patient with type I xanthinuria [13] presents very low serum uric acid because of deficiency in xanthine dehydrogenase, which catalyzes the oxidation of hypoxanthine to xanthine and also of xanthine to uric acid. Affected individuals pass multiple brownish-yellow stones and surgical extraction would be required in some days. Even though, probands with xanthinuria did not have AKI in their medical history. It suggests [14], that hypouricemia alone, probably, could not contribute to renal injury in patients with primary renal hypouricemia. The powerful antioxidant activity of uric acid might not be the fundamental factor for development of AKI in patients with IRHU.
In the present study, no uric acid crystallization was found in the tubular lumen of the kidney sample obtained 9 and 5 days after the first and secondary onset, respectively, of EIAKI. Thus, the role of a tubular obstruction by uric acid crystallization in EIAKI could be excluded in the present case. Acute tubular necrosis, induced by renal vasoconstriction and/or a reduced glomerular filtration rate (because of volume depletion after exercise and vomiting), might have played a role in the pathogenesis of recurrent EIAKI in the present case, according to the results of both renal biopsies. Figure 3 presents the Scr and serum concentration of uric acid simultaneously during the first and second episodes of EIAKI and at follow-up.
SLC2A9 was first identified as a novel member of the facilitative glucose transporter family in 2000 by Phay et al. [15]. In 2008, Matsuo et al. [16] identified two loss-of-function heterozygous mutations in SLC2A9 that caused renal hypouricemia by decreased urate reabsorption on both sides of the proximal renal tubules. Since then, several studies have reported on renal hypouricemia caused by SLC2A9 mutations (summarized in Table 1). Among these studies, six patients were reported as presenting with EIAKI. Dinour [6] reported that three male Israeli-Arab patients with a homozygous L75R mutation presented with EIAKI. Shima [17] reported one female Japanese patient who presented with EIAKI and PRES (posterior reversible encephalopathy syndrome), caused by compound heterozygous G207X/dupExon1a-11 mutations. Stiburkova [18] also reported two renal hypouricemia patients presenting with EIAKI caused by a homozygous G216R mutation or compound heterozygous G216R/N333S mutations in the SLC2A9 gene. In the present study, the patient manifested with EIAKI and carried a homozygous g.68G > A mutation; her brother, mother and father did not present with hypouricemia, elevated fractional excretion of uric acid or EIAKI, although all three carried the same but heterozygous mutation. Above all, we conclude that homozygous or compound heterozygous mutations in the SLC2A9 gene are a prerequisite for presenting with EIAKI in patients with renal hypouricemia. The same phenomenon can also be observed in patients presenting with EIAKI who have SLC22A12 mutations [19–21]. These results imply that not all mutations necessarily lead to EIAKI and that the possible genotype-phenotype correlation is complex and difficult to determine at the present time. Furthermore, mutational analysis could be a useful indicator for the clinical and prognostic evaluation of patients with renal hypouricemia.
When a patient suffering from AKI with renal hypouricemia is first seen, diagnosing renal hypouricemia is generally difficult because of the increased level of UA [26] during the acute phase of the disease. In the present study, the serum concentration of uric acid was 201.8 μmol/L during the first episode of EIAKI; thus, we did not realize that the EIAKI was due to renal hypouricemia. After the second episode of EIAKI, the repeated renal biopsy did not supply any convincing evidence, and as we tried our best to identify the cause of the AKI, we finally realized that there might be a positive relationship in this patient between hypouricemia and the two episodes of EIAKI. Therefore, careful attention should be paid to any signs of hypouricemia during the recovery phase of AKI, especially in patients with recurrent AKI. If anyone can realize the correlation between renal hypouricemia and AKI at an appropriate timing, unnecessary renal biopsies must have been avoided in such kind of patients.
Recently, Bhasin et al. reported [27] an 18-year-old white was diagnosed as iRHUC with recurrent AKI after participating in 400-meter race. Before his next race, he was prescribed allopurinol 300 mg daily for 3 days. He completed this race uneventfully. The underlying mechanism for use of allopurinol is to decrease the production of uric acid, then, reducing the filtered uric acid load and lessening the risk of precipitation of the uric acid in the tubules of kidney. Thus, the value of allopurinol to reduce the risk of exercise induced AKI in patients with iRHUC deserves further study.
Conclusions
Above all, iRHUC is a rare disorder, but it should be considered in patients with EIAKI, especially in those patients who manifest moderately elevated or normal serum concentrations of uric acid during the acute phase of AKI. Mutational screening of the SLC2A9 gene is necessary for the diagnosis of iRHUC, and homozygous mutations of the SLC2A9 alleles can cause severe hypouricemia. Careful attention should be paid to any signs of hypouricemia during the recovery phase of AKI and during long-term follow-up.
Consent
Written informed consent was obtained from the patient’s legal guardian and from the patient for publication of this case report.
For classification according to the rate of decline of glomerular filtration rate, CKD is staged as blow:
Stage 1: GFR ≥ 90 ml/min/1.73 m2
Stage 2: GFR 60 ~ 89 ml/min/1.73 m2
Stage 3a: GFR 45 ~ 59 ml/min/1.73 m2
Stage 3b: GFR 30 ~ 44 ml/min/1.73 m2
Stage 4: GFR 15 ~ 29 ml/min/1.73 m2
Stage 5: GFR < 15 ml/min/1.73 m2 or on dialysis
Abbreviations
- iRHUC:
-
Idiopathic renal hypouricemia
- AKI:
-
Acute kidney injury
- EIAKI:
-
Exercise-induced acute kidney injury
- URAT1:
-
Urate transporter 1
- FEUA:
-
Fractional excretion of uric acid
- eGFR:
-
Estimated glomerular filtration rate
- PRES:
-
Posterior reversible encephalopathy syndrome
- CKD:
-
Chronic kidney disease.
References
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H: Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002, 417 (6887): 447-452.
Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH: Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J Biol Chem. 2004, 279 (16): 16229-16236. 10.1074/jbc.M312226200.
Ichida K, Hosoyamada M, Hisatome I, Enomoto A, Hikita M, Endou H, Hosoya T: Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J Am Soc Nephrol. 2004, 15 (1): 164-173. 10.1097/01.ASN.0000105320.04395.D0.
Stiburkova B, Sebesta I, Ichida K, Nakamura M, Hulkova H, Krylov V, Kryspinova L, Jahnova H: Novel allelic variants and evidence for a prevalent mutation in URAT1 causing renal hypouricemia: biochemical, genetics and functional analysis. Eur J Hum Genet. 2013, 21 (10): 1067-1073. 10.1038/ejhg.2013.3.
Vitart V, Rudan I, Hayward C, Gray NK, Floyd J, Palmer CN, Knott SA, Kolcic I, Polasek O, Graessler J, Wilson JF, Marinaki A, Riches PL, Shu X, Janicijevic B, Smolej-Narancic N, Gorgoni B, Morgan J, Campbell S, Biloglav Z, Barac-Lauc L, Pericic M, Klaric IM, Zgaga L, Skaric-Juric T, Wild SH, Richardson WA, Hohenstein P, Kimber CH, Tenesa A, et al: SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008, 40 (4): 437-442. 10.1038/ng.106.
Dinour D, Gray NK, Campbell S, Shu X, Sawyer L, Richardson W, Rechavi G, Amariglio N, Ganon L, Sela BA, Bahat H, Goldman M, Weissgarten J, Millar MR, Wright AF, Holtzman EJ: Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol. 2010, 21 (1): 64-72. 10.1681/ASN.2009040406.
Sebesta I, Stiburkova B, Bartl J, Ichida K, Hosoyamada M, Taylor J, Marinaki A: Diagnostic tests for primary renal hypouricemia. Nucleosides Nucleotides Nucleic Acids. 2011, 30 (12): 1112-1116. 10.1080/15257770.2011.611483.
Jeannin G, Chiarelli N, Gaggiotti M, Ritelli M, Maiorca P, Quinzani S, Verzeletti F, Possenti S, Colombi M, Cancarini G: Recurrent exercise-induced acute renal failure in a young Pakistani man with severe renal hypouricemia and SLC2A9 compound heterozygosity. BMC Med Genet. 2014, 15 (1): 3-
Schwartz GJ, Brion LP, Spitzer A: The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr Clin North Am. 1987, 34 (3): 571-590.
Erley CM, Hirschberg RR, Hoefer W, Schaefer K: Acute renal failure due to uric acid nephropathy in a patient with renal hypouricemia. Klin Wochenschr. 1989, 67 (5): 308-312. 10.1007/BF01892900.
Yeun JY, Hasbargen JA: Renal hypouricemia: prevention of exercise-induced acute renal failure and a review of the literature. Am J Kidney Dis. 1995, 25 (6): 937-946. 10.1016/0272-6386(95)90579-0.
Murakami T, Kawakami H, Fukuda M, Furukawa S: Patients with renal hypouricemia are prone to develop acute renal failure–why?. Clin Nephrol. 1995, 43 (3): 207-208.
Arikyants N, Sarkissian A, Hesse A, Eggermann T, Leumann E, Steinmann B: Xanthinuria type I: a rare cause of urolithiasis. Pediatr Nephrol. 2007, 22 (2): 310-314.
Stiburkova B, Krijt J, Vyletal P, Bartl J, Gerhatova E, Korinek M, Sebesta I: Novel mutations in xanthine dehydrogenase/oxidase cause severe hypouricemia: biochemical and molecular genetic analysis in two Czech families with xanthinuria type I. Clin Chim Acta. 2012, 413 (1–2): 93-99.
Phay JE, Hussain HB, Moley JF: Cloning and expression analysis of a novel member of the facilitative glucose transporter family, SLC2A9 (GLUT9). Genomics. 2000, 66 (2): 217-220. 10.1006/geno.2000.6195.
Matsuo H, Chiba T, Nagamori S, Nakayama A, Domoto H, Phetdee K, Wiriyasermkul P, Kikuchi Y, Oda T, Nishiyama J, Nakamura T, Morimoto Y, Kamakura K, Sakurai Y, Nonoyama S, Kanai Y, Shinomiya N: Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008, 83 (6): 744-751. 10.1016/j.ajhg.2008.11.001.
Shima Y, Nozu K, Nozu Y, Togawa H, Kaito H, Matsuo M, Iijima K, Nakanishi K, Yoshikawa N: Recurrent EIARF and PRES with severe renal hypouricemia by compound heterozygous SLC2A9 mutation. Pediatrics. 2011, 127 (6): e1621-1625. 10.1542/peds.2010-2592.
Stiburkova B, Taylor J, Marinaki AM, Sebesta I: Acute kidney injury in two children caused by renal hypouricaemia type 2. Pediatr Nephrol. 2012, 27 (8): 1411-1415. 10.1007/s00467-012-2174-0.
Ochi A, Takei T, Ichikawa A, Kojima C, Moriyama T, Itabashi M, Mochizuki T, Taniguchi A, Nitta K: A case of acute renal failure after exercise with renal hypouricemia demonstrated compound heterozygous mutations of uric acid transporter 1. Clin Exp Nephrol. 2011, 16 (2): 316-319.
Kim YH, Cho JT: A case of exercise-induced acute renal failure with G774A mutation in SCL22A12 causing renal hypouricemia. J Korean Med Sci. 2011, 26 (9): 1238-1240. 10.3346/jkms.2011.26.9.1238.
Tanaka M, Itoh K, Matsushita K, Matsushita K, Wakita N, Adachi M, Nonoguchi H, Kitamura K, Hosoyamada M, Endou H, Tomita K: Two male siblings with hereditary renal hypouricemia and exercise-induced ARF. Am J Kidney Dis. 2003, 42 (6): 1287-1292. 10.1053/j.ajkd.2003.08.032.
Anzai N, Ichida K, Jutabha P, Kimura T, Babu E, Jin CJ, Srivastava S, Kitamura K, Hisatome I, Endou H, Sakurai H: Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J Biol Chem. 2008, 283 (40): 26834-26838. 10.1074/jbc.C800156200.
Stiburkova B, Ichida K, Sebesta I: Novel homozygous insertion in SLC2A9 gene caused renal hypouricemia. Mol Genet Metab. 2011, 102 (4): 430-435. 10.1016/j.ymgme.2010.12.016.
Dinour D, Gray NK, Ganon L, Knox AJ, Shalev H, Sela BA, Campbell S, Sawyer L, Shu X, Valsamidou E, Landau D, Wright AF, Holtzman EJ: Two novel homozygous SLC2A9 mutations cause renal hypouricemia type 2. Nephrol Dial Transplant. 2012, 27 (3): 1035-1041. 10.1093/ndt/gfr419.
Finney H, Newman DJ, Thakkar H, Fell JM, Price CP: Reference ranges for plasma cystatin C and creatinine measurements in premature infants, neonates, and older children. Arch Dis Child. 2000, 82 (1): 71-75. 10.1136/adc.82.1.71.
Takeda Y, Abe A, Nakanishi S, Umezu M, Hirano K, Hayakawa H, Ohno I, Ichida K, Yamaguchi Y, Hosoya T, Fukagawa M: Two cases of nephrotic syndrome (NS)-induced acute kidney injury (AKI) associated with renal hypouricemia. Clin Nephrol. 2011, 76 (1): 78-82.
Bhasin B, Stiburkova B, De Castro-Pretelt M, Beck N, Bodurtha JN, Atta MG: Hereditary renal hypouricemia: a new role for allopurinol?. Am J Med. 2014, 127 (1): e3-4. 10.1016/j.amjmed.2013.08.025.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2431/14/73/prepub
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant Nos. 81270792, 81070561 and 81170664), the State "1025" Science and Technology Support Projects (2012BAI03B02), the Research Fund for the Doctoral Program of Higher Education of China (20120101110018), the Zhejiang Provincial Healthy Science Foundation of China (WKJ2010-2-014, 2012KYA119), the Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents and the Zhejiang Provincial Natural Science Foundation of China (LY12H050037), the Zhejiang Provincial Administration of Traditional Chinese Medicine of China (2009CB049), the Zhejiang Provincial Department of Education Foundation of China (Y200804449). We thank the patient and her family members for participating in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SHJ, FCY, JX & FHD carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. WX & LAM participated in the clinical diagnosis of the patient. SQ & DLZ participated in the sequence alignment. GWZ & THF participated in renal biopsy and pathology. SHJ & MJH participated for program design, manuscript drafting. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Shen, H., Feng, C., Jin, X. et al. Recurrent exercise-induced acute kidney injury by idiopathic renal hypouricemia with a novel mutation in the SLC2A9 gene and literature review. BMC Pediatr 14, 73 (2014). https://doi.org/10.1186/1471-2431-14-73
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2431-14-73