
Abstract
Background
Pathogenesis-related proteins belonging to group 10 (PR10) are elevated in response to biotic and abiotic stresses in plants. Previously, we have shown a drastic salinity-induced increase in the levels of ABR17, a member of the PR10 family, in pea. Furthermore, we have also demonstrated that the constitutive expression of pea ABR17 cDNA in Arabidopsis thaliana and Brassica napus enhances their germination and early seedling growth under stress. Although it has been reported that several members of the PR10 family including ABR17 possess RNase activity, the exact mechanism by which the aforementioned characteristics are conferred by ABR17 is unknown at this time. We hypothesized that a study of differences in transcriptome between wild type (WT) and ABR17 transgenic A. thaliana may shed light on this process.
Results
The molecular changes brought about by the expression of pea ABR17 cDNA in A. thaliana in the presence or absence of salt stress were investigated using microarrays consisting of 70-mer oligonucleotide probes representing 23,686 Arabidopsis genes. Statistical analysis identified number of genes which were over represented among up- or down-regulated transcripts in the transgenic line. Our results highlight the important roles of many abscisic acid (ABA) and cytokinin (CK) responsive genes in ABR17 transgenic lines. Although the transcriptional changes followed a general salt response theme in both WT and transgenic seedlings under salt stress, many genes exhibited differential expression patterns when the transgenic and WT lines were compared. These genes include plant defensins, heat shock proteins, other defense related genes, and several transcriptional factors. Our microarray results for selected genes were validated using quantitative real-time PCR.
Conclusion
Transcriptional analysis in ABR17 transgenic Arabidopsis plants, both under normal and saline conditions, revealed significant changes in abundance of transcripts for many stress responsive genes, as well as those related to plant growth and development. Our results also suggest that ABR17 may mediate stress tolerance through the modulation of many ABA- and CK-responsive genes and may further our understanding of the role of ABR17 in mediating plant stress responses.
Similar content being viewed by others

Background
Pathogenesis-related (PR) proteins are part of the plant defense responses that are induced by pathogens as well as by abiotic stresses [1, 2]. To date, 17 different families of PR proteins have been identified, based on their specific structural and functional properties [[3] and references therein]. Among the PR proteins, the PR10 family is composed of intracellular acidic proteins with molecular masses ranging from 15–18 kD and are encoded by multiple genes [1, 3]. PR10 genes were first described in Pisum sativum inoculated with Fusarium solani [4] but have been subsequently described in many species [reviewed in [3]]. In addition to their inducible expression in response to stresses, PR10 genes also exhibit constitutive high expressed levels in roots, flowers and pollen during normal growth and development, suggesting additional roles beyond pathogenesis responses [5].
Based on sequence similarities, PR10 proteins have been suggested to be ribonucleases (RNases) [6]. Indeed, PR10 proteins from a variety of species including two pea PR10 proteins have been demonstrated to possess RNase activity [7, 8]. Although RNase activities have been detected for many PR10 proteins, they have also been shown to interact with molecules such as cytokinins (CKs), brassinosteroids, fatty acids, and flavonoids [9–11]. These observations have led to the suggestion that all PR10 proteins may not be RNases and may be involved during normal plant growth and development as hormone/ligand carriers [10–12]. This suggestion is further supported by the fact that CK-specific binding proteins (CSBPs) exhibit amino acid sequence and predicted secondary-structure similarities with PR10 proteins and, for this reason, have been included in the PR10 family [9].
The pea abscisic acid-responsive protein ABR17, induced by the exogenous application of abscisic acid (ABA) is classified as a member of the PR10 family in pea [13]. ABR17 is produced late in seed development, and is homologous to dehydrins and late embryogenesis abundant (LEA)-related proteins [14, 15]. ABR17 is also significantly homologous to intracellular pathogenesis related (IPR) proteins and major birch pollen allergen Betv1 proteins [16, 17]. Our previous research has demonstrated the expression of ABR17 protein in pea under salt stress [2] and the RNase activity of two members of pea PR10 proteins (PR10.1 and ABR17) [7, 8]. Furthermore, we have also demonstrated that the constitutive expression of pea PR10.1 and ABR17 cDNAs enhance germination and early seedling growth under abiotic stress conditions in B. napus and A. thaliana, respectively [18, 19]. In addition, the transgenic plants also exhibited phenotypic differences when compared to their WT counterparts, which included precocious flowering, a higher number of lateral branches, and increased numbers of seed pods [8]. Many of these characteristics of ABR17-transgenic A. thaliana are suggestive of a role for CKs in ABR17 action, particularly increased lateral branching and early flowering [20, 21]. Our suggestion was further supported by the elevated concentrations of endogenous CKs in PR10.1 transgenic B. napus as well as ABR17-transgenic A. thaliana [7, 8].
These observations led us to hypothesize that PR10 proteins, including ABR17, may mediate the observed phenotypic effects through modulation of endogenous CKs. Additional evidence supporting this hypothesis has been provided by the demonstration that exogenous application of CK enhances germination under abiotic stress conditions [8]. In order to further investigate the ABR17-mediated changes in A. thaliana we investigated global changes in gene expression using microarrays. Microarray analysis was carried out in an ABR17-transgenic line compared to its WT, salt treated ABR17-transgenic line compared to untreated ABR17-transgenic line, and salt treated WT compared to untreated WT seedlings. Our current findings reveal that, even in the absence of stress, the expression of genes involved in plant growth and development are significantly (and approximately 2-fold) increased in the transgenic line. Salt treated ABR17-transgenic A. thaliana seedlings showed general salt response theme comparable to that of the WT counterpart used in this study. However, both the trend as well as the degree of changes in gene expression of many defense related genes including plant defensins and heat shock proteins was different providing additional insights into the possible ways in which ABR17 may mediate plant responses to stress.
Results and discussion
Characterization of ABR17-transgenic plants/seedlings
The appearance of 2-week-old WT and ABR17- transgenic A. thaliana seedlings grown in soil as well as on MS medium (1.5% sucrose, 0.8% agar with pH 5.7) [22] plates are shown in Figure 1. At all growth stages investigated, the ABR17-transgenic line was considerably more developmentally advanced compared to its WT counterpart. For example, in the 5-day-old transgenic seedlings (Figure 1A) cotyledons were more developed than in their WT counterparts and at 14-days the transgenic seedlings possessed more rosette leaves (Figure 1A). Similar developmental differences were also observed at 21 days where many transgenic seedlings had started to bolt whereas very few (if any) WT seedlings had advanced to this developmental stage (Figure 1B). At 28 days, the transgenic seedlings also possessed more lateral branches (Figure 1C, Table 1). The transgenic seedlings also flowered earlier than WT with an average difference of at least 2.5 days (Table 1). Seedlings for microarray experiments were grown on semi-solid MS media in order to maintain sterility and it was evident that under these growth conditions also the transgenic seedlings were more developmentally advanced (Figure 1D). These results are consistent with our previous observations of seedlings from this and other independently derived ABR17-transgenic lines grown on semi solid MS media [19]. In addition, the ABR17-transgenic seedlings grown on MS media with 100 mM NaCl were greener and their roots appeared to be longer compared to the WT seedlings grown under similar conditions (Figure 2A).

Appearance of WT and ABR17 transgenic A. thaliana at various growth stages: Seedlings at 5, 14 days (A), 21 days (B), 28 days (C) and MS-grown 14-day-old seedlings (D) are shown.

Appearance of WT and ABR17 transgenic A. thaliana in response to treatments. (A) Appearance of WT and transgenic ABR17 A. thaliana seedlings grown on MS media with 100 mM NaCl (B) Appearance of 7-day-old WT and ABR17 transgenic A. thaliana seedlings grown under dark.
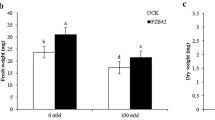
Characteristics like root length, fresh weight, chlorophyll a/b content and carotenoid contents were measured in salt treated- ABR17 and WT seedlings. Roots of ABR17-transgenic seedlings were relatively longer in the absence of salt whereas upon salt treatment, the differences in lengths were not that obvious (Table 1). The fresh weight of ABR17-transgenic seedlings was not significantly different from its WT counterpart in the absence of stress. However, in the presence of 100 mM NaCl, the fresh weights of the transgenic seedlings were significantly (p < 0.05) higher than their WT counterparts (Table 1). Although the chlorophyll and carotenoid contents were almost similar in ABR17 and WT seedlings without any stress, upon NaCl treatment the transgenic seedlings had significantly (p < 0.05) higher levels of chlorophyll (Table 1). Our results indicate that the NaCl treatment had less deleterious effects on the ABR17-transgenic seedlings compared to the WT.
In order to further characterize the differences between the WT and ABR17-transgenic lines, the ability of both WT and ABR17-transgenic seedlings to germinate in the presence or absence of light at RT was compared. In the dark, 85% of ABR17-transgenic A. thaliana had germinated after one week, whereas only 10% of the WT seeds had germinated under the same conditions (Table 1, Figure 2B). In contrast, in the presence of light, 100% of both ABR17-transgenic and WT seeds had germinated in the same period (data not shown). Most Arabidopsis ecotypes require light for germination, which is primarily controlled by a reversible red light dependent equilibrium of the photoreceptors [23]. It is also known that exogenous CKs can substitute for red light and enhance the germination of certain light-requiring species in the dark [24–27]. Furthermore, A. thaliana detiolated (det) mutants exhibits many characteristics of seeds germinated in the presence of light even when germinated in dark [28], a phenotype that has been attributed to CKs because of the fact that even WT seedlings exhibit the same phenotype when germinated in the dark following exogenous CK application [29]. A role for CKs can also be inferred from the observation that coumarin or far-red light, both of which prevent the formation of CK-nucleosides from storage forms of CKs, inhibit germination of lettuce seeds in the dark [30]. Interestingly, amp1 A. thaliana mutants, which possess higher endogenous CKs, also exhibited a photomorphogenic response that in part similar to our ABR17-transgenic seedlings [31]. Taken together, all these results seem to suggest that endogenous CKs play an important role in the germination of light-sensitive seeds and the elevated endogenous CKs in ABR17-transgenic seedlings previously reported [8] may be responsible for the enhanced germination of this genotype in the dark (Table 1, Figure 2B).
Transcriptional profiling using microarrays
In order to characterize the molecular changes brought about by the expression of pea ABR17 cDNA in A. thaliana that resulted in the observed phenotypes, we analyzed gene expression by profiling the transcripts of ABR17-transgenic plants in the absence and presence of 100 mM NaCl. As described earlier, the first set of microarray analysis was the investigation of the differences in gene expression between ABR17-transgenic and WT A. thaliana in the absence of NaCl (ABR17/WT). The second set of microarray analysis was between 100 mM NaCl-treated WT and untreated WT A. thaliana (100 mM NaCl treated WT/WT). The third set of microarray analysis was between 100 mM NaCl treated ABR17-transgenic versus untreated ABR17-transgenic A. thaliana (100 mM NaCl treated ABR17/ABR17).
Microarrays (70-mer oligonucleotide microarrays) consisting of probes presenting 23,686 unique genes identified by Arabidopsis genome initiative (AGI) locus identifiers were used. We identified transcripts as those with mean signal intensities that differed significantly from 0 at α = 0.05 in a Student's t-test in each set of microarrays. The transcripts were categorized based on shared structural elements and/or inferred function. We selected 12 genes representing different functional categories, which according to our microarray analysis showed enhanced or reduced levels of transcript abundance to validate our microarrays. The results from microarrays and qRT-PCR analysis are discussed below.
First set of transcriptional profiling: genes responsive to ABR17
Of the significantly responsive transcripts due the expression of pea ABR17 in A. thaliana, 124 were observed to be modulated in the transgenic line at least 1.5-fold compared to WT with 83 increasing and 41 decreasing in transcript abundance (Additional file 1). Many of these genes had annotations that were associated with either defense or plant growth and development, or both. A total of 16 genes showed significant differences in transcript abundance about 2-fold, where 13 genes exhibited increased transcript abundance and 3 genes showed a decrease in transcript abundance (Table 2).
Among the highly induced transcripts in transgenic seedlings that were putatively related to defense responses (Table 2), we detected 5 members of the plant defensin (PDF) family which exhibited an increased in abundance ~2–3-fold in the transgenic line. PDFs are small (45–54 amino acids), highly basic cysteine-rich peptides belonging to the large defensin family, and are present throughout the plant kingdom. These proteins are known for their involvement in ancestral non-specific innate immune defense system [32]. In addition to being involved in mediating plant responses to pathogens, defensins may also play an important role in plant growth and development. For example, the constitutive expression of AtPep1 induced the expression of PDF1.2 which resulted in better root development in A. thaliana suggesting that plant defensins may regulate root development [32].
Another interesting transcript that exhibited increased abundance (2-fold; Table 2) in ABR17-transgenic plants was a putative mitogen-activated protein kinase (MAPK). MAPK cascades are known to play crucial roles in physiological processes such as cell growth, cell cycle regulation and developmental control as well as plant defense signaling [33]. They are also known to activate WRKY type transcription factors that are involved in transcriptional activation of disease resistance genes [34]. Indeed, we have observed a modest, but elevated expression of four genes belonging to the WRKY family and disease resistance protein (Additional file 1).
We also observed increased transcript abundance for several genes involved in plant growth and development (Table 2). For example, expansins were detected as highly induced transcripts in ABR17-transgenic A. thaliana (Table 2). Expansins are cell wall proteins that are known to induce pH-dependent plant cell wall extension and stress relaxation [35]. The expansins have been related to cell differentiation in tissues such as xylem, leaf primordia and root hairs [36–38]. Previous studies on transgenic plants expressing expansin genes have demonstrated precocious leaf development, longer petioles and larger leaf blades [39, 40].
Glycine-rich proteins (GRPs) were also detected among growth related genes that whose transcripts increased in abundance in ABR17-transgenic plants (Table 2). GRPs consist of quasi-repetitive glycine-rich domains, most commonly GGGX, GGXXXGG or GXGX repeats [41]. Some GRPs have been reported as structural components of the plant cell walls based on their co-localization with cell wall [42]. GRPs have also been reported to be activated by osmotic stress [43], cold shock [44] and wounding [45].
The genes that exhibited significant enhanced expression in ABR17-transgenic plants also included genes for proline-rich protein (PRP) family, xyloglucon endotransglycosylase (XTH), glycosyl hydrolase (GH), phytosulfokine precursor 2 (PSK2), No Apical Meristem (NAM) protein family and glutaredoxins (Additional file 1). PRPs represent a family of structural cell wall proteins that have been implicated in various plant developmental processes [46, 47]. Similarly, XTH and GH family genes are involved in structuring xyloglucan cross-links in plant cell wall and plant development [[37, 48] and [49]]. The PSK2 gene is also involved in cell growth and differentiation [50–52]. Similarly, the NAM gene product is required for shoot apical meristem (SAM) formation during embryogenesis as well as for normal flower development [53–55]. Glutaredoxins have also been demonstrated to be involved in flower development, probably by mediating post-translational modifications of target proteins required for normal petal organ initiation and morphogenesis [56]. Our current observations that the significantly (albeit modest) higher expression of the above mentioned genes related to growth and development including flowering correlates well with the observed phenotypes which include early flowering, increased lateral branching and seed pods as observed in ABR17-transgenic A. thaliana (Figure 1C).
A role for cytokinins in ABR17-induced changes in gene expression?
Interestingly, members of most of the gene families described above (defensin, expansin, MAPK, NAM, WRKY, GRP, PSK2 and Glutaredoxin) that are involved in plant defense as well as growth and development, have been previously reported to be regulated by CKs. For example, genome-wide expression profiling of immediate-early and delayed CK- response genes of A. thaliana, has led to the identification of many genes that are up- regulated by CKs including members of expansin (At1g69530), GRPs (At2g21060), NAM (At4g27410), F-box protein (At3g61060), ERBFs, putative ring zinc finger protein (At1g76410), a member of the bHLH family (At2g18300), blue copper binding protein (At5g20230) and PSK2 [57]. The blue copper binding protein (At5g20230) and putative ring zinc finger protein (At1g76410) identified by Brenner et al. (2005) [57] as CK-induced were observed to be up-regulated in our microarrays analysis. Similarly, gene expression analysis of transgenic A. thaliana seedlings transformed with bacterial isopentenyl transferase (IPT) [58] gene revealed increased transcript abundance for many members of MAPK and WRKY gene families, which included the specific WRKY gene – At1g80840 that has been detected in our microarray studies as being induced by ABR17 expression (Additional file 1). Another investigation into CK action in Arabidopsis has demonstrated increased expression of genes for cytochrome P450, PDF, expansin, patatin, WRKY members and putative disease resistance protein in response to CKs [59]. Therefore, it is apparent that several genes whose transcript levels were modulated by ABR17 expression in A. thaliana have been previously reported in the literature as being CK-responsive, there by suggesting an important role for CK-mediated gene expression in ABR17 action in planta.
Second set of transcriptional profiling: genes responsive to salt stress in WT A. thaliana
Microarray- based analyses of the salt responses in Arabidopsis have been published in several reports. However, most of these studies have investigated responses to very short-term exposure to salt. In this study, we report the transcriptional changes in A. thaliana as a result of long-term, continuous exposure to 100 mM NaCl. Here, we allowed A. thaliana seeds to germinate and grow on semi-solid medium in the presence of 100 mM NaCl for 2 weeks, and the RNA extracted from whole seedlings were used for cDNA synthesis and subsequent microarray analysis. The results from microarray analysis of salt treated WT Arabidopsis seedlings (Additional file 2) agreed with previous studies using similar approaches [60, 61]. We identified 163 genes that showed more than four fold changes in transcript abundance which have been previously reported as being responsive to salt. Our results, therefore, indicate that both short-term "shock" treatments with NaCl as well as long-term treatment used in this study elicit similar responses in A. thaliana at the transcript level (Additional file 2).
Members of protease inhibitor/lipid transfer protein (LTP) family were seen among highly up regulated and/or down regulated genes. At least five members showed increase in transcript abundance and 1 member showed decrease in transcript abundance of more than 4 fold (Additional file 2). LTP genes contain ABA-responsive (ABRE) element (GTACGTGG) and are induced by abscisic acid (ABA) [62, 63]. It has been reported in the literature that NaCl, mannitol or ABA treatments induce the expression of a gene encoding an LTP-like protein in tomato [62, 64]. In addition, the changes in the expression of LTP genes during salt stress have been previously reported [60, 61]. Although, most of the LTP genes were up regulated after short term treatment with salt, they were found to be down regulated after 24 h of salt treatment [60, 61]. From our studies, it appears that many of the LTP genes will get up regulated in response to long term stress as a result of expected increase in ABA levels.
Other major groups of genes with increase in transcript abundance following NaCl treatment included two members of glycosyltransferases (GTs) and five members of glycoside hydrolases (GHs). GTs and GHs are major families of carbohydrate-active that play a primary role in structuring xyloglucan cross-links in plant cell wall [48, 49]. They have been previously reported to be induced by salinity stress in plants and this has been implicated in drought and salt tolerance in A. thaliana [49, 60]. Other genes exhibiting increased transcript abundance included ribonuclease RNS1, osmotin-like protein, hydroxycinnamoyl benzoyltransferase-related, oxidoreductase, 2OG-Fe(II) oxygenase family, glutathione transferase and zinc finger (C3HC4-type RING finger) protein family (Additional file 2). Similarly, the genes which showed decrease in transcript abundance more than 4 fold included many photosynthetic genes, plant defensins, heat shock proteins, auxin-induced proteins, disease resistance protein, Bet v I allergen family and bHLH protein. These results are once again consistent with the previously reported results from microarray-based investigation into salinity stress responses [60, 61].
Third set of transcriptional profiling: genes responsive to salt stress in presence of ABR17
The results from microarray analysis of salt treated ABR17 transgenic A. thaliana seedlings are presented in Tables 3 and 4. We identified 129 genes showing either increase or decrease in transcript abundance more than 4-fold, which included transcription factors (15), stress responsive genes (16), carbohydrate and cell wall metabolism (8), electron transport and oxidoreductases (6), lipid metabolism (3), protein and amino acid metabolism (9), proteins involved in transport across membranes (12) and 60 unknown or unclassified genes.
Transcriptional factors are necessary for the proper transcriptional regulation in response environmental cues [65] and those exhibiting significant increases in transcript abundance included bHLH, 4 members of AP2 related, 2 members of NAM, zinc finger (C3HC4-type RING finger) protein family, ATMYB74 (MYB domain protein 74), ATHB-7 (Arabidopsis thaliana HOMEOBOX 7), and WRKY families. bHLH092 has been indicated among the highly induced transcripts in response to NaCl treatment in the previous transcriptomic studies and are suggested to be important regulators of the NaCl-stress response in Arabidopsis [60]. The APETALA2 (AP2) domain defines a large family of transcription factors which play important roles in plant growth, development as well as stress tolerance [66–71].
Similarly, as previously stated, NAM genes have been found to be induced by abiotic stresses implying roles in stress responses in addition to those in plant growth and development [72, 73]. NAM/NAC proteins belong contain highly conserved NAC (for NAM, ATAF1, 2, and CUC2) domains in their N-terminal regions, that specifically binds target DNA [54]. It has also been demonstrated that NAC transcription factors are ABA-responsive [74, 75] and are also induced by other plant hormones like NAA and ethylene [75–78]. Overexpression of NAC genes has been shown to result in an increase in lateral roots, and tolerance to abiotic stresses like drought and salt stress. NAC genes are believed to exert their stress ameliorating activity through the regulation of stress-inducible genes [77–79]. Similarly, the WRKY family TF genes and myb family genes are known to be biotic and/or abiotic stress responsive [60, 80]. Thus, it is possible that the increased tolerance of ABR17-transgenic seedlings to NaCl is the combined effect of the modulation of the levels of abundance of transcripts for these transcription factors with demonstrated roles in stress tolerance.
The highest transcript abundance of any gene observed in salt treated ABR17 plants was XTR-6, which showed 4.7-fold increase, compared to the untreated ABR17-transgenic line. Xyloglucan endo-transglycosylase (XET) has been suggested to be a key enzyme involved in the modification of the xyloglucan cross-links that controls the strength and extensibility of the plant cell wall [81]. Three members of GH family were also seen among genes which up regulated more than 4-fold change (Table 3). The importance of GHs genes in plant stress [49, 60] has already been discussed in the previous section.
Others salt responsive genes in the ABR17-transgenic line included osmotin, mannitol dehydrogenase, steroid sulfotransferases and RD20 (Table 3) which are known to be regulated by ABA, are expressed in salt-stressed plants and have been used to engineer salinity tolerance [82–86]. In addition, we also observed increase in transcript abundance for ribonuclease- RNS1, peroxidases, copper/zinc superoxidase dismutase (CSD1), cytochrome p450 family, MATE efflux protein and protein kinases which have been previously demonstrated to accumulate in salt treated tissues by others [60, 61]. From our microarray results, it appears that many genes involved in mediating responses to salinity stress are increased in transcript abundance as would be expected.
Comparison of salt responses in WT and ABR17 transgenic seedlings
Although transcriptional changes were almost similar both in salt treated- ABR17 and WT seedlings, the transcript abundance of some genes exhibited significant differences in both the trend as well as the degree of modulation of transcript abundance (Table 5). For instance, as mentioned previously, transcript abundance of xyloglucan endotransglycosylase (XTR-6) (At4g25810) increased 4.7-fold in salt treated ABR17 seedlings, whereas it showed only a 2.4-fold increase in salt-treated WT seedlings (Table 5). Similarly, AP2 domain related transcription factor RAP2.6 (At1g43160) increased 4.5-fold in salt treated ABR17 compared to 1.67-fold in treated WT plants. The expressed proteins- ABA-responsive protein-related (At3g02480) and unknown protein (At5g24640) also showed increase in transcript abundance of at least 4-fold in salt-treated ABR17 transgenic line compared to the 2-fold increase observed for the WT in response to salt. Other genes which exhibited increase in transcript abundance of more than 2 fold in salt-treated ABR17 transgenic but showed less abundance in treated WT included pathogenesis-related protein, (At4g33720) and glutamine-dependent asparagine synthetase (At3g47340). On the other hand, the retroelement pol polyprotein (At2g04460) with unknown function showed a down regulation of more than 2-fold in salt-treated transgenic ABR17 line, but was up regulated in salt treated WT Arabidopsis plants.
Interestingly, many members of heat shock protein (Hsp) family and PDF family showed the opposite response in salt-treated ABR17-transgenic seedlings compared to the salt-treated WT counterparts, with an increase of transcript abundance in salt-treated ABR17-transgenic (Table 5). This difference in the direction of the response in gene expression i.e. induction in the transgenic seedlings versus the repression in the WT may have important consequences with respect to the ability to tolerate salinity (and perhaps other) stress. For example, the Hsp family contains chaperones, which have important roles in protein folding, assembly and in the disposal of unwanted nonfunctional proteins. Hsps are usually induced by environmental stress, and the accumulation of Hsps coincides with enhanced stress tolerance [60, 87–91]. In addition, transgenic Arabidopsis plants overexpressing AtHSP17.7 accumulate high levels of AtHSP17.7 protein and show enhanced tolerance to drought and salinity [89, 90]. The abundance of Hsps in plants and their functional characteristics suggest that Hsps play an important role in plant stress tolerance. Thus, the increased abundance of HSP transcripts in the ABR17-transgenic seedlings may be important for the increased tolerance of this line to the imposed stress.
The up regulation of PDFs in ABR17-transgenic A. thaliana grown under normal conditions (Table 2) and their importance in growth and development has already been discussed earlier. The literature also supports a role for PDFs in stress tolerance [32, 91]. Most of the previously characterized PDFs exhibit anti-fungal, antibacterial, anti- insect and protease inhibitor activity [92]. However, the halophyte salt cress (Thellungiella halophila), a relative of Arabidopsis over expresses PDFs under normal conditions and hence defensins are believed to play a role in salt tolerance [93]. It is therefore possible that the observed relatively tolerant phenotype of ABR17-transgenic seedlings could be due, at least in part, to the elevated expression of XTR6, RAP2.6 transcription factors, unknown proteins (At3g02480, At5g24640), Hsp and PDF gene(s). Additional studies are underway in our laboratory to precisely characterize the role(s) of the above-mentioned genes in stress tolerance of ABR17- transgenic A. thaliana seedlings.
qRT-PCR validation of microarray observations
In order to confirm the fact that CK-responsive genes were indeed up-regulated in the ABR17-transgenic lines, we performed qRT-PCR experiments with the following genes: plant defensin protein (PDF1.2a, At5g44420), expansin (EXPL1, At3g45970), GRP (At1g07135) and putative MAPK 11 (Atg01560) using qRT-PCR. Among the CK- inducible genes identified from our first set of microarray experiments we chose the above 4 genes for qRT-PCR as their transcripts were observed to be at least ~2-fold (1 in log2 ratio) or higher in the transgenic line compared to WT. Our microarray analysis revealed increase in transcripts for defensin, expansin, GRP and MAPK in pea ABR17 seedlings by 2.6, 2.5, 1.9 and 1.9-fold, respectively (Figure 3). Our qRT-PCR results were consistent with the microarray data and showed up-regulation of defensin, expansin, GRP and MAPK by 3.6, 2.5, 2.5 and 2.7-folds, respectively (Figure 3). From these results it is apparent that all the four genes that were up-regulated in our microarray analysis also demonstrated up- regulation in the qRT-PCR relative expression experiments (Figure 3).

Pea ABR17 -modulated transcriptional changes of selected genes. Transcriptional changes of a selected number of transcripts as identified by microarrays, and their validation using qRT- PCR and effects of CK on these genes in WT A. thaliana is given here. The values represented in the graph are fold changes of transcript abundance between transgenic ABR17-A. thaliana seedlings versus WT seedlings grown under normal conditions. Error bars are standard error of fold changes driven from (n = 3) three biological replicates. The AGI annotations are as follows: At5g44420-Plant defensin protein family member PDF1.2, Low-molecular-weight cysteine-rich (LCR77); At3g45970-ATEXLA1 (A. thaliana expansin-like A1); At1g07135-Glycine rich protein; and At1g01560-ATMPK11.
Because of the fact that the specific members of gene families whose transcripts were detected to be modulated by ABR17 cDNA expression in A. thaliana were not exactly identical to those specific members of these families identified by other studies, we wanted to investigate whether those specific members detected in our studies were indeed CK-inducible/repressible. In these experiments, we used WT A. thaliana tissue germinated and grown for 14-days medium supplemented with 5 μM zeatin for additional qRT-PCR experiments. This concentration of zeatin was chosen based on our earlier observations that it induced the largest phenotypic responses in A. thaliana when exogenously applied [8]. It must also be noted that even though 5 μM zeatin was used in our experiments, it is difficult to estimate how much of this exogenously supplied CK actually gets into the seed in order to exert a physiological effect. From the results shown in Figure 3, it is apparent that the expression of EXPL1 (At3g45970), putative MAPK (Atg01560) and GRP (At1g07135) was up-regulated in response to exogenous zeatin by 1.6, 3.8, and 2-fold, respectively (Figure 3). In contrast, the expression of defensin gene was observed to be down-regulated (0.3-fold) in response to the exogenous application of CK (Figure 3). The results for expansin, MAPK and GRP are consistent with our microarray and qRT-PCR results with respect to increased transcript abundance in ABR17-transgenic A. thaliana previously shown to possess higher endogenous CK concentrations [8]. However, in the case of defensin, even though our microarray and qRT-PCR experiments revealed that this gene was up-regulated in the ABR17-transgenic line (Figure 3), its expression was not induced by exogenous CK (Figure 3). The reason for this discrepancy is not immediately clear; however, this may be due to the concentration as well as type of CK used for our exogenous experiments. Furthermore, as indicated previously, the amount of the exogenously supplied CK entering the seed to exert physiological affect may also be different from the concentrations required to elicit induction of this gene.
In order to confirm results from our second and third set of microarray analysis, we performed qRT-PCR experiments with the following 12 genes: unknown proteins (At3g02480; At5g24640; At1g14880), XTR6 (At4g25810), bHLH (At5g43650), AP2 domain transcription factor RAP 2.6 (At1g43160), ATNAC3 (At3g15500), ACD6 (At4g14400), PDF1.2a (At5g44420), EXPL1 (At3g45970), GRP (At1g07135) and MAPK 11 (Atg01560). The unknown proteins were chosen because expression of two of them (At3g02480 and At5g24640) were among highly induced transcripts in salt treated ABR17-trangenic line and also showed comparatively less but high level of transcript abundance in salt treated WT A. thaliana lines (Table 5, Figure 4). Two of them (At1g14880 and At4g14400) were among highly down regulated genes in both salt treated ABR17-transgenic line and WT A. thaliana lines (Table 3 and 4, Figure 4). Our qRT-PCR data showed similar trend as observed by microarrays for all the above-mentioned genes in both salt-treated ABR17-transgenic and salt-treated WT microarrays (Figure 4).

Transcript abundance of selected genes in salt treated-WT and ABR17 transgenic A. thaliana seedlings. The values represented in the graph are fold changes of transcript abundance as identified by microarrays and their validation using qRT-PCR, between salt treated (100 mM) seedlings versus untreated seedlings either in wild type or in ABR17 transgenic A. thaliana. Error bars are standard error of fold changes driven from (n = 3) three biological replicates. The AGI annotations are as follows:At3g02480-ABA-responsive protein-related; At5g43650-basic helix-loop-helix (bHLH) family protein; At5g24640-unknown protein; At4g25810-XTR6: Xyloglucan endotransglycosylase 6; At1g43160-ethylene-responsive transcription factor Related to Apetala 2.6 (Protein RAP2.6); At3g15500-ATNAC3 (A. thaliana NAC domain containing protein 55); At4g14400-ACD6 (Accelerated cell death 6); At1g14880-PLAC8 domain containing protein; At5g44420-Plant defensin protein family member PDF1.2; At3g45970- ATEXLA1 (A. thaliana expansin-like A1); At1g07135-Glycine rich protein; and At1g01560: ATMPK11 (A. thaliana MAP kinase 11). † : the fold change in here is 272.27 ± 58.5.
The genes At3g02480, At5g24640, At1g14880 and At4g14400 showed transcript abundance with fold changes of 5.26, 5.98, 0.27 and 0.06, respectively in our microarray analysis of salt treated WT A. thaliana (Figure 4). Our qRT-PCR analysis of salt treated WT A. thaliana showed transcript abundance of and 18.60, 32.42, 0.03 and 0.06-fold for genes At3g02480, At5g24640, At1g14880 and At4g14400 (Figure 4) compared to qRT-PCR indicated transcript abundance of 23.36, 17.80, 0.14 and 0.04-fold for genes At3g02480, At5g24640, At1g14880 and At4g14400, respectively (Figure 4). Our qRT-PCR analysis of salt treated ABR17 A. thaliana showed transcript abundance of 272.37, 67.49, 0.03 and 0.02-fold for genes At3g02480, At5g24640, At1g14880 and At4g14400, respectively (Figure 4). From these results, it is apparent that all the four genes showed the same trend both in our microarray analysis and qRT-PCR studies (Figure 4) although the absolute values were different with these two experimental methods.
The gene XTR6 (At4g25810) was selected because it was among one of the most highly induced transcripts of any gene on our salt treated ABR17-transgenic A. thalina microarray (Table 3, Figure 4). The genes bHLH (At5g43650), RAP 2.6 (At1g43160) and ATNAC3 (At3g15500) were chosen because their expression was the highest among any other transcription factors identified in response to salt in ABR17- transgenic line (Table 3, Figure 4). The genes At4g25810, At5g43650, At1g43160 and At3g15500 showed transcript abundance of 5.10, 5.29, 3.19 and 3.88-fold, respectively in microarray analysis of salt treated WT A. thaliana, while our qRT-PCR analysis of salt treated WT A. thaliana showed transcript abundance of 32.51, 14.17, 6.58 and 10.23- fold (Figure 4). Similarly, microarray analysis of salt treated ABR17 A. thaliana showed transcript abundance of 25.62, 24.17, 23.00 and 13.96 (Figure 4) and our qRT-PCR analysis values of 54.40, 124.30, 32.27 and 29.88- fold for genes At4g25810, At5g43650, At1g43160 and At3g15500, respectively (Figure 4). Our microarray analysis and qRT-PCR results showed the similar trend in both salt treated-ABR17 and WT samples (Figure 4), The genes PDF1.2a, EXPL1, GRP, and MAPK 11 were chosen as these were validated in our first set of microarrays (ABR17/WT under normal conditions). Once again, a similar trend was observed between microarrays and qRT-PCR analysis thus validating our microarray results.
Relative expression of CK-biosynthetic genes (IPT and CKX) in ABR17-transgenic A. thaliana
As discussed earlier, our observations indicated that many of the genes identified in the transgenic plants as being up-regulated are from families that contain CK-responsive members. We have also previously reported higher endogenous concentrations of CK in this line [8], which suggested the possibility that this may be due to either enhanced de novo CK biosynthesis or decreased degradation. Specifically, the endogenous concentration of total CK in the transgenic line used in this study was ~1–3-fold higher, with the concentration of zeatin (cis and trans combined) being ~1.4-fold and IP being ~2-fold higher in these transgenic lines. However, we did not detect any IPT (involved in CK biosynthesis) or CKX (involved in CK degradation) genes as being significantly up- or down-regulated genes in our microarray experiments suggesting that the elevated endogenous CK concentrations previously reported may not be the result of increased or decreased activities of IPT and CKX genes, respectively. In order to confirm our microarray results and to lend additional support to our above- mentioned hypothesis with respect to the roles (or lack thereof) of IPT and CKX expression in ABR17-transgenic A. thaliana, we also performed qRT-PCR analysis of the expression of IPT and CKX genes using qRT-PCR. There are 9 known IPT genes and 7 known CKX genes but sequence of CKX5 and 7 are very similar therefore we performed qRT-PCR analysis on the 9 IPT and 6 CKX genes. The results from these experiments are summarized in Table 6 and it is apparent that most of the IPT genes exhibit similar expression patterns in both transgenic and WT seedlings. The only exception appears to be IPT 8 where only 0.5-fold expression of this gene was observed in the transgenic line (Table 6). Similarly, CKX expression in the transgenic line was also quite similar to its expression in the WT (Table 6). Our results suggest that the differences in endogenous CK concentrations previously observed in the ABR17-transgenic line may not be the result of increased IPT or decreased CKX levels. However, frequently, there is no correlation between transcript abundance and protein levels and therefore it is possible that IPT and/or CKX protein concentrations may have been affected in the transgenic line resulting in increased endogenous CKs as a result of post-translational processes. However, our previously reported proteome studies on this transgenic line did not reveal any differences between transgenic and WT seedlings with respect to the levels of these proteins [19].
It is possible that the activity of neither IPT nor CKX is responsible for the increased endogenous concentrations of CKs in the ABR17-transgenic lines and the increased endogenous CKs previously reported in the ABR17-transgenic lines may be the result of tRNA degradation by the previously demonstrated RNase activity of pea ABR17 protein [8]. Thus, an increase in free cellular CK would not necessarily involve enhanced IPT or reduced CKX activity; rather it may reflect an increased access to existing, yet tRNA-bound, CK.
Conclusion
We have demonstrated that pea ABR17 cDNA expression modulates the level of a number of transcripts related to plant defense, growth and development, which may explain the observed phenotypic differences between WT and ABR17-transgenic A. thaliana. The gene expression of many transcription factors and defense responsive genes like Hsps and PDFs showed different degree and kind of response between salt treated-ABR17 transgenic and WT A. thaliana, which explains the observed enhanced germination and early seedling vigor in ABR17 transgenic lines, compared to its WT counterpart. Many of the genes exhibiting a 2-fold or higher increase in transcript abundance are known CK-responsive genes providing additional evidence of a role for CKs in ABR17 function. Furthermore, a detailed expression analysis of IPTs and CKXs revealed that the levels of these transcripts were similar in both WT and transgenic seedlings, suggesting the possibility that ABR17 modulates endogenous CKs through an, as of yet, uncharacterized mechanism including the possible degradation of tRNAs which contain CK moieties [94]. These possibilities are currently being investigated in our laboratory.
Methods
Plant material and growth conditions
Transformation of A. thaliana with the pea ABR17 cDNA and the generation of homozygous ABR17-transgenic A. thaliana (line 6.9) have been previously described [19]. This line (6.9) was one of the three independently derived transgenic lines that were characterized in that earlier study. The WT (ecotype WS) and transgenic A. thaliana plants were grown in the green house for observations as previously described [8]. Lateral branches were counted on plants from three independent biological replicates with at least 72 plants per replicate. Average number of days required for the opening of the first flower was also recorded on plants from three biological replicates with 36 plants in each replicate.
In order to measure root lengths of seedlings seeds of A. thaliana (line 6.9) and the WT were surface sterilized [8] and placed on half strength Murashige & Skoog (MS) medium [22] with or without salt (75 mM NaCl or 100 mM NaCl) in square dishes with grids. These dishes were placed vertically in a growth chamber (at 21°C and with light intensity of 250 μmol m-2 s-1) and root lengths were measured after 10 days. The seeds of ABR17 and WT seeds were also grown on half strength MS medium with 0 or 100 mM NaCl to determine their fresh weight and chlorophyll and carotenoid contents in order to assess their ability to grow in the presence of salt. The length of the primary roots of 10-day-old seedlings from three independent biological replicates with at least sixty seedlings per replicate, were calculated using the Image J software (Image J, NIH, MD, USA).
Chlorophyll and carotenoids were extracted from the pooled 2-week-old tissue grown on MS media, using the procedure as described by Srivastava et al., 2006 [7]. Total chlorophyll was estimated using a nomogram [95] and total carotenoid was measured using the formula:
ΔACAR480 = ΔA480+0.114ΔA663-0.638ΔA645
where A is the absorbance and CAR is the carotenoid content [96]. The fresh weight, chlorophyll and carotenoid were calculated using pooled tissue from three independent biological replicates. Percent germination after one week for ABR17-transgenic and WT seeds in the dark and in the presence of light (fluorescent light, 30 μmol m-2 s-1) were compared in Petri dishes at RT. This experiment included three independent biological replicates with at least 45 seeds per replicate. All statistical analyses were performed using the Student's t-test procedure in SAS version 8e (Statistical Analysis System 1985).
Tissue for microarray analysis was obtained by placing surface sterilized seeds of A. thaliana (line 6.9) and the WT on half strength MS medium in Petri dishes with or without 100 mM NaCl at RT (21 ± 2°C) under continuous fluorescent light 30 μmol m-2 s-1 for 14 days. Seedlings (14-day-old) from three independently grown biological replicates in all three set of experiments (comparison of ABR17-transgenic with WT without any stress; comparison of salt treated WT with untreated WT; comparison of salt treated ABR17 transgenic with untreated transgenic) were removed from the MS plates, flash frozen in liquid nitrogen and stored at -80°C until used for RNA extraction.
RNA extraction, cDNA synthesis and microarray analysis
In order to investigate the ABR17-induced gene expression changes under normal and salinity stress conditions, we conducted microarray analysis in three separate hybridization experiments. The first set (set I), consisted of comparison of cDNA samples prepared from ABR17-transgenic and WT seedlings, which were grown in the absence of any stress. Set II consisted of cDNA obtained from salt-treated samples of WT and untreated WT; and set III, cDNA samples of salt treated ABR17- and untreated ABR17-transgenic seedlings for hybridization to the oligo-nucleotide arrays. Each microarray experiment consisted of six hybridizations according to the principles of dye-swap design [97] on tissues across three biological replicates of the experiments.
RNA was isolated using the QIAGEN RNeasy Plant Mini Kit (Qiagen Inc., Mississauga, ON, Canada) from 2-week-old WT and ABR17 seedling tissue from all three set of experiments and the integrity of all RNA samples assessed by agarose gel (1.2%) electrophoresis. For microarray hybridization, 6 μg of total RNA was used to synthesize cDNAs using SuperScript® II RT (Invitrogen Inc., Burlington, ON, Canada) with RT polyA-capture primers in 3D Array 900TM (Genisphere Inc., Hatfield, PA, USA). In these microarray experiments, 70-mer oligonucleotide arrays were used which contained 26,090 probes (Array-Ready Oligo Set for Arabidopsis genome Version 1.0, Qiagen Operon, Alameda, CA, USA), plus additional probes for quality control. Oligonucleotide arrays were spotted on superamine aminosilane-coated slides (TeleChem International Inc., Sunnyvale, CA, USA). Each pair of samples within each of the three biological replicates was labeled in a reciprocal dye-swap design, for a total of 18 hybridizations (overnight, at 52°C) in all three sets of experiments. Slides were scanned using ArrayWoRxe (Applied Precision, Issaquah, WA, USA) and spot intensities were measured, quantified, normalized and analyzed using TM4 [98]. Spots with intensity ratios that differed significantly from 0 (log2 scale) were identified by Student's t-test. This procedure highlights the spots that demonstrated statistically significant differential expression between the different samples. The raw microarray data of 18 hybridizations as well as the protocols used to produce the data were deposited in the ArrayExpress database [ArrayExpress: E-MEXP1024 and E-MEXP1566].
Quantitative real-time PCR (qRT-PCR) validation of microarray data
Primers for qRT-PCR were designed using the Primer Express software (Applied Biosystems Inc., Foster City, CA, USA) to ensure that PCR products of approximately 70–80 bp were generated (Additional file 3). cDNA synthesis and qRT-PCR analysis of gene expression of 19 genes were performed using the Taqman system as described previously [8] on an ABI Prism 7700 Sequence detector (Applied Biosystems Inc., Foster City, CA, USA) and the SNP RT template program or using the SYBR green system as described by Yang and others [99] was used to validate the expression of 8 genes. In both cases, the delta-delta method [100] was used to calculate relative gene expression using actin as the endogenous control. The relative transcript abundance in the controls was normalized to 1 and was used as a basis for comparison to the treatments. Plant tissue from three biological replicates was used in qRT-PCR experiments and reactions for each biological replicate were performed in duplicate (n = 6).
References
Van Loon LC, Pierpoint WS, Boller T, Conejero V: Recommendation for naming plant pathogenesis-related proteins. Plant Mol Biol Rep. 1994, 12 (Suppl 3): 245-264. 10.1007/BF02668748.
Kav NNV, Srivastava S, Goonewardene L, Blade SF: Proteome-level changes in the roots of Pisum sativum in response to salinity. Ann Appl Biol. 2004, 145 (Suppl 2): 217-230. 10.1111/j.1744-7348.2004.tb00378.x.
Liu J-J, Ekramoddoullah AKM: The family 10 of plant pathogenesis-related proteins: Their structure, regulation, and function in response to biotic and abiotic stresses. Physiol Mol Plant Pathol. 2006, 68 (Suppl 1–3): 3-13. 10.1016/j.pmpp.2006.06.004.
Riggleman RC, Fristensky B, Hadwiger LA: The disease resistance response in pea is associated with increased levels of specific mRNAs. Plant Mol Biol. 1985, 4 (Suppl 2–3): 81-86. 10.1007/BF02418753.
Biesiadka J, Bujacz G, Sikorski MM, Jaskolski M: Crystal Structures of Two Homologous Pathogenesis-related Proteins from Yellow Lupine. J Mol Biol. 2002, 319 (Suppl 5): 1223-1234. 10.1016/S0022-2836(02)00385-6.
Moiseyev GP, Beintema JJ, Fedoreyeva LI, Yakovlev GI: High sequence similarity between a ribonuclease from ginsgeng calluses and fungus- elicited proteins from parsley indicates that intracellular pathogenesis-related proteins are ribonucleases. Planta. 1994, 193 (3): 470-472. 10.1007/BF00201828.
Srivastava S, Emery RJN, Kurepin LV, Reid DM, Fristensky B, Kav NNV: Pea PR 10.1 is a ribonuclease and its transgenic expression elevates cytokinin levels. Plant Growth Reg. 2006, 49 (Suppl 1): 17-25. 10.1007/s10725-006-0022-6.
Srivastava S, Emery RJN, Rahman MH, Kav NNV: A crucial role for cytokinins in pea ABR17-mediated enhanced germination and early seedling growth of Arabidopsis thaliana under saline and low temperature stresses. J Plant Growth Reg. 2007, 26 (Suppl 1): 26-37. 10.1007/s00344-006-0046-1.
Fujimoto Y, Nagata R, Fukasawa H, Yano K, Azuma M, Iida A, Sugimoto S, Shudo K, Hashimoto Y: Purification and cDNA cloning of cytokinin-specific binding protein from mung bean (Vigna radiata). Eur J Biochem. 1998, 258 (Suppl 2): 794-802. 10.1046/j.1432-1327.1998.2580794.x.
Mogensen JE, Wimmer R, Larsen JN, Spangfort MD: The major birch allergen, Bet v 1, shows affinity for a broad spectrum of physiological ligands. J Biol Chem. 2002, 277 (26): 23684-23692. 10.1074/jbc.M202065200.
Markovic-Housely Z, Degano M, Lamba D, von Roepenack-Lahaye E, Clemens S, Susani M, Ferreira F, Scheiner O, Breitender H: Crystal structure of a hypoallergenic isoform of the major birch pollen allergen Bet v 1 and its likely biological function as a plant steroid carrier. J Mol Biol. 2003, 325 (Suppl 1): 123-133. 10.1016/S0022-2836(02)01197-X.
Pasternak O, Biesiadka J, Dolot R, Handschuh L, Bujacz G, Sikorski MM, Jaskolski M: Structure of a yellow lupin pathogenesis-related PR-10 protein belonging to a novel subclass. Acta Crystallogr D Biol Crystallogr. 2005, 61 (Pt 1): 99-107.
Iturriaga EA, Leech MJ, Barratt DHP, Wang TL: Two ABA responsive proteins from pea (Pisum sativum L.) are closely related to intracellular pathogenesis-related proteins. Plant Mol Biol. 1994, 24 (Suppl 1): 235-240. 10.1007/BF00040591.
Skriver K, Mundy J: Gene expression in response to abscisic acid and osmotic stress. Plant Cell. 1990, 2 (Suppl 6): 503-512. 10.1105/tpc.2.6.503.
Goday A, Jensen AB, Culianez-Macia FA, Mar AM, Figueras M, Serratosa J, Torrent M, Pages M: The maize abscisic acid-responsive protein Rab17 is located in the nucleus and interacts with nuclear localization signals. Plant Cell. 1994, 6 (Suppl 3): 351-360. 10.1105/tpc.6.3.351.
Fristensky B, Horovitz D, Hadwiger LA: cDNA sequences for pea disease resistance response genes. Plant Mol Biol. 1988, 11 (Suppl 5): 713-715. 10.1007/BF00017470.
Breiteneder H, Pettenburger K, Bito A, Valenta R, Kraft D, Rumpold H, Scheiner O, Breitenbach M: The gene coding for the major birch pollen allergen, Betv1, is highly homologous to a pea disease resistance response gene. EMBO J. 1989, 8 (7): 1935-1938.
Srivastava S, Fristensky B, Kav NNV: Constitutive expression of a PR 10 protein enhances the germination of Brassica napus under saline conditions. Plant Cell Physiol. 2004, 45 (Suppl 9): 1320-1324. 10.1093/pcp/pch137.
Srivastava S, Rahman MH, Shah S, Kav NNV: Constitutive expression of the pea ABA-responsive 17 (ABR17) cDNA confers multiple stress tolerance in Arabidopsis thaliana. Plant Biotechnol J. 2006, 4 (5): 529-549.
Bonhomme F, Kurz B, Melzer S, Bernier G, Jacqmard A: Cytokinin and gibberellin activate SaMADS A, a gene apparently involved in regulation of the floral transition in Sinapis alba. Plant J. 2000, 24 (Suppl 1): 103-111. 10.1046/j.1365-313x.2000.00859.x.
Tanaka M, Takei K, Kojima M, Sakakibara H, Mori H: Auxin controls local cytokinin biosynthesis in the nodal stem in apical dominance. Plant J. 2006, 45 (6): 1028-1036.
Murashige T, Skoog F: A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plantarum. 1962, 15 (Suppl 3): 473-497. 10.1111/j.1399-3054.1962.tb08052.x.
Bentsink L, Koorneef M: Seed dormancy and germination. The Arabidopsis Book. Edited by: Somerville CR, Meyerowitz EM. 2002, Roxkville, MD: Amercan Society of Plant Biologists, [http://www.aspb.org/publications/arabidopsis/]
Miller CO: Similarity of some kinetin and red light effects. Plant Physiol. 1956, 31 (4): 318-319.
Miller CO: The relationship of the kinetin and red light promotions of lettuce seed germination. Plant Physiol. 1958, 33 (2): 115-117.
Skinner CG, Talbert FD, Shive W: Effect of 6-(substituted) purines and gibberellin on the rate of seed germination. Plant Physiol. 1958, 33 (3): 190-194.
Khan AA, Tolbert NE: Reversal of inhibitors of seed germination by red light plus kinetin. Physiol Plantarum. 1965, 18 (Suppl 1): 41-43. 10.1111/j.1399-3054.1965.tb06866.x.
Chory J, Peto C, Feinbaum R, Pratt L, Ausubel F: Arabidopsis thaliana mutant that develops as a light-grown plant in the absence of light. Cell. 1989, 58 (Suppl 5): 991-999. 10.1016/0092-8674(89)90950-1.
Chory J, Reinecke D, Sim S, Washburn T, Brenner M: A role cytokinins in de-etiolation in Arabidopsis. Plant Physiol. 1994, 104 (2): 339-347.
Pietraface WJ, Blaydes DF: Far-red light and coumarin induced changes in 9-methyl-N6-benzyladenine metabolism by Lactuca sativa achenes. Plant Cell Physiol. 1982, 23 (Suppl 8): 1475-1477. [http://pcp.oxfordjournals.org/cgi/content/abstract/23/8/1475]
Chaudhury AM, Letham S, Craig S, Dennis ES: amp1 – a mutant with high cytokinin levels and altered embryonic pattern, faster vegetative growth, constitutive photomorphogenesis and precocious flowering. Plant J. 1993, 4 (Suppl 6): 907-916. 10.1046/j.1365-313X.1993.04060907.x.
Huffaker A, Pearce G, Ryan AC: An endogenous peptide signal in Arabidopsis activates components of the innate immune response. Proc Natl Acad Sci USA. 2006, 103 (Suppl 26): 10098-10103. 10.1073/pnas.0603727103.
Tena G, Asai T, Chiu W, Sheen J: Plant mitogen-activated protein kinase signaling cascades. Curr Opin Plant Biol. 2001, 4 (Suppl 5): 392-400. 10.1016/S1369-5266(00)00191-6.
Kim YC, Zhang S: Activation of a mitogen-activated protein kinase cascade induces WRKY family of transcription factors and defense genes in tobacco. Plant J. 2004, 38 (Suppl 10): 142-151. 10.1111/j.1365-313X.2004.02033.x.
McQueen-Mason S, Cosgrove DJ: Expansin mode of action on cell walls: analysis of wall hydrolysis, stress relaxation, and binding. Plant Physiol. 1995, 107 (1): 87-100.
Reinhardt D, Wittwer F, Mandel T, Kuhlemeier C: Localized upregulation of a new expansin gene predicts the site of leaf formation in the tomato meristem. Plant Cell. 1998, 10 (Suppl 9): 1427-1437. 10.1105/tpc.10.9.1427.
Reidy B, McQueen-Mason S, Nösberger J, Fleming A: Differential expression of α- and β-expansin genes in the elongating leaf of Festuca pratensis. J Plant Mol Biol. 2001, 46 (Suppl 4): 491-504. 10.1023/A:1010621417854.
Cho H-T, Cosgrove JD: Regulation of root hair initiation and expansin gene expression in Arabidopsis. Plant Cell. 2002, 14 (Suppl 12): 3237-3253. 10.1105/tpc.006437.
Cho H-T, Cosgrove DJ: Altered expression of expansin modulates leaf growth and pedicel abscission in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2000, 97 (Suppl 17): 9783-9788. 10.1073/pnas.160276997.
Pien S, Wyrzykowska J, McQueen-Mason S, Smart C, Fleming A: Local expression of expansin induces the entire process of leaf development and modifies leaf shape. Proc Natl Acad Sci. 2001, 98 (Suppl 20): 11812-11817. 10.1073/pnas.191380498.
Sachetto-Martins G, Franco LO, Oliveira de DE: Plant glycine-rich proteins: a family or just proteins with a common motif?. Biochim Biophys Acta. 2000, 1492 (1): 1-14.
Cassab GI: Plant cell wall proteins. Ann Rev Plant Physiol Plant Mol Biol. 1998, 49: 281-309. 10.1146/annurev.arplant.49.1.281.
Xu D, Lei M, Wu R: Expression of the rice Osgrp1 promoter- Gus reporter gene is specifically associated with cell elongation/expansion and differentiation. Plant Molecular Biology. 1995, 28 (Suppl 3): 455-471. 10.1007/BF00020394.
Carpenter CD, Kreps JA, Simon AE: Genes encoding glycine-rich Arabidopsis thaliana proteins with RNA-binding motifs are influenced by cold treatment and an endogenous circadian rhythm. Plant Physiol. 1994, 104 (3): 1015-25. 10.1104/pp.104.3.1015. [http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=160700]
Showalter AM, Zhou J, Rumeau D, Worst SG, Varner JE: Tomato extensin and extensin-like cDNAs: Structure and expression in response to wounding. Plant Mol Biol. 1991, 16 (Suppl 4): 547-565. 10.1007/BF00023421.
Chen J, Varner JE: Isolation and characterization of cDNA clones for carrot extensin and proline-rich 33-kDa proteins. Proc Natl Acad Sci USA. 1985, 82 (Suppl 13): 4399-4403. 10.1073/pnas.82.13.4399.
Bernhardt C, Tierney ML: Expression of AtPRP3, a proline-rich structural cell wall protein from Arabidopsis, is regulated by cell-type-specific developmental pathways involved in root hair formation. Plant Physiol. 2000, 122 (3): 705-714. 10.1104/pp.122.3.705.
Goujon T, Minic Z, Amrani AE, Lerouxel O, Aletti E, Lapierre C, Joseleau J-P, Jouanin L: AtBXL1, a novel higher plant (Arabidopsis thaliana) putative beta-xylosidase gene, is involved in secondary cell wall metabolism and plant development. Plant J. 2003, 33 (Suppl 4): 677-690. 10.1046/j.1365-313X.2003.01654.x.
Vissenberg K, Fry CS, Pauly M, Höfte H, Verbelen J-P: XTH acts at the microfibril-matrix interface during cell elongation. J Exp Bot. 2005, 56 (Suppl 412): 673-683. 10.1093/jxb/eri048.
Yamakawa S, Sakuta1 C, Matsubayashi Y, Sakagami Y, Kamada H, Satoh S: The promotive effects of a peptidyl plant growth factor, phytosulfokine-α, on the formation of adventitious roots and expression of a gene for a root-specific cystatin in cucumber hypocotyls. J Plant Res. 1998, 111 (Suppl 3): 453-458. 10.1007/BF02507810.
Matsubayashi Y, Takagi L, Omura N, Morita A, Sakagami Y: The endogenous sulfated pentapeptide phytosulfokine-α stimulates tracheary element differentiation of isolated mesophyll cells of Zinnia. Plant Physiol. 1999, 120 (Suppl 4): 1043-1048. 10.1104/pp.120.4.1043.
Igasaki T, Akashi N, Ujino-Ihara T, Matsubayashi Y, Sakagami Y, Shinohara K: Phytosulfokine stimulates somatic embryogenesis in Cryptomeria japonica. Plant Cell Physiol. 2003, 44 (Suppl 5): 1412-1416. 10.1093/pcp/pcg161.
Souer E, van Houwelingen A, Kloos D, Mol J, Koes R: The no apical meristem gene of Petunia is required for pattern formation in embryos and flowers and is expressed at meristem and primordia boundaries. Cell. 1996, 85 (Suppl 2): 159-170. 10.1016/S0092-8674(00)81093-4.
Aida M, Ishida T, Fukaki H, Fujisawa H, Tasaka M: Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell. 1997, 9 (Suppl 6): 841-857. 10.1105/tpc.9.6.841.
Sablowski RW, Meyerowitz EM: A homolog of NO APICAL MERISTEM is an immediate target of the floral homeotic genes APETALA3/PISTILLATA. Cell. 1998, 92 (Suppl 1): 93-103. 10.1016/S0092-8674(00)80902-2.
Xing S, Rosso GM, Zachgo S: ROXY1, a member of the plant glutaredoxin family, is required for petal development in Arabidopsis thaliana. Development. 2005, 132 (Suppl 7): 1555-1565. 10.1242/dev.01725.
Brenner GW, Romanov AG, Köllmer I, Bürkle L, Schmülling T: Immediate-early and delayed cytokinin response genes of Arabidopsis thaliana identified by genome-wide expression profiling reveal novel cytokinin-sensitive processes and suggest cytokinin action through transcriptional cascades. Plant J. 2005, 44 (Suppl 2): 314-333. 10.1111/j.1365-313X.2005.02530.x.
Hoth S, Ikeda Y, Morgante M, Wang X, Zuo J, Hanafey MK, Gaasterland T, Tingey SV, Chua NH: Monitoring genome-wide changes in gene expression in response to endogenous cytokinin reveals targets in Arabidopsis thaliana. FEBS Lett. 2003, 554 (3): 373-380. 10.1016/S0014-5793(03)01194-3.
Rashotte AM, Carson DBS, To PCJ, Kieber JJ: Expression profiling of cytokinin action in Arabidopsis. Plant Physiol. 2003, 132 (Suppl 4): 1998-2011. 10.1104/pp.103.021436.
Jiang Y, Deyholos MK: Comprehensive transcriptional profiling of NaCl-stressed Arabidopsis roots reveals novel classes of responsive genes. BMC Plant Biol. 2006, 6 (25):
Ma SS, Gong QQ, Bohnert HJ: Dissecting salt stress pathways. J Exp Bot. 2006, 57 (Suppl 5): 1097-1107. 10.1093/jxb/erj098.
Hughes MA, Dunn MA, Pearce RS, White AJ, Zhang L: An abscisic-acid-responsive, low temperature barley gene has homology with a maize phospholipid transfer protein. Plant Cell Environ. 1992, 15 (Suppl 7): 861-865. 10.1111/j.1365-3040.1992.tb02155.x.
White AJ, Dunn MA, Brown K, Hughes MA: Comparative analysis of genomic sequence and expression of a lipid transfer protein gene family in winter barley. J Exp Bot. 1994, 45 (Suppl 12): 1885-1892. 10.1093/jxb/45.12.1885.
Torres-Schumann S, Godoy JA, Pintor-Toro JA: A probable lipid transfer protein gene is induced by NaCl in stems of tomato plants. Plant Mol Biol. 1992, 18 (Suppl 4): 749-757. 10.1007/BF00020016.
Riechmann J, Heard G, Martin L, Reuber CZ, Jiang J, Keddie L, Adam O, Pineda OJ, Ratcliffe RR, Samaha R, Creelman M, Pilgrim P, Broun JZ, Zhang D, Ghandehari BK, Sherman G, Yu L: Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science. 2000, 290 (Suppl 5499): 2105-2110. 10.1126/science.290.5499.2105.
Ohmetakagi M, Shinshi H: Ethylene inducible DNAbinding proteins that interact with an ethylene-responsive element. Plant Cell. 1995, 7 (Suppl 1): 173-182. 10.1105/tpc.7.2.173.
Gilmour SJ, Zarka DG, Stockinger EJ, Salazar MP, Houghton JM, Thomashow MF: Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold-induced COR gene expression. Plant J. 1998, 16 (Suppl 4): 433-442. 10.1046/j.1365-313x.1998.00310.x.
Kizis D, Lumbreras V, Pages M: Role of AP2/EREBP transcription factors in gene regulation during abiotic stress. FEBS Lett. 2001, 498 (Suppl 2–3): 187-189. 10.1016/S0014-5793(01)02460-7.
Ma S, Bohnert H: Integration of Arabidopsis thaliana stress-related transcript profiles, promoter structures, and cell-specific expression. Genome Biol. 2007, 8 (4): R49-10.1186/gb-2007-8-4-r49.
Guo ZJ, Chen XJ, Wu XL, Ling JQ, Xu P: Overexpression of the AP2/EREBP transcription factor OPBP1 enhances disease resistance and salt tolerance in tobacco. Plant Mol Biol. 2004, 55 (Suppl 4): 607-618. 10.1007/s11103-004-1521-3.
Shukla RK, Raha S, Tripathi V, Chattopadhyay D: Expression of CAP2, an APETALA2-family transcription factor from chickpea, enhances growth and tolerance to dehydration and salt stress in transgenic tobacco. Plant Physiol. 2006, 142 (Suppl 1): 113-123. 10.1104/pp.106.081752.
Xie Q, Frugis G, Colgan D, Chua N-H: Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Develop. 2000, 14 (Suppl 23): 3024-3036. 10.1101/gad.852200.
Olsen AN, Ernst HA, Leggio LL, Skriver K: NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005, 10 (Suppl 2): 79-87. 10.1016/j.tplants.2004.12.010.
Greve K, La Cour T, Jensen MK, Poulsen FM, Skriver K: Interactions between plant RING-H2 and plant-specific NAC (NAM/ATAF1/1/CUC2) proteins: RING-H2 molecular specificity and cellular localization. Biochem J. 2003, 371 (Suppl 1): 97-108. 10.1042/BJ20021123.
Fujita M, Fujita Y, Maruyama K, Seki M, Hiratsu K, Ohme-Takagi M, Tran LS, Yamaguchi-Shinozaki K, Shinozaki K: A dehydration-induced NAC protein, RD26, is involved in a novel ABA-dependent stress-signaling pathway. Plant J. 2004, 39 (Suppl 6): 863-876. 10.1111/j.1365-313X.2004.02171.x.
Hegedus D, Yu M, Baldwin D, Gruber M, Sharpe A, Parkin I, Whitwill S, Lydiate D: Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol Biol. 2003, 53 (Suppl 3): 383-97. 10.1023/B:PLAN.0000006944.61384.11.
Tran LS, Nakashima K, Sakuma Y, Simpson SD, Fujita Y, Maruyama K, et al: Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell. 2004, 16 (Suppl 11): 2481-98. 10.1105/tpc.104.022699.
He XJ, Mu RL, Cao WH, Zhang ZG, Zhang JS, Chen SY: AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J. 2005, 44 (Suppl 6): 903-916. 10.1111/j.1365-313X.2005.02575.x.
Hu H, Dai M, Yao J, Xiao B, Li X, Zhang Q, Xiong L: Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc Natl Acad Sci USA. 2006, 103 (Suppl 35): 12987-12992. 10.1073/pnas.0604882103.
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, Satou M, Akiyama K, Taji T, Yamaguchi-Shinozaki K, Carninci P, Kawai J, Hayashizaki Y, Shinozaki K: Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002, 31 (Suppl 3): 279-292. 10.1046/j.1365-313X.2002.01359.x.
Silva J, Arrowsmith D, Hellyer A, Whiteman S, Robinson S: Xyloglucan endotransglycosylase and plant growth. J Exp Bot. 1994, 45 (Suppl 280): 1693-1701.
Singh NK, Bracker CA, Hasegawa PM, Handa AK, Buckel S, Hermodson MA, Pfankoch E, Regnier FE, Bressan RA: Characterization of osmotin. A thaumatin-like protein associated with osmotic adaptation in plant cells. Plant Physiol. 1987, 85 (2): 529-536.
Takahashi S, Katagiri T, Yamaguchi-Shinozaki K, Shinozaki K: An Arabidopsis gene encoding a Ca2+-binding protein is induced by abscisic acid during dehydration. Plant Cell Physiol. 2000, 41 (Suppl 7): 898-903. 10.1093/pcp/pcd010.
Abede T, Guenzi AC, Martin B, Cushman JC: Tolerance of mannitol-accumulating transgenic wheat to water stress and salinity. Plant Physiol. 2003, 131 (4): 1748-1755. 10.1104/pp.102.003616.
King GJ, Ce Hausey JR, Turner VA: A protein induced by NaCl in suspension cultures of Nicotiana tabacum accumulates in whole plant roots. Plant Mol Biol. 1986, 7 (Suppl 6): 441-449. 10.1007/BF00020328.
Klein M, Papenbrock J: Sulfotransfersases and their role in glucosinolate biosynthesis. Sulfur Assimilation and Abiotic Stress in Plants. Edited by: Khan NA, Umar S, Singh. 2008, 189-166. [http://www.springerlink.com/content/u70054228570462p/]
Coca MA, Almoguera C, Thomas TL, Jordano J: Differential regulation of small heat-shock genes in plants: analysis of a water-stress-inducible and developmentally activated sunflower promoter. Plant Mol Biol. 1996, 31 (Suppl 4): 863-876. 10.1007/BF00019473.
Campalans A, Pages M, Messeguer R: Identification of differentially expressed genes by the cDNA-AFLP technique during dehydration of almond (Prunus amygdalus). Tree Physiol. 2001, 21 (10): 633-643.
Kuznetsov VV, Rakutin VY, Borisova NN, Rotschupkin BV: Why does heat shock increase salt resistance in cotton plants?. Plant Physiol Biochem. 1993, 31 (Suppl 2): 181-188.
Sun W, Bernard C, Cotte van de B, van Montagu M, Verbruggen N: At-HSP17.6A, encoding a small heat-shock protein in Arabidopsis, can enhance osmotolerance upon overexpression. Plant J. 2001, 27 (Suppl 5): 407-415. 10.1046/j.1365-313X.2001.01107.x.
Koike M, Okamoto T, Tsuda S, Imai R: A novel plant defensin-like gene of winter wheat is specifically induced during cold acclimation. Biochem Biophys Res Commun. 2002, 298 (Suppl 1): 46-53. 10.1016/S0006-291X(02)02391-4.
Spelbrink GR, Dilmac N, Allen A, Smith JT, Shah MD, Hockerman HG: Differential antifungal and calcium channel-blocking activity among structurally related plant defensins. Plant Physiol. 2004, 135 (Suppl 4): 2055-2067. 10.1104/pp.104.040873.
Taji T, Seki M, Satou M, Sakurai T, Kobayashi M, Ishiyama K, Narusaka Y, Narusaka M, Zhu JK, Shinozaki K: Comparative genomics in salt tolerance between Arabidopsis and Arabidopsis-related halophyte salt cress using Arabidopsis microarray. Plant Physiol. 2004, 135 (Suppl 3): 1697-1709. 10.1104/pp.104.039909.
Prinsen E, Kaminek M, van Onckelen HA: Cytokininbiosynthesis: a black box?. J Plant Growth Reg. 1997, 23 (Suppl 1–2): 3-15. [http://www.springerlink.com/content/mw363804rl2p3633/]
Kirk JTO: Studies on the dependence of chlorophyll synthesis on protein synthesis in Euglena gracilis together with a nomogram for determination of chlorophyll concentration. Planta. 1968, 78 (Suppl 2): 200-202. [http://www.springerlink.com/content/k71714x102743547/]
Kirk JT, Allen RL: Dependence of pigment synthesis on protein synthesis. Biochem Biophys Res Commun. 1965, 21 (Suppl 6): 523-530. 10.1016/0006-291X(65)90516-4.
Martin-Magniette M, Aubert J, Cabannes E, Daudin J: Evaluation of gene-specific dye bias in cDNA microarray experiments. Bioinform. 2005, 21 (Suppl 9): 1995-2000. 10.1093/bioinformatics/bti302.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J: TM4: A free, open-source system for microarray data management and analysis. Biotechniques. 2003, 34 (2): 374-378.
Yang H, Matsubayashi Y, Nakamura K, Sakagami Y: Diversity of Arabidopsis genes encoding precursors for phytosulfokine, a peptide growth factor. Plant Physiol. 2001, 127 (Suppl 3): 842-851. 10.1104/pp.127.3.842. [http://www.plantphysiol.org/cgi/content/abstract/127/3/842]
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real time quantitative PCR and the 2ΔΔCt method. Methods. 2001, 25 (Suppl 4): 402-408. 10.1006/meth.2001.1262.
Acknowledgements
Financial support from Natural Sciences and Engineering Research Council (NSERC) of Canada, the Alberta Agricultural Research Institute (AARI) and the University of Alberta is gratefully acknowledged. We thank Dr. Neil Emery for critically reading the manuscript and providing valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SSK designed and carried out all the experiments with the assistance from SS, MM and MHR and drafted the manuscript. NNVK and MKD supervised all research and contributed to the writing and editing of the manuscript.
Electronic supplementary material
12870_2008_299_MOESM1_ESM.pdf
Additional file 1: Transcriptional profiling: genes exhibiting more than 1.5-fold increase/decrease in transcript abundance in ABR17 transgenic A. thaliana seedlings. The data shows the genes that exhibited more than 2-fold increase/decrease in transcript abundance in two week old ABR17 transgenic Arabidopsis seedlings over two week old wild type Arabidopsis seedlings, both grown on normal media. (PDF 93 KB)
12870_2008_299_MOESM2_ESM.pdf
Additional file 2: Transcriptional profiling: genes exhibiting more than 4-fold increase/decrease in transcript abundance in salt- treated wild type A. thaliana seedlings. The data shows the genes that exhibited more than 4-fold increase/decrease in transcript abundance in two week old salt- treated wild type Arabidopsis seedlings over two week old wild type Arabidopsis seedlings that were grown on normal media. (PDF 106 KB)
12870_2008_299_MOESM3_ESM.pdf
Additional file 3: List of primers used in qRT-PCR. The table lists the primers used in qRT-PCR for validating microarray data (PDF 75 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Krishnaswamy, S.S., Srivastava, S., Mohammadi, M. et al. Transcriptional profiling of pea ABR17 mediated changes in gene expression in Arabidopsis thaliana. BMC Plant Biol 8, 91 (2008). https://doi.org/10.1186/1471-2229-8-91
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-8-91