
Abstract
Background
Cassava (Manihot esculenta Crantz), a starchy root crop grown in tropical and subtropical climates, is the sixth most important crop in the world after wheat, rice, maize, potato and barley. The repertoire of simple sequence repeat (SSR) markers for cassava is limited and warrants a need for a larger number of polymorphic SSRs for germplasm characterization and breeding applications.
Results
A total of 846 putative microsatellites were identified in silico from an 8,577 cassava unigene set with an average density of one SSR every 7 kb. One hundred and ninety-two candidate SSRs were screened for polymorphism among a panel of cassava cultivars from Africa, Latin America and Asia, four wild Manihot species as well as two other important taxa in the Euphorbiaceae, leafy spurge (Euphorbia esula) and castor bean (Ricinus communis). Of 168 markers with clean amplification products, 124 (73.8%) displayed polymorphism based on high resolution agarose gels. Of 85 EST-SSR markers screened, 80 (94.1%) amplified alleles from one or more wild species (M epruinosa, M glaziovii, M brachyandra, M tripartita) whereas 13 (15.3%) amplified alleles from castor bean and 9 (10.6%) amplified alleles from leafy spurge; hence nearly all markers were transferable to wild relatives of M esculenta while only a fraction was transferable to the more distantly related taxa. In a subset of 20 EST-SSRs assessed by fluorescence-based genotyping the number of alleles per locus ranged from 2 to 10 with an average of 4.55 per locus. These markers had a polymorphism information content (PIC) from 0.19 to 0.75 with an average value of 0.55 and showed genetic relationships consistent with existing information on these genotypes.
Conclusion
A set of 124 new, unique polymorphic EST-SSRs was developed and characterized which extends the repertoire of SSR markers for cultivated cassava and its wild relatives. The markers show high PIC values and therefore will be useful for cultivar identification, taxonomic studies, and genetic mapping. The study further shows that mining ESTs is a highly efficient strategy for polymorphism detection within the cultivated cassava gene pool.
Similar content being viewed by others

Background
Cassava, Manihot esculenta subsp esculenta Crantz (2n = 36), is a starchy root crop and the primary staple for over 800 million people worldwide [1]. Native to the Neotropics [2], cassava is now cultivated in tropical and subtropical regions in Asia, Latin America and Africa. In 2007, Africa was the world's largest producer with 118 million tons out of a global production of 228 million tons [3]. Cassava can grow under adverse climatic conditions with erratic rainfall and on marginal land under low intensity management [4]. Because of these attributes, cassava plays an essential food security role in Africa. In addition to food, cassava also has important industrial uses for the production of starch-based products, alcohol and animal feed [5, 6].
Like most vegetatively propagated crops, cassava is highly heterozygous. Low rates of flowering and the long breeding cycle further render conventional breeding of cassava arduous [7]. Breeders have used controlled wide crosses with wild Manihot species for cassava improvement: for example, resistance to mosaic virus and bacterial blight has been successfully transferred from Manihot glaziovii to locally adapted cassava cultivars in Africa [8, 9]. Because the genus Manihot is largely inter-fertile, wild species could further broaden the genetic base of cultivated cassava [10]. Natural hybridization between wild Manihot and cultivated cassava has also been reported [11].
Molecular marker technology is an efficient tool for the characterization and management of plant genetic resources and also in crop improvement programs. Among the different classes of molecular markers, microsatellites or simple sequence repeats (SSRs) are widely utilized for genetic diversity and genetic mapping because they are largely co-dominant, multi-allelic, have good genome coverage and can be multiplexed on semi-automated systems [12, 13]. In the case of cassava, SSR markers have been utilized for the characterization of genetic resources [14–16] and an SSR-based molecular genetic map for cassava comprising 100 markers was described [17]. Despite these advances, the development and utilization of molecular markers for cassava lags behind the achievements of other major crop plants. For example, in case of barley, a high density microsatellite consensus map with 775 SSR loci has been described [18].
In recent years, EST and cDNA resources for cassava have increased substantially [19–21]. Cassava genomic and EST resources will further vastly increase with the recent completion of a draft cassava genome sequence and associated EST projects http://www.jgi.doe.gov/genome-projects/. These datasets constitute valuable and inexpensive sources of molecular markers such as SNPs [22] and microsatellites, since these can be readily detected computationally. Thus, mining these resources provides an opportunity to greatly expand the database of molecular markers for cassava at minimal cost.
Cassava is a member of the Euphorbiaceae family. The invasive weed leafy spurge (Euphorbia esula) and the oilseed plant castor bean (Ricinus communis) are other important members of this family. A draft genomic sequence for castor bean is available and both castor bean and leafy spurge have extensive EST resources (http://castorbean.tigr.org; [23]). With the recent completion of the cassava genome sequence, cassava and castor bean will constitute a valuable comparative genomics system.
The objectives of this study were to (i) increase the number of SSR markers by mining a previously developed cassava 8,577 unigene set; (ii) characterize EST-derived microsatellite markers in terms of abundance and level of polymorphism; and (iii) assess their transferability to other taxa in the Euphorbiaceae. The approach taken has been to characterize and validate 192 microsatellites identified in silico from a cassava unigene set by DNA fingerprinting a sample set consisting of: cultivated cassava from Africa, Latin America and Asia; four wild Manihot relatives; and leafy spurge and castor bean, two more distantly related taxa within the Euphorbiaceae.
Results and Discussion
Frequency and distribution of microsatellites in the cassava transcriptome
A total of 18,177 ESTs were used in this study, which were clustered in 8,577 unigenes [20]. In total, 836 microsatellite repeats were identified in the 8,577 unigenes (Table 1). Perfect repeats and imperfect repeats, i.e. repeats with small insertions or deletions within the repeat, were identified. Mononucleotide repeats were not included due to difficulties in accurate sizing of polymorphisms. Of 836 EST-SSRs, 316 (37.8%) were perfect repeats while 520 (62.2%) were imperfect repeats. A total of 137 ESTs contained perfect as well as imperfect repeats. Also, several ESTs contained 2 or more perfect or imperfect repeats. Overall, 596 ESTs were identified in the 8,577 unigene set that contained microsatellite repeats.
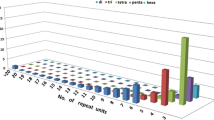
SSR loci were classified by repeat type and structure (Table 1). The most frequent number of repeats per motif was between 5 and 9, thus microsatellites in cassava ESTs are rather small, in agreement with previous findings in coffee [24] eggplant [25] and barley [26]. Since the unigene set represented 5.8 Mb of the cassava transcriptome, the average distance between SSRs is approximately 7.0 kb or one SSR-containing EST every 10.3 ESTs. This number corresponds well with barley where the average distance between SSRs was 6.3 kb or one SSR-containing EST every 13.3 ESTs [26], Coffea which has an average density of one microsatellite every 7.73 kb [27], but is lower compared to pepper (Capsicum) where one SSR was found in every 3.8 kb EST [28].
The highest proportion of repeat motifs comprised di-nucleotide (68.7%) repeats, followed by tri-nucleotide repeats (30.4%). Only five tetra- (0.6%) and three penta-nucleotides (0.3%) were found. This result is in contrast with the majority of earlier studies which report tri-nucleotide repeats as the most abundant class of repeats [28–31] but in agreement with studies in Picea species [32] and Coffea [24] where di-nucleotide repeats were found to be the most abundant class. Reported frequencies and distribution of EST-SSRs is variable for different plant and animal species. This can be partly attributed to the microsatellite search criteria and the characteristics of the EST database analyzed in these studies. Since the cassava EST dataset used in this study was sequenced from the 5'end, a greater proportion of the EST-SSRs could be located in the 5'untranslated region rather than in the coding region where tri-nucleotide repeats are expected to be the predominant repeat type.
EST-SSRs were identified in 9.7% of the total number of contigs in cassava, compared to 10.2% for pepper [28], 6.8% for coffee [27], and 7.5% for wheat [31]. In this study, the SSR search was conducted on a unigene set which will return non-redundant SSRs. This approach results in a realistic estimate of microsatellite repeat frequency and ensures that EST-SSR markers correspond to unique loci in the genome.
EST-SSR marker development
Out of the 596 unique ESTs containing microsatellites, primer pairs were designed for 346; 167 with perfect repeats (48.3%) and 179 (51.7%) containing imperfect repeats. The remaining sequences were inappropriate for primer design mainly because of insufficient DNA sequence flanking the microsatellite core. In addition, duplicates (21) with an existing cassava SSR dataset [33] were eliminated. One hundred and ninety-two randomly chosen primer pairs flanking di-, tri-, or tetrameric SSRs were optimized for PCR amplification using one or two cassava genotypes, TMS30572 and/or TME3. Details of the primer sequences and expected product size with SSR motif are described [see Additional file 1]. Of the 192 primer pairs tested, 168 (87.5%) yielded scorable amplicons. This percentage of working primers (mono- and polymorphic) is in the range previously observed for zoysiagrass (1,044 out of 1,163 or 89.7%) [34], clementine (41 out of 48 or 85.4%) [35], eggplant (39 out of 50 or 78%) [25] and Coffea (18 out of 24 or 75%) [24]. Twenty-seven primer pairs produced weak or no amplicons leading to exclusion from further analysis. Nine microsatellites produced amplicons that were significantly longer than expected and most likely indicate the presence of introns. Five of these produced fragments larger than 500 bp and could not be scored accurately for small differences in fragment size. These were also excluded from further analysis.
Next, primer pairs with scorable amplicons were assayed for polymorphism among cassava cultivars from Africa, Asia and Latin America (see Table 2). This panel included cassava landraces, elite breeding lines and some putative interspecific hybrids. To assess transferability of the EST-SSRs to other taxa within the Euphorbiaceae, the panel also included four wild Manihot species, castor bean and leafy spurge. In order to assess the potential of the newly developed EST-SSR markers for detecting polymorphism, all 168 primer pairs yielding PCR products were tested on this diversity panel or on a subset thereof, hereafter referred to as panel 2. Panel 2 is a subset of 24 genotypes, intended to represent the larger sample set; it includes cassava cultivars from 14 different countries in Africa, South America and Asia, three wild Manihot species, plus castor bean and leafy spurge (Table 2). Overall, 124 primer pairs (73.8%) showed polymorphisms in the germplasm analyzed, as illustrated for MeESSR119 (Figure 1A). Of the 124 polymorphic primers, 78 (62.9%) were for perfect SSRs while 46 (37.1%) were for imperfect SSRs. Thus perfect SSR repeats were more polymorphic than imperfect repeat types. When only cultivated cassava was considered, 40 out of 79 markers tested on panel 2 were polymorphic, corresponding to a frequency of 50.6%. The difference in level of polymorphism between cultivated cassava and all genotypes in panel 2 is due to the presence of additional, unique alleles in the wild Manihot species.

Allelic variation of microsatellites MeESSR119 (A) and MeESSR106 (B) among genotypes of diversity panel 2 as detected on high resolution agarose gels. Lanes: M is the size marker in bp; lane 1: Nachinyaya; 2: TME1738; 3: CM6740-7; 4: MCol1468; 5: MBra12; 6: MEcu72; 7: MPer183; 8: MTai18; 9: M epruinosa; 10: M brachyandra; 11: M glaziovii; 12: castor bean; 13: leafy spurge; 14: TMS30572; 15: TME419; 16: TME117; 17: TME3; 18: TME7; 19: TMS4(2)1425; 20: TME388; 21: TME1786; 22: TME530; 23: TME225; 24: TME638.
Marker polymorphism for cultivated cassava in this study is about 2.7-fold higher compared to a study on cassava EST-SSRs where 18.4% polymorphism was detected (9 out of 49 primers tested were polymorphic) [36]. In citrus, more than 90% of EST-SSR markers were polymorphic which is higher compared to our results [35]. Fluctuations in level of polymorphism detected in EST-SSRs can be explained in part by the level of diversity among the germplasm surveyed. The higher frequency of polymorphism observed here compared to the study of Tangphatsornruang et al. [36] is possibly due to the inclusion of diverse landraces from 14 different countries in Africa, Latin America, Asia as well as elite cassava genotypes in this panel. Since polymorphism was scored on high resolution agarose gels, it is also possible that the level of polymorphism is higher than the frequency reported here since allelic variants with small size differences may not have been resolved on this system. Overall, our results show that mining ESTs for the development of SSRs is a highly effective strategy to increase the SSR database for cassava.
Out of 85 markers with clear amplicons tested on the 19 cassava cultivars of panel 2, 25 (29.4%) showed non-amplifying or null alleles in one or more genotypes. Therefore null alleles were common among the diverse cassava cultivars examined here. Null alleles for genic SSRs have also been described in other plant species such as for example kiwifruit [37], amongst others. The frequency of null alleles is influenced by several factors, including the diversity of the germplasm tested and technical aspects such as quality of the sequence data for primer development or PCR conditions. In a study on sunflower and related wild relatives, a clear relationship was found between genetic distance and amplification failures, with the frequency of null alleles ranging from 11.2% for an inbred sunflower accession to 34.5% among a wild sunflower species [38]. In a sample of wheat germplasm consisting of 52 diverse genotypes, null alleles for EST-SSRs occurred at a frequency of 45% [39] which is higher compared to the result obtained here. In view of the highly heterozygous nature of cassava and the occurrence of point mutations and/or insertion-deletions in the EST-SSR alleles outside the microsatellite repeat region (unpublished results), most null alleles observed in the present study are likely due to sequence variation in the primer binding sites.
SSRs are assumed to be locus specific. Cassava is considered an allotetraploid with disomic inheritance [40, 41]. Therefore we expect amplification of one or two bands per locus. Out of 79 markers tested on panel 2, 49 (62%) amplified one or two alleles per locus while the remaining 30 (38%) markers amplified more than two bands for at least one genotype (see Figure 1B). In a previous study on genomic microsatellites in cassava [42], approximately 80% of the markers detected one or two alleles per accession, thus suggesting a fairly low degree of genomic microsatellite locus duplication. Our results show about a two-fold higher frequency of markers amplifying more than two bands per genotype. This is surprising in view of the fact that genic SSRs are generally considered to be more conserved compared to genomic SSRs [12]. However, multiple bands could also be due to the presence of multigene families. Therefore, in case of genic SSRs, the multiple bands could be due to cross-amplification of conserved members of multigene families. It should be noted that the frequency of multiple loci reported here represents a minimum estimate, considering that this result is based on high resolution agarose gel electrophoresis which does not resolve all allelic variants (unpublished observations). Similarly, the frequency of polymorphism reported here is a conservative estimate as markers with an allelic size range of 3-5 bp or less may have escaped detection. While the resolution of the agarose gel system is limited to about 3-5 bp, it does allow for a rapid screening of candidate microsatellite markers.
Cross-species and cross-genera transferability
Cross-species transferability of the new cassava-derived SSR markers was tested for three wild Manihot species and two other Euphorbiaceae taxa; leafy spurge and castor bean. In general, the markers resulted in robust cross-species amplification with alleles of comparable sizes in cassava cultivars and the wild Manihot species. Out of 85 amplicons tested, 85 amplified for TMS30572 (100%); 82 for TME117 (97%); 80 for TME419 (94%) which are cassava cultivars, while 78 amplified for M glaziovii (91%); 74 for M brachyandra (87%) and 72 for M epruinosa (84.7%) which are wild Manihot species. Overall, a transferability of ~94% was observed for the wild Manihot relatives, i.e. 80 markers amplified alleles from one or more wild species. In agreement with this, it was previously demonstrated in cassava that genomic SSRs can be used to assess its wild relatives [14]. By contrast, only 13 (15%) and 9 (11%) respectively, of the EST-SSR markers yielded amplicons in castor bean and leafy spurge. Moreover, the sizes of the amplicons from castor bean and leafy spurge were often outside the allelic range observed in cultivated cassava and the wild Manihot species (see Figure 1), suggesting that these may not represent orthologous sequences. These numbers are very similar to those obtained in a study on the Compositae where transferability from sunflower (Helianthus annuus) to wild species was 88.6% while transferability to the more distantly related genera safflower (Carthamus tinctorius) and lettuce (Lactuca sativa) was 14.8% and 14.4% respectively [38]. Thus, the cassava EST-SSR markers display broad utility within the genus Manihot but limited utility beyond the genus level, in agreement with previous studies in plants [43–45]. It should be pointed out that amplification of the genic SSRs in the wild species does not necessarily imply that these loci are polymorphic in these species, since only a single accession was analyzed per taxon.
Allelic polymorphism and genetic relationships
A subset of 20 randomly selected polymorphic EST-SSRs producing one or two amplicons per genotype was tested for their potential in genetic studies by ascertaining the genetic diversity in panel 2. Alleles were resolved by semi-automated fluorescence-based genotyping to attain a more accurate estimation of the allelic diversity and size range. Good amplification was obtained for all the markers across the tested genotypes. Leafy spurge and castor bean produced fewer amplicons compared to cassava and the wild Manihot species, in agreement with the results obtained from high resolution agarose gel electrophoresis.
The 20 polymorphic EST-SSR markers generated 94 alleles and discriminated all 24 accessions into different clusters according to their phylogenetic relationship. The principal coordinate plot (Figure 2) shows the genetic relationship and distribution of the panel. The first and second principal coordinates accounted for 60.79% of the total variation, and showed an effective clustering among the species. From the principal coordinate plot, three groups were clearly shown: (i) the cassava accessions (ii) the wild Manihot species and (iii) castor bean and leafy spurge. Next, a dendrogram was constructed to show the genetic differentiation of the cassava accessions and wild Manihot species (Figure 3). The dendrogram grouping showed the expected separation of the cultivated cassava accessions from their wild relatives, in agreement with the cluster grouping obtained from the principal coordinate plot. In addition, the dendrogram revealed expected genetic relationships between cassava cultivars based on their pedigree or previous diversity studies. For example, TMS 4(2)1425 and TMS 30572 are two progeny from TMS 58308 and group together in the dendrogram, in agreement with their shared ancestry [46]. Genetic similarity was also detected between TME225 and TME419, landraces from Benin and Togo respectively, in agreement with a previous genetic diversity study based on 18 genomic SSRs [47]. Overall, this analysis supports the botanical classification of the germplasm surveyed and suggests the usefulness of the EST-SSR markers for genetic diversity studies and other genotyping applications in cassava and its wild relatives.

Genetic differentiation by principal coordinate analysis showing distinct variation between cultivated cassava (A), wild Manihot species (B), castor bean and leafy spurge (C). The germplasm accessions are numbered according to the lanes in Figure 1.

Neighbor-joining tree showing relationships between 19 cassava cultivars and three wild Manihot species based on the allelic diversity generated using 20 new EST-SSR markers.
The allelic diversity and extent of polymorphism for panel 2 accessions were also determined and are summarized in Table 3. The number of alleles per locus ranged from 2 to 10 with an average of 4.55 alleles per marker. The allele size of the original TME117 EST sequences all fall within the allele size range determined by fluorescence-based genotyping as summarized in Table 3, as expected. The number of alleles per locus is higher compared to a previous study on cassava EST-SSRs where this ranged from 2 to 4 [36]. In the latter study, alleles were detected on polyacrylamide gels after silver staining. The higher number of alleles observed in our study is likely due to the higher resolution of fluorescence-based genotyping as well as the inclusion of three wild species, leafy spurge and castor bean in the panel. In previous studies on genomic SSRs in cassava, an average of 8.6 alleles per locus [42] and 9.0 alleles per locus [15] was detected which is higher compared to the value 4.55 found here for the genic SSRs. The average allelic size range for the cassava EST-SSRs described here is 16.7 bp which is lower compared to the value of 25.4 for seven cassava genomic SSRs previously analyzed using fluorescence-based genotyping [42]. EST-SSR markers were found to be less polymorphic compared to genomic SSRs in other crop plants such as rice and barley [48, 49]. In cultivated durum wheat, EST-SSR markers were less powerful in discriminating genotypes compared to genomic sources, though they produced higher quality markers, i.e. with fewer stutter bands [50]. The polymorphism information content (PIC) values of the EST-SSR markers identified here varied from 0.19 to 0.75 with an average of 0.55 (Table 3). These values are within the range of those reported for other plant species where average PIC values of EST-SSR markers varied from 0.32 to 0.66 (see [12] and references therein). In terms of allelic polymorphism, our results in cassava seem to support the findings in other species such as rice and maize where microsatellites from coding regions tended to have a narrower allele size range and be less polymorphic than those derived from genomic clones. It will be interesting to compare the EST-SSRs described here to the genomic SSRs previously developed for cassava in terms of discriminating power and quality.
EST-SSRs as functional markers
To assess functionality of the new markers, BlastX searches were performed for 123 ESTs containing polymorphic SSR. These results are summarized [see Additional file 2] and showed that a putative function could be assigned to 50 ESTs (40.7%) assuming a threshold of E<1.10-4, while 54 (43.9%) corresponded to predicted or hypothetical proteins. Ten ESTs (8.1%) produced no significant match. Cassava ESTs that produced no specific matches, or matches to hypothetical or unknowns, were then compared to annotation of the original cassava EST-database http://titan.biotec.uiuc.edu/cgi-bin/ESTWebsite/estima_start?seqSet=cassava to identify potential Arabidopsis homologues. This search resulted in annotation of an additional 24 EST sequences, bringing the total number of matches to proteins with a putative function to 74 (60.2%). As expected, most searches retrieved sequences from castor bean (70) followed by poplar (22), two plant species whose complete genomic sequence has been determined and that are most closely related to cassava.
Conclusion
In cassava, the number of published SSR markers is limited compared to other major crops. In this study, an in depth analysis of genic microsatellites in terms of density, level of polymorphism, and informativeness is presented. A new set of 124 genic SSR markers with PIC values comparable to genic SSRs in other plant species was developed. These resources will be of interest to the Euphorbiaceae research community in general and the cassava research community in particular, where research focuses on basic and applied aspects of cassava biology, including germplasm characterization, breeding applications, and phylogenetic studies of Manihot species. We have shown that mining ESTs is an effective strategy to identify functional microsatellites in the cultivated cassava gene pool with perfect repeats being more polymorphic that imperfect repeats. Nearly all genic SSRs are readily transferable to other species within Manihot but much less so with two other genera of the Euphorbiaceae examined here, Ricinus communis and Euphorbia esula.
Methods
Plant material
The cassava cultivars, wild Manihot relatives along with other Manihot accessions and castor bean summarized in Table 2 were collected from the Cassava Breeding Unit and the Genetic Resources Center of the International Institute for Tropical Agriculture, Ibadan, Nigeria. Leafy spurge DNA was provided by USDA-ARS, Fargo, ND, USA. DNA was extracted from leaf tissue as described [51].
Searching for microsatellites and primer design
The identification and localization of microsatellites in ESTs was accomplished by a microsatellite search as described previously [52]. For in silico identification of the perfect repeats, the minimum number of repeat motifs was set at 6 for the di-nucleotide repeats and at 5 for all other repeats. For the imperfect repeats, the minimum number of repeats motifs was set at 7 for the di- and tri-nucleotide repeats and 6 for the tetra- and penta-nucleotide repeats. The repeats in Table 1 are the combined number of perfect and imperfect repeats; all possible combinations are listed for the di- and tri-nucleotide repeats while for the tetra- and penta-nucleotide repeats, only repeats for which microsatellites were retrieved are listed. The number of repeat units for the imperfect and perfect repeats separately can be obtained upon request. Primer pairs were designed using Primer3 http://frodo.wi.mit.edu/primer3. The major primer design parameters were set as follows: primer length from 18 to 28 (optimum 20), PCR product from 100 to 300 bp, optimum annealing temperature between 57 and 60°C. Primer sequences and annealing temperatures for polymorphic SSRs reported in this paper are given [see Additional file 1]. Information on additional primer pairs that failed to amplify, could not be easily scored or were monomorphic, can be provided upon request.
Polymerase chain reaction conditions and product electrophoresis
PCR was performed in a 10 μl volume containing 10 ng DNA in 1.5 mM MgCl2, 1× PCR buffer (Bioline, United Kingdom), 0.2 mM of each dNTP, 0.2 μM of forward and reverse primer and 1.0 u Biotaq DNA polymerase (Bioline, United Kingdom). Reactions were performed in 96-well plates in MJ thermocyclers (PCT-100; MJ Research, Watertown, Mass, USA) under the following touchdown profile: (I) 3-min initial denaturation at 94°C; (II) 10 cycles of: 30 s at 94°C; 30 s at 60°C with 0.5°C decrease per cycle; 60 s at 72°C; (III) 25 cycles of: 30 s at 94°C, 30 s at 55°C and 1-min extension at 72°C; (IV) final extension was 5-min at 72°C. PCR reactions were optimized using 3 annealing temperatures: 55°C, 58°C and 60°C. Primer pairs that did not amplify under these conditions were subsequently tested using 1, 1.5 and 4 mM MgCl2 concentration for the 3 touchdown profiles. Success of amplification was checked on 2% agarose gels run in 1× TBE (89 mM Tris-Borate, 20 mM ethylenediaminetetraacetic acid) and stained with EtBr. Primer pairs with clean amplification products were then screened for polymorphism using the fingerprinting panels in Table 2. PCR products were run on 3.5% MetaPhor Agarose (Lonza, Rockland, ME, USA) or Super Fine Resolution Agarose (Amresco, Solon, OH, USA) in 1× TBE buffer and visualized by EtBr staining. Band sizing was estimated by comparison with the size marker HyperLadder V™ (Bioline, United Kingdom). A subset of polymorphic markers was resolved on ABI3100 DNA Genetic Analyzer and analyzed using GeneMapper v 3.7 (Applied Biosystems). The allelic data were used to determine the number and sizes of amplified alleles.
Data analysis
Principal coordinate and cluster analyses were performed to estimate the discriminatory ability of 20 EST-SSR markers to assess inter and intra species relationships. Genetic similarity coefficient was generated from 94 EST-SSR alleles using the Nei and Li [53] method. The similarity matrix was used to construct a dendrogram using the neighbor joining method and the TREE VIEW sub program of the NTSYS-PC software [54]. Principal coordinate analysis was performed to show genetic relationships among the accessions. The polymorphism information content (PIC) of the markers was determined to measure the informativeness of the markers using the equation:

Where PICi is the polymorphic information content of a marker i; Pij is the frequency of the jth pattern for marker i and the summation extends over n patterns [55, 56]. For functional analysis, the translated EST sequences were searched against the GenBank non-redundant CDS translations + PDB + SwissProt + PIR + PRF excluding environmental samples from WGS project using the BLASTX algorithm (GenBank release May 2009; [57]).
References
Lebot V: Tropical root and tuber crops cassava, sweet potato, yams and aroids 17th edition. Wallingford: CABI; 2009.
Olsen K, Schaal B: Microsatellite variation in cassava (Manihot esculenta, Euphorbiaceae) and its wild relatives: further evidence for a southern Amazonian origin of domestication. Am J Bot. 2001, 88: 131-142. 10.2307/2657133.
FAO (Food and Agriculture Organization of the United Nations). 2008, [http://faostat.fao.org/site/567/DesktopDefault.aspx?PageID=567#ancor]
El Sharkawy MA: Cassava biology and physiology. Plant Mol Biol. 2004, 56: 481-501. 10.1007/s11103-005-2270-7.
Tonukari NJ: Cassava and the future of starch. Electronic Journal of Biotechnology. 2004, 7: 5-8.
Sriroth K, Piyachomkwan K, Wanlapatit S, Oates C: Cassava starch technology: the Thai experience. Starch. 2000, 52: 439-449. 10.1002/1521-379X(200012)52:12<439::AID-STAR439>3.0.CO;2-E.
Jennings DL, Iglesias C: Breeding for Crop Improvement. Cassava Biology, Production and Utilization. Edited by: Hillocks RJ, Thresh JM. 2001, Oxon: CAB International, 149-166.
Hahn SK, Howland AK, Terry ER: Correlated resistance of cassava to mosaic and bacterial blight diseases. Euphytica. 1980, 29: 305-311. 10.1007/BF00025127.
Jennings DL: Breeding for resistance to African cassava mosaic geminivirus in East Africa. Tropical Science. 1994, 34: 110-122.
Nassar NM: Gene flow between cassava, Manihot esculenta Crantz, and wild relatives. Genet Mol Res. 2003, 2: 334-347.
Duputié A, David P, Debain C, McKey D: Natural hybridization between a clonally propagated crop, cassava (Manihot esculenta Crantz) and a wild relative in French Guiana. Mol Ecol. 2007, 16: 3025-3038. 10.1111/j.1365-294X.2007.03340.x.
Varshney RK, Graner A, Sorrells ME: Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005, 23: 48-55. 10.1016/j.tibtech.2004.11.005.
Zwart RS, Muylle H, Van Bockstaele E, Roldán-Ruiz I: Evaluation of genetic diversity of Fusarium head blight resistance in European winter wheat. Theor Appl Genet. 2008, 117: 813-828. 10.1007/s00122-008-0822-3.
Roa AC, Chavarriaga-Aguirre P, Duque MC, Maya MM, Bonierbale MW, Iglesias C, Tohme J: Cross-species amplification of cassava (Manihot esculenta) (Euphorbiaceae) microsatellites: allelic polymorphism and degree of relationship. Am J Bot. 2000, 87: 1647-1655. 10.2307/2656741.
Fregene MA, Suarez M, Mkumbira J, Kulembeka H, Ndedya E, Kulaya A, Mitchel S, Gullberg U, Rosling H, Dixon AG, Dean R, Kresovich S: Simple sequence repeat marker diversity in cassava landraces: genetic diversity and differentiation in an asexually propagated crop. Theor Appl Genet. 2003, 107: 1083-1093. 10.1007/s00122-003-1348-3.
Raji AAJ, Fawole I, Gedil M, Dixon AGO: Genetic differentiation analysis of African cassava (Manihot esculenta) landraces and elite germplasm using amplified fragment length polymorphism and simple sequence repeat markers. Ann App Biol. 2009,
Okogbenin E, Marin J, Fregene MA: An SSR-based molecular genetic map of cassava. Euphytica. 2006, 147: 433-440. 10.1007/s10681-005-9042-y.
Varshney RK, Marcel TC, Ramsay L, Russell J, Roder MS, Stein N, Waugh R, Langridge P, Niks RE, Graner A: A high density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet. 2007, 114: 1091-1103. 10.1007/s00122-007-0503-7.
Anderson JV, Delseny M, Fregene MA, Jorge V, Mba C, Lopez C, Restrepo S, Soto M, Piegu B, Verdier V, Cooke R, Tohme J, Horvath DP: An EST resource for cassava and other species of Euphorbiaceae. Plant Mol Biol. 2004, 56: 527-539. 10.1007/s11103-004-5046-6.
Lokko Y, Anderson JV, Rudd S, Raji AAJ, Horvath D, Mikel MA, Kim R, Liu L, Hernandez A, Dixon AGO, Ingelbrecht IL: Characterization of an 18,166 EST dataset for cassava (Manihot esculenta Crantz) enriched for drought-responsive genes. Plant Cell Rep. 2007, 26: 1605-1618. 10.1007/s00299-007-0378-8.
Sakurai T, Plata G, Rodriguez-Zapata F, Seki M, Salcedo A, Toyoda A, Ishiwata A, Tohme J, Sakaki Y, Shinozaki K, Ishitani M: Sequencing analysis of 20,000 full-length cDNA clones from cassava reveals lineage specific expansions in gene families related to stress response. BMC Plant Biol. 2007, 7: 66-10.1186/1471-2229-7-66.
López C, Piegu B, Cooke R, Delseny M, Tohme J, Verdier V: Using cDNA and genomic sequences as tools to develop SNP strategies in cassava (Manihot esculenta Crantz). Theor Appl Genet. 2005, 110: 425-431. 10.1007/s00122-004-1833-3.
Anderson JV, Horvath D, Chao WS, Foley ME, Hernandez A, Thimmapuram J, Lie L, Gong GL, Band M, Kim R, Mikel MA: Characterization of an EST database for the perennial weed leafy spurge: An important resource for weed biology research. Weed Science. 2007, 55: 193-203. 10.1614/WS-06-138.1.
Aggarwal RK, Hendre PS, Varshney RK, Bhat PR, Krishnakumar V, Singh L: Identification, characterization and utilization of EST-derived genic microsatellite markers for genome analyses of coffee and related species. Theor Appl Genet. 2007, 114: 359-372. 10.1007/s00122-006-0440-x.
Stàgel A, Portis E, Toppino L, Rotino GL, Lanteri S: Gene-based microsatellite development for mapping and phylogeny studies in eggplant. BMC Genomics. 2008, 9: 357-10.1186/1471-2164-9-357.
Thiel T, Michalek W, Varshney RK, Graner A: Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003, 106: 411-422.
Poncet V, Rondeau M, Tranchant C, Cayrel A, Hamon S, de Kochko A, Hamon P: SSR mining in coffee tree EST databases: potential use of EST-SSRs as markers for the Coffea genus. Mol Genet Genomics. 2006, 276: 436-449. 10.1007/s00438-006-0153-5.
Yi G, Lee JM, Lee S, Choi D, Kim BD: Exploitation of pepper EST-SSRs and an SSR-based linkage map. Theor Appl Genet. 2006, 114: 113-130. 10.1007/s00122-006-0415-y.
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ: Analysis of SSRs derived from grape ESTs. Theor Appl Genet. 2000, 100: 723-726. 10.1007/s001220051344.
Kantety RV, La Rota M, Matthews DE, Sorrells ME: Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol Biol. 2002, 48: 501-510. 10.1023/A:1014875206165.
Nicot N, Chiquet V, Gandon B, Amilhat L, Legeai F, Leroy P, Bernard M, Sourdille P: Study of simple sequence repeat (SSR) markers from wheat expressed sequence tags (ESTs). Theor Appl Genet. 2004, 109: 800-805. 10.1007/s00122-004-1685-x.
Rungis D, Berube Y, Zhang J, Ralph S, Ritland CE, Ellis BE, Douglas C, Bohlmann J, Ritland K: Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor Appl Genet. 2004, 109: 1283-1294. 10.1007/s00122-004-1742-5.
Mba REC, Stephenson P, Edwards K, Melzer S, Mkumbira J, Gullberg U, Apel K, Gale M, Tohme J, Fregene M: Simple sequence repeat (SSR) markers survey of the cassava (Manihot esculenta Crantz) genome: towards an SSR-based molecular genetic map of cassava. Theor Appl Genet. 2001, 102: 22-31. 10.1007/s001220051614.
Cai HW, Inoue M, Yuyama N, Takahashi W, Hirata M, Sasaki T: Isolation, characterization and mapping of simple sequence repeat markers in zoysiagrass (Zoysia spp.). Theor Appl Genet. 2005, 112: 158-166. 10.1007/s00122-005-0118-9.
Luro FL, Costantino G, Terol J, Argout X, Allario T, Wincker P, Talon M, Ollitrault P, Morillon R: Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genomics. 2008, 9: 287-10.1186/1471-2164-9-287.
Tangphatsornruang S, Sraphet S, Singh R, Okogbenin E, Fregene M, Triwitayakorn K: Development of polymorphic markers from expressed sequence tags of Manihot esculenta Crantz. Mol Ecol Res. 2008, 8: 682-685. 10.1111/j.1471-8286.2007.02047.x.
Fraser LG, Harvey CF, Crowhurst RN, De Silva HN: EST-derived microsatellites from Actinidia species and their potential for mapping. Theor Appl Genet. 2004, 108: 1010-1016. 10.1007/s00122-003-1517-4.
Heesacker A, Kishore VK, Gao W, Tang S, Kolkman JM, Gingle A, Matvienko M, Kozik A, Michelmore RM, Lai Z, Rieseberg LH, Knapp SJ: SSRs and INDELs mined from the sunflower EST database: abundance, polymorphisms, and cross-taxa utility. Theor Appl Genet. 2008, 117: 1021-1029. 10.1007/s00122-008-0841-0.
Gupta PK, Rustgi S, Sharma S, Singh R, Kumar N, Balyan HS: Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol Genet Genomics. 2003, 270: 315-323. 10.1007/s00438-003-0921-4.
Jennings DL: Variation in pollen and ovule fertility in varieties of cassava and the effect of interspecific crossing on fertility. Euphytica. 1963, 12: 69-76. 10.1007/BF00022358.
Umanah E, Hartmann R: Chromosome numbers of karyotypes of some Manihot species. J Am Soc Hor Sci. 1973, 98: 272-274.
Chavarriaga-Aguirre PP, Maya MM, Bonierbale M, Kresovich S, Fregene MA, Tohme J, Kochert G: Microsatellites in Cassava (Manihot esculenta Crantz): discovery, inheritance and variability. Theor Appl Genet. 1998, 97: 493-501. 10.1007/s001220050922.
Guo W, Wang W, Zhou B, Zhang T: Cross-species transferability of G. arboreum-derived EST-SSRs in the diploid species of Gossypium. Theor Appl Genet. 2006, 112: 1573-1581. 10.1007/s00122-006-0261-y.
Saha MC, Cooper JD, Mian MA, Chekhovskiy K, May GD: Tall fescue genomic SSR markers: development and transferability across multiple grass species. Theor Appl Genet. 2006, 113: 1449-1458. 10.1007/s00122-006-0391-2.
Barbará T, Palma-Silva C, Paggi GM, Bered F, Fay MF, Lexer C: Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol. 2007, 16: 3759-3767. 10.1111/j.1365-294X.2007.03439.x.
Hahn SK, Bai KV, Asiedu R: Tetraploids, triploids, and 2n pollen from diploid interspecific crosses with cassava. Theor Appl Genet. 1990, 79: 433-439. 10.1007/BF00226148.
Lokko Y, Dixon AG, Offei S, Danquah E, Fregene MA: Assessment of genetic diversity among African cassava Manihot esculenta Grantz accessions resistant to the cassava mosaic virus disease using SSR markers. Genet Resour Crop Evol. 2009, 53: 1441-1453. 10.1007/s10722-005-6841-x.
Chen X, Cho YG, McCouch SR: Sequence divergence of rice microsatellites in Oryza and other plant species. Mol Genet Genomics. 2002, 268: 331-343. 10.1007/s00438-002-0739-5.
Chabane K, Ablett GA, Cordeiro GM, Valkoun J, Henry RJ: EST versus Genomic Derived Microsatellite Markers for Genotyping Wild and Cultivated Barley. Genet Resour Crop Evol. 2005, 52: 903-909. 10.1007/s10722-003-6112-7.
Eujayl I, Sorrells M, Baum M, Wolters P, Powell W: Assessment of genotypic variation among cultivated durum wheat based on EST-SSRS and genomic SSRS. Euphytica. 2001, 119: 39-43. 10.1023/A:1017537720475.
Raji AAJ, Ladeinde O, Dixon AGO: Screening landraces for additional sources of field resistance to cassava mosaic disease and green mite for integration into the cassava improvement program. J Integr Plant Biol. 2008, 50: 311-318. 10.1111/j.1744-7909.2007.00606.x.
Rudd S: openSputnik--a database to ESTablish comparative plant genomics using unsaturated sequence collections. Nucleic Acids Res. 2005, 33: D622-D627. 10.1093/nar/gki040.
Nei M, Li WH: Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979, 76: 5269-5273. 10.1073/pnas.76.10.5269.
Rohlf FJ: NTYSYS-pc Numerical taxonomy and Multivariate Analysis System. In Version 2.02 Exeter Publ Setauket New York;1998.
Botstein D, White RL, Skolnick M, Davis RW: Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 1980, 32: 314-331.
Anderson JA, Churchill GA, Autrique JE, Tanksley SD, Sorrells ME: Optimizing parental selection for genetic linkage maps. Genome. 1993, 36: 181-186. 10.1139/g93-024.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Acknowledgements
The authors gratefully acknowledge Mrs J Plancarte for preparing the figures, tables and bibliography and Mr B Odeseye for expert technical assistance. The authors also want to thank an anonymous reviewer for helpful comments on the manuscript. This research was supported by the US Agency for International Development (USAID) and the IITA core fund. The opinions expressed herein are those of the authors and do not necessarily reflect the views of USAID. This work is dedicated to the memory of Mr Chike Ugwu, an MSc student who had started to work on this project and whose young life suddenly ended during the course of his MSc program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
AAJR and OAK carried out all aspects of marker optimization and screening with contributions from CDU. AAJR and ILI were responsible for data analysis. ILI, AAJR, JVA, and AGOD co-wrote the manuscript. ILI conceived the study and provided overall coordination of the project. All authors critically reviewed the manuscript.
Electronic supplementary material
12870_2009_460_MOESM1_ESM.xls
Additional file 1: Features of new set of polymorphic EST-SSR markers developed. The data provided represent details of the new SSR markers e.g. marker name, primer sequence, PCR conditions, indication of perfect (P) or imperfect (I) repeat type, and expected product size. (XLS 42 KB)
12870_2009_460_MOESM2_ESM.xls
Additional file 2: Identifiers of the polymorphic EST-SSR markers with their annotation following BlastX searches.(XLS 128 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Raji, A.A., Anderson, J.V., Kolade, O.A. et al. Gene-based microsatellites for cassava (Manihot esculentaCrantz): prevalence, polymorphisms, and cross-taxa utility. BMC Plant Biol 9, 118 (2009). https://doi.org/10.1186/1471-2229-9-118
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-9-118