
Abstract
Background
Alpha proteobacteria are one of the largest and most extensively studied groups within bacteria. However, for these bacteria as a whole and for all of its major subgroups (viz. Rhizobiales, Rhodobacterales, Rhodospirillales, Rickettsiales, Sphingomonadales and Caulobacterales), very few or no distinctive molecular or biochemical characteristics are known.
Results
We have carried out comprehensive phylogenomic analyses by means of Blastp and PSI-Blast searches on the open reading frames in the genomes of several α-proteobacteria (viz. Bradyrhizobium japonicum, Brucella suis, Caulobacter crescentus, Gluconobacter oxydans, Mesorhizobium loti, Nitrobacter winogradskyi, Novosphingobium aromaticivorans, Rhodobacter sphaeroides 2.4.1, Silicibacter sp. TM1040, Rhodospirillum rubrum and Wolbachia (Drosophila) endosymbiont). These studies have identified several proteins that are distinctive characteristics of all α-proteobacteria, as well as numerous proteins that are unique repertoires of all of its main orders (viz. Rhizobiales, Rhodobacterales, Rhodospirillales, Rickettsiales, Sphingomonadales and Caulobacterales) and many families (viz. Rickettsiaceae, Anaplasmataceae, Rhodospirillaceae, Acetobacteraceae, Bradyrhiozobiaceae, Brucellaceae and Bartonellaceae). Many other proteins that are present at different phylogenetic depths in α-proteobacteria provide important information regarding their evolution. The evolutionary relationships among α-proteobacteria as deduced from these studies are in excellent agreement with their branching pattern in the phylogenetic trees and character compatibility cliques based on concatenated sequences for many conserved proteins. These studies provide evidence that the major groups within α-proteobacteria have diverged in the following order: (Rickettsiales(Rhodospirillales (Sphingomonadales (Rhodobacterales (Caulobacterales-Parvularculales (Rhizobiales)))))). We also describe two conserved inserts in DNA Gyrase B and RNA polymerase beta subunit that are distinctive characteristics of the Sphingomonadales and Rhodosprilllales species, respectively. The results presented here also provide support for the grouping of Hyphomonadacea e and Parvularcula species with the Caulobacterales and the placement of Stappia aggregata with the Rhizobiaceae group.
Conclusion
The α-proteobacteria-specific proteins and indels described here provide novel and powerful means for the taxonomic, biochemical and molecular biological studies on these bacteria. Their functional studies should prove helpful in identifying novel biochemical and physiological characteristics that are unique to these bacteria.
Similar content being viewed by others


Background
The α-proteobacteria form one of the largest groups within bacteria that includes numerous phototrophs, chemolithotrophs, chemoorganotrophs and aerobic photoheterotrophs [1]. They are abundant constituents of various terrestrial and marine environments [2]. Pelagibacter oblique, which is the smallest known free-living bacteria, is believed to be the most numerous bacteria on this planet (about 1028 cells) comprising about 25% of all microbial cells in the oceans [2]. The intimate association that many α-proteobacteria exhibit with the eukaryotic organisms is of central importance from agricultural and medical perspectives [3, 4]. Symbiotic association of the Rhizobiaceae family members with the plant root nodules is responsible for most of the atmospheric nitrogen fixation by plants [4–6]. Many other α-proteobacteria such as Rickettsiales, Brucella and Bartonella have adopted intracellular life styles and they constitute important human and animal pathogens [3, 7–9]. Additionally, the α-proteobacteria have also played a seminal role in the origin of the eukaryotic cell [10, 11].
In the current taxonomic scheme based on 16S rRNA, α-proteobacteria are recognized as a Class within the phylum Proteobacteria [1, 12, 13]. They are subdivided into 7 main subgroups or orders (viz. Caulobacterales, Rhizobiales, Rhodobacterales, Rhodospirillales, Rickettsiales, Sphingomonadales and Parvularculales) [12]. The α-proteobacteria and their main subgroups are presently distinguished from each other and from other bacteria primarily on the basis of their branching in phylogenetic trees [14, 15]. However, we have previously described several conserved inserts and deletions (indels), as well as whole proteins, that are specific for these bacteria [16, 17].
In the past 3–4 years, the numbers of α-proteobacterial genomes that have been sequenced has increased markedly. In addition to > 60 complete genomes (Table 1), sequence information is available for a large number of other species. These genomes cover all of the main groups within α-proteobacteria (Table 1) and provide an enormously valuable resource for identifying molecular characteristics that are unique to them. This information can be used to identify unique sets of genes or proteins that are distinctive characteristics of various higher taxonomic groups (e.g. families, orders, etc.) within α-proteobacteria. Such genes/proteins provide valuable tools for taxonomic, biochemical and molecular biological studies [17–26]. With this goal in mind, in the present work, we have performed comprehensive phylogenomic analyses of α-proteobacterial genomes to identify proteins/ORFs that are distinctive characteristics of the various higher taxonomic groups within α-proteobacteria. The phylogenomic distribution of these proteins is also compared with the branching patterns of these species in phylogenetic trees and in the character compatibility cliques to develop a reliable picture of α-proteobacterial evolution.
Results and discussion
Phylogeny of alpha proteobacteria
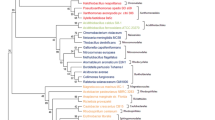
For comparing and interpreting the results of phylogenomic analysis, it was necessary at first to examine the evolutionary relationships among α-proteobacteria in phylogenetic trees. Phylogenetic analyses for α-proteobacteria was carried out based on concatenated sequences for 12 highly conserved proteins (see Methods). The relationships among these species were examined by both traditional phylogenetic methods (viz. neighbour-joining (NJ), maximum parsimony (MP) and maximum-likelihood (ML)) and by the character compatibility approach [27]. Figure 1 presents a neighbour-joining distance tree for α-proteobacteria, showing the bootstrap scores for various nodes using the NJ, MP and ML methods. In this tree, all of the main groups or orders within α-proteobacteria (viz. Caulobacterales, Rhizobiales, Rhodobacterales, Rhodospirillales and Sphingomonadales), except the Rickettsiales, formed well-resolved clades by different methods. For the Parvularculales, sequence information was available from a single species and it branched with the Caulobacterales. The two main families of the Rickettsiales i.e. Rickettsiaceae and Anaplasmataceae did not form a monophyletic clade, although they constituted the deepest branching lineages within α-proteobacteria. Except for the NJ method, the relative branching of different main groups within α-proteobacteria was not resolved by other methods. The branching orders of different groups as seen here is similar to that observed previously in the rRNA trees [1, 15, 16, 28]. Recently, after this work was completed, Williams et al. [29] have reported similar results based on phylogenetic analysis of a different large set of protein sequences.

A neighbour-joining distance tree based on concatenated sequences for 12 conserved proteins. The numbers on the nodes indicate bootstrap scores (out of 100) observed in the neighbour-joining (NJ), maximum parsimony (MP) and maximum-likelihood (ML) analyses (NJ/MP/ML). The species marked with * are presently not part of the Caulobacterales order, but the results of phylogenetic and phylogenomic studies presented here suggest their placement in this group.
The evolutionary relationships among α-proteobacteria were also examined using the character compatibility approach [27]. This method removes all homoplasic and fast-evolving characters from the dataset [27, 30, 31] and it has proven useful in obtaining correct topology in cases which have proven difficult to resolve by other means [31–33]. These analyses were carried out on a smaller set of 27 species containing all main groups of α-proteobacteria and two ε-proteobacteria. The concatenated sequence alignment for the 12 proteins contained 896 positions that were useful for these studies (i.e. those sites where only two amino acids were found with each present in at least two species). The compatibility analysis of these sites resulted in 12 largest cliques each containing 350 mutually compatible characters. The two main relationships observed in these cliques are shown in Fig. 2. The other cliques differed from those shown only in the relative branching positions of various Rhizobiaceae species, which varied from each other by a single character and are shown as unresolved in Fig. 2. In both these cliques, the species from all main orders within α-proteobacteria were clearly distinguished by multiple unique characters. Further, in contrast to the phylogenetic trees where Rickettsiaceae and Anaplasmataceae did not form a distinct clade (Fig. 1), their monophyletic grouping was strongly supported by 9 unique shared characters (Fig. 2). The Rickettsiales and Rhodospirillales formed the deepest branching lineages in these cliques and other groups within α-proteobacteria branched in the following order: (Sphingomonadales (Rhodobacterales (Caulobacterales (Rhizobiales)))). This branching order was supported by multiple unique characters at each node giving confidence in the results.

Character compatibility cliques showing the two largest cliques of mutually compatible characters based on the two states sites in the concatenated sequence alignment for 12 conserved proteins. The cliques consisted of 350 mutually compatible characters. The numbers of characters that distinguished different clades are indicated on the nodes. Rooting was done using the sequences for Helicobacter pylori and Campylobacter jejuni. *, as in Figure 1.
The two main cliques obtained in these analyses differed from each other in terms of the branching position of the species belonging to the order Rhodospirillales. In one clique (Fig. 2A), the Rhodospirillales branched with the Rickettsiales, whereas in the other this group of species was found to branch after the Rickettsiales and it formed outgroup of the other α-proteobacteria (Fig. 2B). However, only a single character supported the former relationship indicating that it was not reliable. The two families within the Rhodospirillales order (Rhodospirillaceae and Acetobacteraceae), although they branched close to each other, no unique character common to them was identified, indicating that they are highly divergent. The exact branching position of the Xanthobacter was also not resolved in these cliques. In some cliques, it appeared as an outgroup of the Bradyrhizobiaceae (as shown by the dotted lines in Fig. 2), whereas in others it was placed in the middle of the Rhizobiaceae and Bradyrhizobiaceae families (as seen for the Aurantiomonas).
Phylogenomic analyses of alpha proteobacteria
Table 1 lists some characteristics of various α-proteobacterial genomes that have been sequenced. The genomes vary in size from less than 1 Mb for Neorickettsia sennetsu to more than 9.0 Mb for Bradyrhizobium japonicum. To identify proteins that are distinguishing features of various higher taxonomic groups within α-proteobacteria, systematic Blastp searches were performed on each ORF in the genomes of B. japonicum USDA 110, Brucella suis 1330, Caulobacter crescentus CB15, Gluconobacter oxydans 621H, Mesorhizobium loti MAFF303099, Nitrobacter winogradskyi Nb-255, Novosphingobium aromaticivorans DSM 12444, Rhodobacter sphaeroides 2.4.1, Silicibacter sp. TM1040, Rhodospirillum rubrum ATCC 11170 and Wolbachia (Drosophila melanogaster) endosymbiont (see Methods section). The genomes chosen covered all main taxonomic groups within the sequenced α-proteobacteria. These analyses have identified large numbers of proteins that are uniquely found in particular groups of α-proteobacteria. A brief description of these results is given below.
Proteins that are distinguishing features of all (or most) alpha proteobacteria
We previously described 6 proteins (viz. CC1365, CC1725, CC1887, CC2102, CC3292 and CC3319) that appeared distinctive characteristics of α-proteobacteria [17]. The α-proteobacterial specificity of these proteins in earlier work was only assessed by means of Blastp searches and it was not confirmed by PSI-Blast, as in the present work (see Methods). Further, since the earlier work, the number of sequenced α-proteobacteria and other genomes has more than doubled. Hence, it was important to confirm the α-proteobacteria specificity of these proteins. Our results reveal that four of these proteins viz. CC1365, CC2102, CC3292 and CC3319, are indeed specific for the α-proteobacteria as a whole, whereas for the remaining two proteins homologs showing significant similarities are also found in other bacteria. Of these four proteins, CC3292 is present in all sequenced α-proteobacteria including Candidatus Pelagibacter ubique, which is the smallest known free-living bacterium [2]. The protein CC2102 is only missing in P. ubique, while the other two proteins are only missing in 1–2 rickettsiae species (CC1365) and P. ubique (CC3319). These proteins, which are uniquely present in virtually all α-proteobacteria, provide distinguishing markers for this Class of bacteria (Fig. 3).

Summary of the phylogenomic analyses showing the species distribution of various α-proteobacteria-specific proteins and the suggested evolutionary stages where the genes for these proteins have likely evolved. The genes IDs for some proteins described in earlier work are indicated [17]. The information for all other proteins can be found in the indicated Tables or Additional files. A large numbers of conserved indels that are specific for different groups or clades within α-proteobacteria shown here have also been identified in our earlier work [16] (not shown here). The branching order of α-proteobacteria relative to other bacteria has been established in earlier work [32,58].
In earlier work, 9 proteins were identified that were present in nearly all α-proteobacteria, except the Rickettsiales [17]. These latter species are all intracellular bacteria that have lost many genes that are not required under these conditions [3, 34]. The Blastp and PSI-Blast reexamination of these proteins have confirmed that 7 of these proteins (CC0100, CC0520, CC1211, CC1886, CC2245, CC3010 and CC3470) exhibit the indicated specificity. Except for CC0520 and CC3470, the other five proteins are also found in P. ubique, providing evidence for its placement within α-proteobacteria [2]. These results also provide evidence that P. ubique is not specifically related to the Rickettsiales. The phylogenomic distributions of these genes/proteins can be explained by either their evolution after the divergence of the Rickettsiales (Fig. 3), or by gene loss from this lineage.
Proteins that are distinguishing features of the Rhizobiales species
The Rhizobiales species comprise more than 1/3rd of the sequenced α-proteobacterial genomes (see Table 1). This order includes a wide assortment of species many of which interact with the eukaryotic organisms to produce diverse effects. This group includes various rhizobia (Rhizobium, Mesorhizobium, Sinorhizobium) and Bradyrhizobia species that induce root nodules in plants and live symbiotically within them to enable nitrogen fixation [4–6, 35, 36]. Another Rhizobiaceae species, A. tumefaciens, induces Crown gall disease (tumors) in plants [37]. Bartonella and Brucella species are intracellular pathogens responsible for a number of diseases in human and animals including trench fever and brucellosis [7, 38–40]. Other members of this order exhibit enormous versatility in terms of their metabolic capabilities and life styles [1, 41, 42]. Earlier studies identified six proteins that appeared distinctive characteristics of the Rhizobiales species [17]. Reexamination of these proteins by the more stringent criteria used in the present work confirmed that three of these proteins (BQ00720, BQ07670 and BQ12030) are indeed specific for the Rhizobiales and they are present in virtually all sequenced species from this large order. In addition to the Rhizobiales, these proteins are also present in Stappia aggregata, which is presently grouped with the Rhodobacterales [12], but was originally known as a strain of Agrobacterium [43].
New blast searches on the genomes of B. japonicum, B. suis, M. loti and N. winogradskyi have identified large number of other proteins that are specific for different subgroups within the Rhizobiales order. Seven proteins listed in Table 2A are uniquely present in most of the sequenced species belonging to the Rhizobiaceae (Rhizobium, Sinorhizobium, Agrobacterium), Brucellaceae, Phyllobacteriaceae (Mesorhizobium) and Aurantimonadaceae families. The absence of many of these proteins in Bartonella species is probably due to gene loss [7, 34]. These groups form a well-defined clade with high bootstrap scores in the NJ, MP and ML trees (see Fig. 1) and the genes for them likely evolved in a common ancestor of these genera/families (Fig. 3). Another 9 proteins (Table 2B) are uniquely present in most of the above species except Aurantimonas, which forms outgroup of the Rhizobiaceae, Brucellaceae, Bartonellaceae and Phyllobacteriaceae families (Figs. 1 and 2) [29]. Thus, the genes for these proteins have evolved after the branching of Aurantimonadaceae (Fig. 3). For six of the proteins in Tables 2A and 2B (marked with *), homologs with low E values are also found in S. aggregata, providing further evidence for its relatedness to the Rhizobiaceae family. Nine other proteins in Table 2C are found in most Rhizobiaceae species and M. loti, but they are missing in Bartonella and Brucella species. Although these proteins suggest a closer relationship between the Rhizobiaceae and Phyllobacteriaceae families, it is more likely that their genes have been lost from the Bartonella and Brucella species [34], which are intracellular bacteria. Additionally, some proteins were only found in either Mesorhizobium and Rhizobium, or Mesorhizobium and Sinorhizobium (Table 2D). These analyses have also identified 43 proteins that are uniquely present in all four sequenced Brucella species and many other proteins that are present in either three or two of the sequenced Brucella species (see Additional file 1).
The analyses of proteins in the genomes of B. japonicum and N. winogradskyi have identified 12 proteins that are uniquely present in either all (or most) of the sequenced Bradyrhizobiaceae species as well as X. autotrophicus (Table 3A). The species from these two families form a strongly supported clade in the phylogenetic tree (Fig. 1)[29]. Sixty-two additional proteins in Table 3B are uniquely present in various species belonging to the Bradyrhizobiaceae family (i.e. Bradyrhizobium, Nitrobacter, Rhodopseudomonas). Many other proteins (see Additional file 2) are only found in two of the three Bradyrhizobiaceae genera and their distributions can result from gene losses, lateral gene transfers (LGTs), or other mechanisms.
Proteins that are distinguishing features of the Rhodobacterales species
The order Rhodobacterales is a heterogeneous lineage of bacteria that exhibit much diversity in terms of their metabolism and cell division cycles [1, 13]. This group includes many photosynthetic bacteria that are capable of CO2 as well as nitrogen fixation and also many chemoorganotrophs that can metabolize various sulfur-containing compounds [44, 45]. A number of budding, stalk forming and prosthecate bacteria also belong to this group [46]. In addition to many completely sequenced genomes (see Table 1), information for several other species belonging to this order (e.g. Sulfitibacter, Oceanicola, Loktanella, Jannaschia, Dinoroseobacter, Roseovarius and Sagittula) is available in the NCBI database. To identify proteins that are specific for the Rhodobacterales, phylogenomic analyses were carried out on various ORFs in the genomes of R. sphaeroides 2.4.1 and Silicibacter sp. TM1040.
These studies have identified 29 proteins that are present in all-available Rhodobacterales species (Table 4A), but these proteins as well as those listed in Table 4B and 4C are not found in H. neptunium, Oceanicaulis alexandrii, Maricaulis maris or Stappia aggregata. These latter species are presently grouped with the Rhodobacterales [12], however, the absence of various Rhodobacterales-specific proteins in them and phylogenetic studies indicate that the placement of these species within this order is incorrect and needs be revised. In phylogenetic trees based on concatenated sequences for many proteins, O. alexandrii and M. maris consistently branched with the Caulobacter rather than the well-defined clade of Rhodobacterales (Figs. 1 and 2) [29]. The studies by Badger et al. [47] also provide strong evidence for the grouping of H. neptunium with the Caulobacterales.
Six additional proteins in Table 4B are present in most of the Rhodobacterales species, but they are missing in R. sphaeroides and P. denitrificans, which form a distinct clade that appears as the outgroup of other Rhodobacterales species (Fig. 1)[29]. Thus, the genes for these proteins have likely evolved after the branching of these two genera. Thirteen additional proteins, which are only found in Silicibacter and Roseobacter genera (Table 4C) support a close relationship among them, as seen in phylogenetic trees (Fig. 1).
Of the proteins that are specific for Rhodobacterales, two of them (YP_613058 and YP_613059 in Table 4A, corresponding to proteins ABK27256 and ABK27255 in R. capsulatus genome) were previously identified as part of a complex referred to as gene transfer agent [48], based on similarity to certain virus-like elements. Another protein in the same category (viz. ABK27253) is specific for the Rhodobacter genus. The significance of these results is presently unclear.
Proteins that are distinctive characteristics of the Caulobacterales
The order Caulobacterales is comprised of a single family with only four genera [12, 49]. These chemoorganotrophic bacteria are commonly found in marine aerobic environments and they are distinguished by their ability to form stalked cells and unusual cell division cycle [49, 50]. The complete genome of only C. crescentus, which is the best-studied organism from this group, is presently available [51]. However, as discussed above, a number of other species which are presently classified as Rhodobacterales viz. O. alexandrii, M. maris, H. neptunium and also Parvularcula bermudensis, consistently branch with C. crescentus in different phylogenetic trees (Figs. 1 and 2) [29, 47]. Thus, phylogenomic analysis of the ORFs from C. crescentus genome was of much interest.
These analyses have identified 2 proteins (CC0486 and CC2480), which are uniquely present in this species as well as O. alexandrii, M. maris, H. neptunium and P. bermudensis (Table 5) One additional protein, CC2764 is present in all of these species except H. neptunium. The remaining eight proteins in Table 5 are only found in C. crescentus, O. alexandrii and M. maris, indicating that the latter two species are more closely related to C. crescentus in comparison to either H. neptunium or P. bermudensis. These results are strongly supported by the branching patterns of these species in phylogenetic trees (Figs. 1 and 2) [29, 47]. Previously, Badger et al. [46] have reported identification of 62 proteins that were only found in C. crescentus and H. neptunium. However, the blast threshold used in this study to infer the absence of these proteins in other species was very high i.e. 1e-10. By the criteria used in the present work (see Methods), none of these 62 proteins was found to be unique to these two species.
Proteins that are distinguishing characteristics of the Sphingomonadales
The species belonging to the order Sphingomonadales are present in both terrestrial and aquatic environments [52]. A distinguishing characteristic of many species from this group is the presence of glycosphingolipids in their cell envelope rather than lipopolysaccharides [52, 53]. Several species from this group (e.g. N. aromaticivorans) can degrade a wide variety of aromatic hydrocarbons [52], whereas others such as Zymomonas mobilis, can highly efficiently ferment sugar to ethanol [53], making them of much interest and importance from biotechnological standpoints. This group also includes phototrophic organisms (e.g. Erythrobacter litoralis), which contain bacteriochlorophyll a and can derive significant fraction of their metabolic energy via anaerobic photosynthesis [54]. The complete genomes of 5 species from this order are now available (see Table 1). In addition, large numbers of sequences for Sphingomonas sp. SKA58, are also available in the NCBI database. Blast searches on the ORFs in the genome of N. aromaticivorans have identified 16 proteins (Table 6A) that are uniquely present in all 6 of the Sphingomonadales species for which information is available. Thirteen additional proteins (Table 6B) are present in all other Sphingomonadales species, except Z. mobilis, which is the deepest branching species within this group (Fig. 1). The genes for these proteins likely evolved after the branching of Z. mobilis. Many other proteins are present in only 3 or 4 of these species (viz. N. aromaticivorans, E. litoralis, S. alaskensis, S. wittichiii and Sphingomonas sp. SKA58) (see Additional file 3) and their phylogenomic distribution can result from a variety of mechanisms including shared ancestry, gene loss and LGTs among these species.
We have also identified a 4 aa insert in a highly conserved region of Gyrase B that is mainly specific for the species from this order (Fig. 4). This indel is present in all available Sphingomonadales species, but it is not found in most other alpha proteobacteria or other bacteria (results not shown). Besides Sphingomonadales, a similar size indel is also present in three other species (viz. C. leidyia, R. blasticus, and Pesudorhodobacter incheonensis). Because other Rhodobacterales or Caulobacter species lack this insert, the presence of this indel in these three species could be either due to LGTs or possibly due to taxonomic misclassification of these species.

Partial sequence alignments of DNA Gyrase B showing a 4 aa insert that is mainly specific for the Sphingomonadales species. A 4–5 aa insert present in some other α-proteobacteria could be due to either LGTs or taxonomic anomalies. The dashes (-) denote identity with the amino acid on the top line. Sequence information for other groups of bacteria (which do not contain this insert) is not shown.
Proteins and indels that are specific for the Rhodospirillales species
The order Rhodospirillales is comprised of diverse species including some photosynthetic and magnetotactic bacteria, some acidophiles as well as other bacteria commonly associated with flowers, fruits and fermented beverages that are involved in the partial oxidation of carbohydrates and alcohols [55]. The order is made up of two main families, Rhodospirillaceae and Acetobacteraceae [12, 55]. The complete genomes of four species, two from each family, Rhodospirillaceae (Magnetospirillum magnticum, R. rubrum) and Acetobacteraceae (Acidiphilium cryptum and G. oxydans), are available (Table 1) [56, 57]. Phylogenomic analyses of various ORFs in the genomes of G. oxydans and R. rubrum have led to identification of one proteins, GOX0963, which is uniquely found in all of these species (Table 7A). Three other proteins in this Table are present in at least 3 of the 4 species from this order. This table also lists 14 proteins each that are distinctive characteristics of either the Acetobacteraceae (Table 7B) or the Rhodospirillaceae (Table 7C) families, providing molecular markers for these families. We have also identified a 25 aa insert in a conserved region of the RNA polymerase beta subunit (RpoB) that is unique to various sequenced Rhodospirillales species, but not found in any other bacteria (Fig. 5).

Partial sequence alignments of RNA polymerase β subunit (RpoB) showing a large insert (boxed) that is a distinctive characteristic of various Rhodospirillales species and not found in any other bacteria. There are two homologs of RpoB in Magentospirillum (Mag.) magneticum and only one of these contains the insert. The dashes (-) denote identity with the amino acid on the top line.
Proteins that are specific for the Rickettsiales
The Rickettsiales species are intracellular pathogens responsible for a number of diseases in humans and other animals [3, 34]. This order is comprised of three families, Rickettsiaceae, Anaplasmataceae and Holosporaceae. All of the sequenced genomes are from the first two families. Phylogenomic analysis of various ORFs from the genome of Wolbachia (D. melanogaster) endosymbiont has identified 3 proteins viz. WD0161, WD0715 and WD0771 (Table 8A) that are specific for the entire Rickettsiales order. Five other proteins in Table 8B (viz. WD0083, WD0157, WD0821, WD0827 and WD0863) are present in all sequenced Ehrlichia, Anaplasma, Wolbachia and Neorickettsia species (belonging to the Anaplasmataceae family), but they are not found in any of the Rickettsiaceae or other bacteria. Ten additional proteins (Table 8C) are uniquely present in the Ehrlichia, Anaplasma and Wolbachia species, but are absent in Neorickettsia. In view of the deep branching of Neorickettsia in comparison to these other genera (Fig. 1), the genes for these proteins have likely evolved after the branching of Neorickettsia. In earlier work, a number of proteins that appeared specific for the Rickettsiaceae family were also identified [17]. Additional Blastp and PSI-Blast searches on these proteins confirm that three of these proteins viz. RP030, RP187 are RP192, are indeed specific for the Rickettsiaceae family. Of these, RP030 is also found in Orientia tsutsugamushi.
Conclusion
In this work, we have used a combined phylogenetic and phylogenomic approach to examine the evolutionary relationships among α-proteobacteria. Our analyses have identified large numbers of genes/proteins that are uniquely found in α-proteobacteria at various phylogenetic depths (Fig. 3). These include several proteins that are distinctive characteristics of all α-proteobacteria, as well as many proteins that constitute the unique repertoires of either all of the main orders of α-proteobacteria (viz. Rhizobiales, Rhodobacterales, Rhodospirillales, Rickettsiales, Sphingomonadales and Caulobacterales) or its different families (viz. Rickettsiaceae, Anaplasmataceae, Rhodospirillaceae, Acetobacteraceae, Bradyrhizobiaceae, Brucellaceae and Bartonellaceae). In addition, numerous other α-proteobacteria-specific proteins are present at different phylogenetic depths and they provide important information regarding the evolution of these bacteria. This work also describes two novel conserved indels in important housekeeping genes (viz. Gyrase B and RpoB) that are distinctive characteristics of the Sphingomonadales and Rhodospirillales orders, respectively. These indels are in addition to many other α-proteobacteria-specific indels that have been described in earlier work [16, 58].
Based upon these α-proteobacteria-specific proteins and conserved indels, it is now possible to define nearly all of the higher taxonomic groups (i.e. most orders and many families) within α-proteobacteria in clear and definitive molecular terms based upon multiple characteristics (Fig. 3). The species distribution profiles of these α-proteobacteria-specific proteins and indels also provide important information regarding their branching order and interrelationships, which are highly concordant with each other (c.f. Fig. 26 in ref. [16] with Fig. 3 in this work). Importantly, the relationships that emerge from these phylogenomic analyses (Fig. 3) are in excellent agreement with the branching patterns of these species in different phylogenetic trees (Figs. 1 and 2) [29], giving high degree of confidence in the derived inferences. It should be noted that both in our work (Fig. 1) and that by Williams et al. [29], when analyses were performed using the traditional phylogenetic methods (viz. NJ, MP or ML analyses), the branching of Caulobacterales with respect to Rhizobiales and Rhodobacterales was not resolved. In contrast, using the character compatibility approach, the Caulobacterales and related species were found to consistently branch in between the Rhizobiales and the Rhodobacterales (Fig. 2). Previously, this approach has also proven useful in clarifying the phylogenetic placement of Salinibacter ruber, which was not resolved by other methods [33, 59].
The phylogenetic studies presented here reveal that a number of species belonging to the Hyphomonadaceae family (viz. M. maris, O. alexandrii and H. neptunium), for which sequence information is available and that are presently grouped with the Rhodobacterales, branch reliably with the Caulobacterales (Figs 1 and 2)[29, 47]. The same is also true for P. bermudensis, which is the only species from Parvularculales order, for which sequence information is available. The grouping of these species with the Caulobacterales is independently strongly supported by phylogenomic studies, where a number of proteins that are unique for C. crescentus were present in these species and at the same time many proteins that are distinctive characteristics of the Rhodobacterales were absent in them. These results make a strong case for the transfer of these Hyphomonadaceae species and also P. bermudensis to an expanded Caulobacterales order [29, 47]. Another taxonomic anomaly identified by the present study concerns the phylogenetic position of Stappia aggregata. This species was originally identified as an Agrobacterium-related species, but later transferred to the Rhodobacterales order [43]. However, the shared presence of many Rhizobiales -specific proteins by S. aggregata and the absence of various Rhodobacterales-specific proteins in it, strongly suggest that it should be regrouped with the Rhizobiaceae-Phyllobacteriacea species.
The overwhelming majority of the identified α-proteobacteria-specific proteins do not have a homolog showing significant similarity in any other bacteria. The group-specificities of these proteins indicate that their genes have evolved in a common ancestor of these particular groups or clades of α-proteobacteria (Fig. 3). The clade specificities of these proteins also provide evidence that following their evolution, their genes have been transmitted primarily in a vertical manner, and that non-specific mechanisms such as LGTs have not played a significant role in their species distribution. Similar inferences have been reached in earlier studies for proteins that are specific for other higher taxa of bacteria [20, 22–24, 33, 60]. Most of the α-proteobacteria-specific proteins identified in the present work are of unknown function. A number of these proteins are present in the genomes in clusters of two or three, suggesting that they may form functional units and could be involved in related functions [18, 26, 48, 61]. The retention of these α-proteobacteria-specific proteins and conserved indels by the indicated clades of α-proteobacteria over long evolutionary periods strongly suggests that they serve essential functions in these groups of bacteria. Hence, studies on their cellular functions may lead to the discovery of novel biochemical and physiological characteristics that are distinctive characteristics of either all α-proteobacteria or their particular subgroups. Lastly, the primary sequences of many of these genes/proteins are highly conserved and they provide novel means for the identification and characterization of these bacteria by PCR-based and immunological methods.
Methods
Identification of proteins that are specific for alpha proteobacteria
The Blastp searches were carried out on each ORF in the genomes of B. japonicum USDA 110, B. suis 1330, C. crescentus CB15, G. oxydans 621H, M. loti MAFF303099, N. winogradskyi Nb-255, N. aromaticivorans DSM 12444, R. sphaeroides 2.4.1, Silicibacter sp. TM1040, R. rubrum ATCC 11170 and Wolbachia (D. melanogaster) endosymbiont to identify proteins that are uniquely present in α-proteobacteria species at different phylogenetic depths. The blast searches were performed against all organisms (i.e. non-redundant (nr) database) using the default parameters, without the low complexity filter [62]. The proteins that were of interest were those where either all significant hits were from the indicated groups (or orders) of α-proteobacteria, or which involved a large increase in E values from the last hit belonging to a particular group to the first hit from any other group and the E values for the latter hits were > 1e -4, indicating weak similarity that could occur by chance. However, higher E values were often considered significant for smaller proteins as the magnitude of the E value depends upon the length of the query sequence [62]. All promising proteins were further analyzed using the position-specific iterated (PSI)-Blast program [62]. In this study, we have also retained a few proteins where 1 or 2 isolated species from other groups had acceptable E values, as they provide possible cases of LGTs. For all of the proteins that are specific for α-proteobacteria, their protein ID's, accession numbers and any information regarding cellular functions (such as COG number or presence of any conserved domain) were tabulated and are presented. In describing various proteins in the text, "bll, bsl, blr", "BQ", "BR or BRA" "CC," "GOX" "ml", "Nwi", "Saro", "RSP", "TM1040", "Rru" and "WD" indicate the identification numbers of the proteins in the genomes of B. japonicum, Bartonella quintana, B. suis, C. crescentus, G. oxydans, M. loti, N. winogradskyi Nb-255, N. aromaticivorans, R. sphaeroides 2.4.1, Silicibacter sp. TM1040, R. rubrum and Wolbachia (Dros.) endosymbiont, respectively.
Phylogenetic analysis
The amino acid sequences for 12 conserved proteins (viz. RNA polymerase β and β' subunits, alanyl-tRNA synthetase, phenyalanyl-tRNA synthetase, arginyl-tRNA synthetase, protein synthesis elongation factors EF-Tu and EF-G, RecA, Gyrase A, Gyrase B, Hsp60 and Hsp70) for different species were downloaded from the NCBI database and aligned using the CLUSTAL × program [63]. In addition to the sequences for 50 α-proteobacteria species, sequences for two deep-branching species viz. Helicobacter pylori and Campylobacter jejuni [58], were also included for rooting purposes. The sequence alignments for all 12 proteins were concatenated into a single large file and poorly aligned regions were removed using the Gblocks 0.91b program [64]. The final sequence alignment that was used for phylogenetic analyses contained 7652 aligned positions. A neighbour-joining tree based on this alignment was constructed based on Kimura's two parameter model distances using the TREECON program [65]. Maximum-likelihood and MP trees were computed using the WAG+F model plus a gamma distribution with four categories using the TREE-PUZZLE [66] and Mega 3.1 program [67], respectively. All trees were bootstrapped 100 times [68], unless otherwise indicated.
The character compatibility analysis was performed on a concatenated sequences for the above 12 proteins for 25 α-proteobacteria species representing all its main orders plus two outgroup species (i.e. H. pylori and C. jejuni) [32]. Using the program "DUALSITE" [32], all sites in the sequence alignments where only two amino acid states were found, with each state present in at least two species, were selected. All columns where any gap was present in any of the species were omitted. The useful two state sites were converted into a binary file of "0, 1" characters using the DUALSITE program and this file was used for compatibility analysis [32]. The compatibility analysis was carried out using the CLIQUE program from the PHYLIP (ver. 3.5c) program package [68] to identify the largest clique(s) of compatible characters. The cliques were drawn and the numbers of characters that distinguished different nodes were indicated.
Identification of conserved indels specific for α-proteobacteria subgroups
Multiple sequence alignments for various proteins constructed in this work were visually inspected to search for any indels in a conserved region that was unique to particular subgroups or orders of α-proteobacteria. The group-specificity of any indel was evaluated by carrying out blast searches on a short segment of the sequence (between 80–120 aa) containing the indel and flanking conserved regions against the non-redundant database. The sequence information for various α-proteobacteria was compiled into signature files that are presented. Sequence information for all other groups of bacteria, which lack these inserts, is not shown.
References
Kersters K, Devos P, Gillis M, Swings J, Vandamme P, Stackebrandt E: Introduction to the Proteobacteria. The Prokaryotes: A Handbook on the Biology of Bacteria. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH and Stackebrandt E. 2006, New York, Springer, 3-37. 3rd edition, Release 3.12
Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappe MS, Short JM, Carrington JC, Mathur EJ: Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005, 309: 1242-1245.
Fredricks DN: Introduction to the Rickettsiales and other intracellular prokaryotes. The Prokaryotes: A Handbook on the Biology of Bacteria. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH and Stackebrandt E. 2006, New York, Springer, 457-466. 3rd edition
Skorpil P, Broughton WJ: Molecular interactions between Rhizobium and legumes. Prog Mol Subcell Biol. 2006, 41: 143-164.
Kaneko T, Nakamura Y, Sato S, Minamisawa K, Uchiumi T, Sasamoto S, Watanabe A, Idesawa K, Iriguchi M, Kawashima K, Kohara M, Matsumoto M, Shimpo S, Tsuruoka H, Wada T, Yamada M, Tabata S: Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 2002, 9: 189-197.
Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S: Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000, 7: 331-338.
Alsmark CM, Frank AC, Karlberg EO, Legault BA, Ardell DH, Canback B, Eriksson AS, Naslund AK, Handley SA, Huvet M, La Scola B, Holmberg M, Andersson SG: The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc Natl Acad Sci U S A. 2004, 101: 9716-9721.
Andersson SG, Alsmark C, Canback B, Davids W, Frank C, Karlberg O, Klasson L, Antoine-Legault B, Mira A, Tamas I: Comparative genomics of microbial pathogens and symbionts. Bioinformatics. 2002, 18 Suppl 2: S17-
Moreno E, Moriyon I: The Genus Brucella. The Prokaryotes: A Handbook on the Biology of Bacteria. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH and Stackebrandt E. 2006, New York, Springer, 315-456. 3rd edition
Gray MW, Burger G, Lang BF: Mitochondrial evolution. Science. 1999, 283: 1476-1481.
Andersson SG, Karlberg O, Canbäck B, Kurland CG: On the origin of mitochondria: a genomics perspective. Philos Trans R Soc Lond B Biol Sci. 2003, 358 (1429): 165-177.
Garrity GM, Bell JA, Lilburn TG: The Revised Road Map to the Manual. Bergey's Manual of Systematic Bacteriology, Volume 2, Part A, Introductory Essays. Edited by: Brenner DJ, Krieg NR and Staley JT. 2005, New York, Springer, 159-220.
Bergey's Manual of Systematic Bacteriology. Edited by: Brenner DJ, Krieg NR and Staley JT. 2005, New York, Springer, 2, Part C, The Alpha-, Beta-, Delta-, and Epsilonproteobacteria: 2nd Edition
Woese CR, Stackebrandt E, Weisburg WG, Paster BJ, Madigan MT, Fowler CMR, Hahn CM, Blanz P, Gupta R, Nealson KH, Fox GE: The phylogeny of purple bacteria: the alpha subdivision. System Appl Microbiol. 1984, 5: 315-326.
Ludwig W, Klenk HP: Overview: A phylogenetic backbone and taxonomic framework for prokaryotic systamatics. Bergey's Manual of Systematic Bacteriology. Edited by: Boone DR and Castenholz RW. 2001, Berlin, Springer-Verlag, 49-65. 2nd
Gupta RS: Protein signatures distinctive of alpha proteobacteria and its subgroups and a model for alpha-proteobacterial evolution. Crit Rev Microbiol. 2005, 31 (2): 101-135.
Kainth P, Gupta RS: Signature Proteins that are Distinctive of Alpha Proteobacteria. BMC Genomics. 2005, 6: 94-
Siew N, Fischer D: Analysis of singleton ORFans in fully sequenced microbial genomes. Proteins. 2003, 53: 241-251.
Gao B, Gupta RS: Phylogenomic analysis of proteins that are distinctive of Archaea and its main subgroups and the origin of methanogenesis. BMC Genomics. 2007, 8: 86-
Gupta RS: Molecular signatures (unique proteins and conserved Indels) that are specific for the epsilon proteobacteria (Campylobacterales). BMC Genomics. 2006, 7: 167-
Gupta RS, Griffiths E: Chlamydiae-specific proteins and indels: novel tools for studies. Trends Microbiol. 2006, 14: 527-535.
Daubin V, Gouy M, Perriere G: A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 2002, 12: 1080-1090.
Lerat E, Daubin V, Ochman H, Moran NA: Evolutionary Origins of Genomic Repertoires in Bacteria. PLoS Biol. 2005, 3: e130-
Santos SR, Ochman H: Identification and phylogenetic sorting of bacterial lineages with universally conserved genes and proteins. Environ Microbiol. 2004, 6: 754-759.
Kolker E, Makarova KS, Shabalina S, Picone AF, Purvine S, Holzman T, Cherny T, Armbruster D, Munson RS, Kolesov G, Frishman D, Galperin MY: Identification and functional analysis of 'hypothetical' genes expressed in Haemophilus influenzae. Nucleic Acids Res. 2004, 32: 2353-2361.
Galperin MY, Koonin EV: 'Conserved hypothetical' proteins: prioritization of targets for experimental study. Nucleic Acids Res. 2004, 32: 5452-5463.
Felsenstein J: Inferring Phylogenies. 2004, Sunderland, Mass., Sinauer Associates, Inc.
Olsen GJ, Woese CR, Overbeek R: The winds of (evolutionary) change: breathing new life into microbiology. J Bacteriol. 1994, 176: 1-6.
Williams KP, Sobral BW, Dickerman AW: A robust species tree for the alphaproteobacteria. J Bacteriol. 2007, 189 (13): 4578-4586.
Le Quesne WJ: The uniquely evolved character concept and its cladistic application. Systematic Zoology. 1975, 23: 513-517.
Meacham CA, Estabrook GF: Compatibility methods in Systematics. Ann Rev Ecol Syst. 1985, 16: 431-446.
Gupta RS, Sneath PHA: Application of the character compatibility approach to generalized molecular sequence data: Branching order of the proteobacterial subdivisions. J Mol Evol. 2007, 64: 90-100.
Gupta RS, Lorenzini E: Phylogeny and molecular signatures (conserved proteins and indels) that are specific for the Bacteroidetes and Chlorobi species. BMC Evol Biol. 2007, 7: 71-
Sallstrom B, Andersson SG: Genome reduction in the alpha-Proteobacteria. Curr Opin Microbiol. 2005, 8: 579-585.
Gonzalez V, Santamaria RI, Bustos P, Hernandez-Gonzalez I, Medrano-Soto A, Moreno-Hagelsieb G, Janga SC, Ramirez MA, Jimenez-Jacinto V, Collado-Vides J, Davila G: The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc Natl Acad Sci U S A. 2006, 103: 3834-3839.
Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, Barloy-Hubler F, Barnett MJ, Becker A, Boistard P, Bothe G, Boutry M, Bowser L, Buhrmester J, Cadieu E, Capela D, Chain P, Cowie A, Davis RW, Dreano S, Federspiel NA, Fisher RF, Gloux S, Godrie T, Goffeau A, Golding B, Gouzy J, Gurjal M, Hernandez-Lucas I, Hong A, Huizar L, Hyman RW, Jones T, Kahn D, Kahn ML, Kalman S, Keating DH, Kiss E, Komp C, Lelaure V, Masuy D, Palm C, Peck MC, Pohl TM, Portetelle D, Purnelle B, Ramsperger U, Surzycki R, Thebault P, Vandenbol M, Vorholter FJ, Weidner S, Wells DH, Wong K, Yeh KC, Batut J: The composite genome of the legume symbiont Sinorhizobium meliloti. Science. 2001, 293: 668-672.
Wood DW, Setubal JC, Kaul R, Monks DE, Kitajima JP, Okura VK, Zhou Y, Chen L, Wood GE, Almeida NF, Woo L, Chen Y, Paulsen IT, Eisen JA, Karp PD, Bovee D, Chapman P, Clendenning J, Deatherage G, Gillet W, Grant C, Kutyavin T, Levy R, Li MJ, McClelland E, Palmieri A, Raymond C, Rouse G, Saenphimmachak C, Wu Z, Romero P, Gordon D, Zhang S, Yoo H, Tao Y, Biddle P, Jung M, Krespan W, Perry M, Gordon-Kamm B, Liao L, Kim S, Hendrick C, Zhao ZY, Dolan M, Chumley F, Tingey SV, Tomb JF, Gordon MP, Olson MV, Nester EW: The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science. 2001, 294: 2317-2323.
DelVecchio VG, Kapatral V, Redkar RJ, Patra G, Mujer C, Los T, Ivanova N, Anderson I, Bhattacharyya A, Lykidis A, Reznik G, Jablonski L, Larsen N, D'Souza M, Bernal A, Mazur M, Goltsman E, Selkov E, Elzer PH, Hagius S, O'Callaghan D, Letesson JJ, Haselkorn R, Kyrpides N: The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc Natl Acad Sci USA. 2002, 99: 443-448.
Chain PS, Comerci DJ, Tolmasky ME, Larimer FW, Malfatti SA, Vergez LM, Aguero F, Land ML, Ugalde RA, Garcia E: Whole-genome analyses of speciation events in pathogenic Brucellae. Infect Immun. 2005, 73: 8353-8361.
Paulsen IT, Seshadri R, Nelson KE, Eisen JA, Heidelberg JF, Read TD, Dodson RJ, Umayam L, Brinkac LM, Beanan MJ, Daugherty SC, DeBoy RT, Durkin AS, Kolonay JF, Madupu R, Nelson WC, Ayodeji B, Kraul M, Shetty J, Malek J, Van Aken SE, Riedmuller S, Tettelin H, Gill SR, White O, Salzberg SL, Hoover DL, Lindler LE, Halling SM, Boyle SM, Fraser CM: The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci U S A. 2002, 99: 13148-13153.
Larimer FW, Chain P, Hauser L, Lamerdin J, Malfatti S, Do L, Land ML, Pelletier DA, Beatty JT, Lang AS, Tabita FR, Gibson JL, Hanson TE, Bobst C, Torres JL, Peres C, Harrison FH, Gibson J, Harwood CS: Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotechnol. 2004, 22: 55-61.
Starkenburg SR, Chain PS, Sayavedra-Soto LA, Hauser L, Land ML, Larimer FW, Malfatti SA, Klotz MG, Bottomley PJ, Arp DJ, Hickey WJ: Genome sequence of the chemolithoautotrophic nitrite-oxidizing bacterium Nitrobacter winogradskyi Nb-255. Appl Environ Microbiol. 2006, 72: 2050-2063.
Uchino Y, Hirata A, Yokota A, Sugiyama J: Reclassification of marine Agrobacterium species: Proposals of Stappia stellulata gen. nov., comb. nov., Stappia aggregata sp. nov., nom. rev., Ruegeria atlantica gen. nov., comb. nov., Ruegeria gelatinovora comb. nov., Ruegeria algicola comb. nov., and Ahrensia kieliense gen. nov., sp. nov., nom. rev. J Gen Appl Microbiol. 1998, 44: 201-210.
Swingley WD, Sadekar S, Mastrian SD, Matthies HJ, Hao J, Ramos H, Acharya CR, Conrad AL, Taylor HL, Dejesa LC, Shah MK, O'huallachain ME, Lince MT, Blankenship RE, Beatty JT, Touchman JW: The complete genome sequence of Roseobacter denitrificans reveals a mixotrophic rather than photosynthetic metabolism. J Bacteriol. 2007, 189: 683-690.
Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP, Henriksen JR, King GM, Belas R, Fuqua C, Brinkac L, Lewis M, Johri S, Weaver B, Pai G, Eisen JA, Rahe E, Sheldon WM, Ye W, Miller TR, Carlton J, Rasko DA, Paulsen IT, Ren Q, Daugherty SC, DeBoy RT, Dodson RJ, Durkin AS, Madupu R, Nelson WC, Sullivan SA, Rosovitz MJ, Haft DH, Selengut J, Ward N: Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature. 2004, 432: 910-913.
Badger JH, Hoover TR, Brun YV, Weiner RM, Laub MT, Alexandre G, Mrazek J, Ren Q, Paulsen IT, Nelson KE, Khouri HM, Radune D, Sosa J, Dodson RJ, Sullivan SA, Rosovitz MJ, Madupu R, Brinkac LM, Durkin AS, Daugherty SC, Kothari SP, Giglio MG, Zhou L, Haft DH, Selengut JD, Davidsen TM, Yang Q, Zafar N, Ward NL: Comparative genomic evidence for a close relationship between the dimorphic prosthecate bacteria Hyphomonas neptunium and Caulobacter crescentus. J Bacteriol. 2006, 188: 6841-6850.
Badger JH, Eisen JA, Ward NL: Genomic analysis of Hyphomonas neptunium contradicts 16S rRNA gene-based phylogenetic analysis: implications for the taxonomy of the orders 'Rhodobacterales' and Caulobacterales. Int J Syst Evol Microbiol. 2005, 55: 1021-1026.
Lang AS, Beatty JT: Importance of widespread gene transfer agent genes in alpha proteobacteria. Trends Microbiol. 2007, 15: 54-62.
Poindexter JS: Dimorphic prosthecate bacteria: The genera Caulobacter, Asticcacaulis, Hyphomicrobium, Pedomicrobium, Hyphomonas and Thiodendron. The Prokaryotes: A Handbook on the Biology of Bacteria. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH and Stackebrandt E. 2006, New York, Springer, 72-90.
Brun YV, Marczynski G, Shapiro L: The expression of asymmetry during Caulobacter cell differentiation. Annu Rev Biochem. 1994, 63: 419-450.
Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen J, Heidelberg JF, Alley MR, Ohta N, Maddock JR, Potocka I, Nelson WC, Newton A, Stephens C, Phadke ND, Ely B, DeBoy RT, Dodson RJ, Durkin AS, Gwinn ML, Haft DH, Kolonay JF, Smit J, Craven MB, Khouri H, Shetty J, Berry K, Utterback T, Tran K, Wolf A, Vamathevan J, Ermolaeva M, White O, Salzberg SL, Venter JC, Shapiro L, Fraser CM: Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci U S A. 2001, 98: 4136-4141.
Yabuuchi E, Kosako Y: Order IV. Sphingomonadalesord. nov. Bergey's Manual of Systematic Bacteriology. Edited by: Brenner DJ, Krieg NR and Staley JT. 2005, New York, Springer, Volume Two, Part C, The Alpha-, Beta-, Delta-, and Epsilonproteobacteria: 230-258. 2nd edition
Seo JS, Chong H, Park HS, Yoon KO, Jung C, Kim JJ, Hong JH, Kim H, Kim JH, Kil JI, Park CJ, Oh HM, Lee JS, Jin SJ, Um HW, Lee HJ, Oh SJ, Kim JY, Kang HL, Lee SY, Lee KJ, Kang HS: The genome sequence of the ethanologenic bacterium Zymomonas mobilis ZM4. Nat Biotechnol. 2005, 23: 63-68.
Yurkov VV, Beatty JT: Aerobic anoxygenic phototrophic bacteria. Microbiol Mol Biol Rev. 1998, 62: 695-724.
Kersters K, Lisdiyanti P, Komagata K, Swings J: The Family Acetobacteraceae: The Genera Acetobacter, Acidomonas, Asaia, Gluconoacetobacter, Gluconobacter and Kozakia. The Prokaryotes: A Handbook on the Biology of Bacteria. Edited by: Dworkin M, Falkow S, Rosenberg E, Schleifer KH and Stackebrandt E. 2006, New York, Springer, 163-200. 3rd edition
Prust C, Hoffmeister M, Liesegang H, Wiezer A, Fricke WF, Ehrenreich A, Gottschalk G, Deppenmeier U: Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat Biotechnol. 2005, 23: 195-200.
Matsunaga T, Okamura Y, Fukuda Y, Wahyudi AT, Murase Y, Takeyama H: Complete Genome Sequence of the Facultative Anaerobic Magnetotactic Bacterium Magnetospirillum sp. strain AMB-1. DNA Res. 2005, 12: 157-166.
Gupta RS: The phylogeny of Proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol Rev. 2000, 24: 367-402.
Soria-Carrasco V, Valens-Vadell M, Pena A, Anton J, Amann R, Castresana J, Rossello-Mora R: Phylogenetic position of Salinibacter ruber based on concatenated protein alignments. Syst Appl Microbiol. 2007, 30: 171-179.
Gao B, Parmanathan R, Gupta RS: Signature proteins that are distinctive characteristics of Actinobacteria and their subgroups. Antonie van Leeuwenhoek. 2006, 90: 69-91.
Danchin A: From protein sequence to function. Curr Opin Struct Biol. 1999, 9: 363-367.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein databases search programs. Nucleic Acids Research. 1997, 25: 3389-3402.
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ: Multiple sequence alignment with Clustal x. Trends Biochem Sci. 1998, 23: 403-405.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, 17: 540-552.
Van de PY, De Wachter R: Construction of evolutionary distance trees with TREECON for Windows: accounting for variation in nucleotide substitution rate among sites. Comput Appl Biosci. 1997, 13: 227-230.
Schmidt HA, Strimmer K, Vingron M, von Haeseler A: TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002, 18: 502-504.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163.
Felsenstein J: PHYLIP, version 3.5c. 1993, Seattle, WA, University of Washington
Halling SM, Peterson-Burch BD, Bricker BJ, Zuerner RL, Qing Z, Li LL, Kapur V, Alt DP, Olsen SC: Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J Bacteriol. 2005, 187: 2715-2726.
Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, Palmer GH, McGuire TC, Knowles DP: Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci U S A. 2005, 102: 844-849.
Hotopp JC, Lin M, Madupu R, Crabtree J, Angiuoli SV, Eisen J, Seshadri R, Ren Q, Wu M, Utterback TR, Smith S, Lewis M, Khouri H, Zhang C, Niu H, Lin Q, Ohashi N, Zhi N, Nelson W, Brinkac LM, Dodson RJ, Rosovitz MJ, Sundaram J, Daugherty SC, Davidsen T, Durkin AS, Gwinn M, Haft DH, Selengut JD, Sullivan SA, Zafar N, Zhou L, Benahmed F, Forberger H, Halpin R, Mulligan S, Robinson J, White O, Rikihisa Y, Tettelin H: Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006, 2: e21-
Frutos R, Viari A, Ferraz C, Morgat A, Eychenie S, Kandassamy Y, Chantal I, Bensaid A, Coissac E, Vachiery N, Demaille J, Martinez D: Comparative genomic analysis of three strains of Ehrlichia ruminantium reveals an active process of genome size plasticity. J Bacteriol. 2006, 188: 2533-2542.
Collins NE, Liebenberg J, de Villiers EP, Brayton KA, Louw E, Pretorius A, Faber FE, van Heerden H, Josemans A, van Kleef M, Steyn HC, van Strijp MF, Zweygarth E, Jongejan F, Maillard JC, Berthier D, Botha M, Joubert F, Corton CH, Thomson NR, Allsopp MT, Allsopp BA: The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc Natl Acad Sci U S A. 2005, 102: 838-843.
Ogata H, La Scola B, Audic S, Renesto P, Blanc G, Robert C, Fournier PE, Claverie JM, Raoult D: Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2006, 2: e76-
Ogata H, Audic S, Renesto-Audiffren P, Fournier PE, Barbe V, Samson D, Roux V, Cossart P, Weissenbach J, Claverie JM, Raoult D: Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science. 2001, 293: 2093-2098.
Ogata H, Renesto P, Audic S, Robert C, Blanc G, Fournier PE, Parinello H, Claverie JM, Raoult D: The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 2005, 3: e248-
Andersson SGE, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UCM, Podowski RM, Näslund AK, Eriksson AS, Winkler HH, Kurland CG: The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998, 396: 133-140.
McLeod MP, Qin X, Karpathy SE, Gioia J, Highlander SK, Fox GE, McNeill TZ, Jiang H, Muzny D, Jacob LS, Hawes AC, Sodergren E, Gill R, Hume J, Morgan M, Fan G, Amin AG, Gibbs RA, Hong C, Yu XJ, Walker DH, Weinstock GM: Complete genome sequence of Rickettsia typhi and comparison with sequences of other rickettsiae. J Bacteriol. 2004, 186: 5842-5855.
Wu M, Sun LV, Vamathevan J, Riegler M, DeBoy R, Brownlie JC, McGraw EA, Martin W, Esser C, Ahmadinejad N, Wiegand C, Madupu R, Beanan MJ, Brinkac LM, Daugherty SC, Durkin AS, Kolonay JF, Nelson WC, Mohamoud Y, Lee P, Berry K, Young MB, Utterback T, Weidman J, Nierman WC, Paulsen IT, Nelson KE, Tettelin H, O'Neill SL, Eisen JA: Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004, 2: E69-
Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B: The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005, 3: e121-
Young JP, Crossman LC, Johnston AW, Thomson NR, Ghazoui ZF, Hull KH, Wexler M, Curson AR, Todd JD, Poole PS, Mauchline TH, East AK, Quail MA, Churcher C, Arrowsmith C, Cherevach I, Chillingworth T, Clarke K, Cronin A, Davis P, Fraser A, Hance Z, Hauser H, Jagels K D, Moule S, Mungall K, Norbertczak H, Rabbinowitsch E, Sanders M, Simmonds M, Whitehead S, parkhill J: The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7 (4): R34-
Acknowledgements
We thank Venus Wong and Yan Li for providing computer support for blast analyses and Beile Gao for help with phylogenetic analysis. We thank the investigators at US DOE Joint Genome Institute, The Institute of Genomic Research, Gordon and Betty Moore Foundation Marine Biotechnology Initiative and Rocky Mountain Laboratory, NIH, for making genome data for a number of alpha proteobacteria available in public databases prior to publication, which was very helpful in these studies. This work was supported by a research grant from the Canadian Institute of Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
The initial blastp searches on various genomes were carried out by RSG with the computer assistance provided by Venus Wong. AM analyzed the results of these blast searches to identify various group-specific proteins and confirmed their specificities by means of PSI-blast and genomic blasts. Phylogenetic analyses and identification of conserved indels was done by RSG. RSG was also responsible for directing this study, for final evaluation of the results, and for writing this manuscript. All authors have read and approved the submitted manuscript.
Electronic supplementary material
12866_2007_423_MOESM1_ESM.pdf
Additional file 1: Proteins that are specific for the Brucella species. Many of these proteins are specific for all sequenced Brucella species (viz. B. abortus, B. melitensis, B. ovis and B. suis), whereas others are present in only 2 or 3 of these species. The proteins only found in a single Brucella species are not listed here. (PDF 16 KB)
12866_2007_423_MOESM2_ESM.pdf
Additional file 2: Bradyrhizobiaceae-specific proteins that are missing in some species. For the proteins listed in this table, all significant hits in Blastp and PSI-Blast searches are from Bradyrhizobiaceae species. However, unlike the proteins listed in Table 3, which are present in all sequenced Bradyrhizobiaceae species belonging to the genera Bradyrhizobium, Nitrobacter and Rhodopseudomonas, these proteins are generally missing in species from one of these three genera. (PDF 9 KB)
12866_2007_423_MOESM3_ESM.pdf
Additional file 3: Proteins that are specific for the Sphingomonadales but missing in one or more species. The proteins listed in part (A) of this table are present in three of the following four Sphingomonadales species (Novosphingobium, Erythrobacter, Sphingomonas and Sphingopyxis), where as those listed in part (B) are present in Novosphingobium and either Sphingomonas or Erythrobacter. (PDF 8 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gupta, R.S., Mok, A. Phylogenomics and signature proteins for the alpha Proteobacteria and its main groups. BMC Microbiol 7, 106 (2007). https://doi.org/10.1186/1471-2180-7-106
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-7-106