
Abstract
Background
Corynebacterium glutamicum cg1790/pgk encodes an enzyme active as a 3-phosphoglycerate kinase (PGK) (EC 2.7.2.3) catalyzing phosphoryl transfer from 1,3-biphosphoglycerate (bPG) to ADP to yield 3-phosphoglycerate (3-PG) and ATP in substrate chain phosphorylation.
Results
C. glutamicum 3-phosphoglycerate kinase was purified to homogeneity from the soluble fraction of recombinant E. coli. PGKHis was found to be active as a homodimer with molecular weight of 104 kDa. The enzyme preferred conditions of pH 7.0 to 7.4 and required Mg2+ for its activity. PGKHis is thermo labile and it has shown maximal activity at 50–65°C. The maximal activity of PGKHis was estimated to be 220 and 150 U mg-1 with KM values of 0.26 and 0.11 mM for 3-phosphoglycerate and ATP, respectively. A 3-phosphoglycerate kinase negative C. glutamicum strain ∆pgk was constructed and shown to lack the ability to grow under glycolytic or gluconeogenic conditions unless PGK was expressed from a plasmid to restore growth. When pgk was overexpressed in L-arginine and L-ornithine production strains the production increased by 8% and by 17.5%, respectively.
Conclusion
Unlike many bacterial PGKs, C. glutamicum PGK is active as a homodimer. PGK is essential for growth of C. glutamicum with carbon sources requiring glycolysis and gluconeogenesis. Competitive inhibition by ADP reveals the critical role of PGK in gluconeogenesis by energy charge. Pgk overexpression improved the productivity in L-arginine and L-ornithine production strains.
Similar content being viewed by others
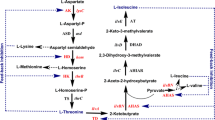

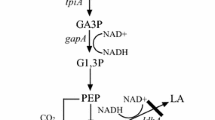
Background
Central carbon metabolism uses a complex series of enzymatic steps to convert sugars into metabolic precursors [1]. When terminal electron acceptors are not available, glycolysis supplies all of the ATP molecules required for cellular activity through substrate chain phosphorylation and the glycolytic intermediates are direct precursors of many cellular building blocks. In glycolysis, substrate chain phosphorylation has a net yield of two moles of ATP per mole of glucose that are generated in the reactions catalyzed by 3-phosphoglycerate kinase (PGK) and pyruvate kinase. PGK catalyzes the phosphoryl transfer from 1,3-biphosphoglycerate (bPG) to ADP yielding 3-phosphoglycerate (3-PG) and ATP [2].
C. glutamicum is a Gram-positive soil bacterium belonging to the order Corynebateriales within the class of Actinobacteria[3]. Since the discovery of this organism, it has been used for the industrial production of L-amino acids, and strains have been developed for the production of D-amino acids, organic acids, diamines or biofuels from different carbon sources [4–8]. C. glutamicum was metabolically engineered for the production of amino acids from alternative substrates, such as starch, the hemicellulose components xylose and arabinose, cellobiose, the whey components galactose and lactose, and glycerol [9–11]. The phosphotransferase system substrates such as glucose, fructose and sucrose are used for production of amino acids on an industrial scale in the form of starch hydrolysate or molasses [12].
C. glutamicum has been extensively studied and the central carbon metabolism genes in C. glutamicum are under the control of a transcriptional regulatory network composed of several global regulators and various transcriptional regulators which have been characterized [13, 14]. The genes of glycolytic enzymes are not clustered except for the gapA-pgk-tpi-ppc cluster encoding glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, triose phosphate isomerase and phosphoenolpyruvate carboxykinase, respectively [14]. The transcription of the gapA-pgk-tpi-ppc cluster involves mono-, di- and tricistronic mRNAs [15]. Expression of the gapA-pgk-tpi operon is coordinately regulated by SugR, RamA, and GlxR [16–18].
However, despite of the importance as an energy generating metabolic reaction in glycolysis in C. glutamicum and other bacteria, limited information is available on PGK. In this work, we aimed to identify and enzymatically characterize the PGK from C. glutamicum and to analyze its physiological role.
Results
Phylogenetic analysis of PGK from C. glutamicum
Databank searches with the amino acid sequences for PGK of C. glutamicum revealed similarities to biochemically characterized PGKs of different multimerization states such as monomeric PGK from E. coli, dimeric PGKs from Methanothermus fervidus[19] and Pyrococcus woesi, and tetrameric PGKs from Sulfolobus solfataricus and Trypanosoma brucei, the latter shown to be a dimer of dimers [20–23]. The N-terminal sequences of all PGKs showed high amino acid sequence similarity and a proline residue mostly conserved in all proteins in the hinge region. Based on its amino acid sequence, PGK from C. glutamicum is closely related to the monomeric enzyme from E. coli (Data not shown).
Purification of PGK of C. glutamicum from recombinant E. coli expressing cg1790/pgk
For purification, PGK from C. glutamicum was overproduced as an N-terminally His-tagged protein in recombinant E. coli. Metal chelate chromatography allowed to purify the enzyme to homogeneity as determined by SDS-PAGE (Figure 1) revealing a molecular mass of 47 kDa for the monomer. To determine the multimeric state of C. glutamicum PGKHis, gel filtration chromatography was performed revealing a single peak with a molecular mass of about 104 kDa and only this fraction showed activity as PGK (data not shown). Thus, unlike the monomeric PGK from E. coli, PGK from C. glutamicum appears to be active as a homodimer as also reported for several archaeal PGKs [22].

Sodium dodecyl sulfate-polyacrylamide gel electrophoreses of expression and purification of PGK His of C. glutamicum from E. coli BL21 (DE3; pET16b- pgk ). The gel was loaded with cell extract from E. coli (pET16b-pgk) before (lane 1) and 4 h after induction with 0.5 mM IPTG (lane 2), the flow through after Ni-NTA chromatography (lane 3), and the eluate of Ni-NTA chromatography after rebuffering on a PD-10 column (lane 4). Lanes M1 shows protein standards SeaBlue Plus2 prestained standard (Invitrogen) containing protein of the indicated masses.
Characterization of PGK from C. glutamicum
The pH and metal ion concentrations were varied to determine the optimal buffer conditions for activity of PGKHis. Within a pH range of 4.5 to 9.8, PGK activity was optimal at pH 7.0-7.4 in 100 mM TEA-Cl buffer with only 10% of PGK activity remaining at pH 4.5 and 33% at pH 8.9. PGK required bivalent cations with Mg2+ being the most effective. In the presence of 1 mM Ni2+, Co2+, Mn2+, Cd2+, Ca2+ or Zn2+, PGKHis activity was reduced to between 80% and 40% in comparison to the presence of Mg2+ only. To determine its thermal stability, PGKHis was incubated at temperatures ranging from 25 to 65°C prior to activity measurements at 30°C. A temperature optimum between 50-65°C was observed. Protein precipitation was observed above 65°C due to the instability of rabbit muscle GAPDH used as indicator enzyme in the assay.
Kinetic parameters of PGK from C. glutamicum
The kinetic parameters of PGKHis for the substrates 3-phosphoglycerate and ATP were determined at 30°C, the optimal growth temperature of the bacterium. The activity of PGKHis with 3-phosphoglycerate and ATP in the gluconeogenetic direction followed Michaelis-Menten kinetics (data not shown). The KM values of C. glutamicum PGK for 3-phosphoglycerate and ATP, respectively, were determined to be 0.26 mM and 0.11 mM, respectively. The Vmax values were 220 U mg-1 and 150 U mg-1, respectively (Table 1). Catalytic efficiencies were about 733 s-1 mM-1 and 592 s-1 mM-1, respectively (Table 1).
Metabolites such as glycerol-2-phosphate, glycerol-3-phosphate, glucose-6-phosphate, fructose-6-phosphate, fructose-1, 6-bisphosphate, phosphoenolpyruvate, pyruvate, acetate, L-lysine, L-alanine, L-glutamate, GTP, AMP and ADP were tested as potential effectors of PGK activity in the gluconeogenic direction (data not shown). Only ADP affected PGK activity as competitive inhibitor with 0.1 mM resulting in half-maximal activity (Figure 2).

Effect of ADP on the activity of 3-phosphoglycerate kinase at variable ATP concentrations. The ATP concentrations were 0.25 mM (open diamonds), 0.5 mM (open squares), 1 mM (open triangles), 2 mM (open circles). The concentration of 3-PG was 10 mM through out the experiment. 1/v corresponds to reciprocal of the velocity of the reaction.
Physiological studies of pgk deletion mutants
pgk was deleted to study the role of PGK in C. glutamicum. Since pgk is part of the gapA-pgk-tpi-ppc operon [15] the specific activities of GapA, Tpi and Ppc were determined in crude extracts of the deletion mutant. While GapA activity in ∆pgk was comparable to wild type, ∆pgk showed decreased Tpi activity (0.1 U/mg as compared to 0.4 U/mg in wild type) and Ppc activity (0.03 U/mg as compared to 0.05 U/mg in wild type) which might be explained by polar effects of pgk deletion on the downstream genes tpi and ppc. Thus, plasmid pEKEx3-pgk was constructed for complementation analysis. C. glutamicum ∆pgk showed no growth in CgXII minimal medium with glucose or pyruvate as sole source of carbon and energy, but grew with blends of glucose plus pyruvate (Table 2). Complementation of C. glutamicum ∆pgk led to comparable growth with either glucose or pyruvate as sole carbon source (Table 2) revealing the requirement of PGK for growth with glycolytic as well as with gluconeogenic carbon sources.
Effect of pgk overexpression on amino acid production
pgk was overexpressed in the L-lysine producing strain DM1933 [7], the L-arginine production strain ARG1 [6], and the L-ornithine production strain ORN1 [6] to assay the affects of pgk overexpression on amino acid production rates. In each strain, PGK specific activities increased upon pgk overexpression to similar levels as when pgk was overexpressed in the wild type (Table 3). Amino acid production in CgXII minimal medium with 4% (w/v) glucose was performed and amino acid accumulation was followed. While pgk overexpression did not increase the L-lysine production rate, pgk overexpression accelerated L-arginine and L-ornithine production by 8% and 17.5%, respectively (Figure 3). Glucose consumption was comparable between control and pgk overexpression strains.

L-lysine, L-arginine and L-ornithine production by C. glutamicum DM1933, ARG1 and ORN1 strains either with empty vector (white column represents pVWEx1) or overproducing phosphoglycerate kinase (black column represents pVWEx1- pgk ). Cells were grown in CgXII minimal medium with 4% glucose as a carbon source. Cell pellets of cultures grown in glucose CgXII minimal medium after consumption of the carbon source. Data represent mean values and standard deviations of six replicates from two independent cultivations with three flasks per strain each. Significant differences (p < 0.01 in a student’s t-test) between empty vector control (pVWEx1) and strains carrying pVWEx1-pgk are highlighted by an asterisk.
Discussion
Phosphoglycerate kinase from C. glutamicum was purified to homogeneity and shown to be active as homodimer. This is unusual and was hitherto only found in the thermophilic archaea Pyrococcus and Methanothermus[19, 22]. Most prokaryotes and eukaryotes possess monomeric PGKs [24, 25]. Tetramers have been reported in rare cases as in Sulfolobus solfataricus[26] and Trypanosoma[21]. C. glutamicum PGKHis showed higher temperature stability than monomeric PGKs, e.g., 40°C for Mus musculus[27] and 24°C for S. cervisiae[28]. Possibly, dimerization of PGK from C. glutamicum contributes to the stability of the protein by favoring hydrophobic interactions via the subunit contacts and by reducing the surface area exposed to the solvent in a similar manner as reported for dimeric PGKs [29]. Other characteristics are shared by many PGKs. An optimum pH 7.0 to 7.4 is found for phosphoglycerate kinases from Escherichia coli and Homo sapiens, while Saccharomyces cerivisiae shows optimum activity at pH 7.5 [24, 30, 31]. The KM for ATP of PGKHis of 0.11 mM is similar to those reported for PGK from Mus musculus and Homo sapiens[32, 33] and within the range reported for prokaryotic PGKs, e.g. 0.21 mM for PGK from Pseudomonas sp. and 0.31 mM for yeast PGK [33, 34]. The KM value of 0.26 mM for 3-PG of PGKHis is comparable to that of PGK from Mus musculus[33] and slightly lower than that of PGK from Pseudomonas sp. which is 0.48 mM [35]. The enzyme is very specific for Mg+2 ions for catalysis as reported for several other phosphoglycerate kinases. Other divalent cations such as Ni2+, Co2+, Mn2+, Cd2+, Ca2+ or Zn2+ could not replace Mg2+ and were inhibitory at higher concentrations. Zn2+ is a strong inhibitor with Ki of 0.45 mM. The monovalent cations Na+ and K+ activated the enzyme whereas sulfate ions showed no influence as reported for multimeric PGKs from archaea [22].
PGK from C. glutamicum was shown to be subject to allosteric regulation. Of the cellular metabolites tested only ADP affected the activity of PGKHis. ADP inhibited PGK from C. glutamicum as competitive inhibitor with a low Ki value (0.1 mM). Since ADP concentrations in C. glutamicum typically range from 0.5 to 1 mM in glucose batch cultures [36], this indicates that gluconeogenesis via PGK is tightly regulated by the energy charge. Regulation by the energy charge has also been described for eukaryotic PGKs, such as mouse PGK which has a very low Ki of ≈ 0.08 mM [32] and yeast PGK which has a Ki of ≈ 0.22 mM [37].
PGK from C. glutamicum was shown here to be required for growth with different glycolytic as well as with different gluconeogenic carbon sources. Besides PGK, other enzymes of glycolysis are expected to be essential for both glycolysis and gluconeogenesis in C. glutamicum, however, experimental evidence has only been obtained for fructose-1,6-bisphosphate aldolase [38] and phosphoglycerate mutase [39]. Deletion of the genes encoding phosphofructokinase and glyceraldehyde-3-phosphate dehydrogenase (GapA) [40] prevented growth with glycolytic substrates, while growth with gluconeogenic substrates was still possible. On the other hand, growth of C. glutamicum on gluconeogenic carbon sources has been shown to be dependent on PEPCk [41] and FBPase [42], but the lack of these enzymes did not preclude growth with glycolytic substrates. Deletion of the phosphoglucoisomerase gene pgi perturbed growth on glucose indirectly since the glucose PTS permease gene ptsG was hardly expressed in the deletion mutant. The lack of pyruvate kinase cannot be compensated during growth with some glycolytic or gluconeogenic carbon sources [43]. Pyruvate kinase is essential for growth with glycolytic non-PTS carbon substrates such as maltose but not for PTS substrates such as glucose, fructose and sucrose. Similarly, pyruvate kinase is essential for growth on gluconeogenic carbon sources that do not enter the central carbon metabolism via pyruvate, such as acetate or citrate, but is not required for growth with pyruvate or lactate [43].
Overproduction of PGK did not notably accelerate growth of C. glutamicum with glucose (data not shown). However, ornithine and arginine production rates were increased upon pgk overexpression in the respective amino acid producing strains (Figure 3), while lysine production was not accelerated. It is tempting to speculate that arginine and ornithine biosynthesis are positively affected while lysine biosynthesis is not because the latter has a lower ATP requirement (1 ATP per lysine) than the former (2 ATP per ornithine or arginine which in addition requires carbamoylphosphate). Most metabolic engineering approaches of glycolysis in C. glutamicum focused on increasing the product yield e.g. by redirecting carbon flux to reduce glycolysis and increase pentose phosphate pathway flux and NADPH provision [44–46]. Overexpression of the gene encoding gluconeogenic enzyme FBPase during growth and production with sucrose increased gluconeogenic flux from fructose-1, 6-bisphosphate to glucose-6-phosphate and into the pentose phosphate pathway [47]. Production of D-lactic acid [48], succinic acid [49] and alanine [50] by C. glutamicum under oxygen-deprivation conditions is characterized by a high glycolytic flux which could be enhanced by overexpression of gapA. Also overexpression of the genes coding for phosphofructokinase, triosephosphate isomerase, and fructose-1,6-bisphosphate aldolase accelerated D-lactate production under oxygen-deprivation conditions [48].
Conclusions
C. glutamicum 3-phosphoglycerate kinase encoded by pgk was shown to be essential for growth with glycolytic as well as with gluconeogenetic carbon sources. ADP was shown to be a competitive inhibitor of PGK, which unlike most bacterial PGKs is active as a dimer. Since overexpression of pgk increased amino acid productivity for ornithine and arginine, but not for lysine, PGK may be a promising target to accelerate production processes requiring high glycolytic flux but needs careful testing.
Methods
Microorganisms and cultivation conditions
Microorganisms are listed in Table 4. ATCC 13032 was used as C. glutamicum wild type (WT) [51] along with the amino acid producing strains DM1933, ORN1, ARG1 [6], PUT21 [52]. C. glutamicum strains were pre-cultured in lysogeny broth (LB) medium [53] with antibiotics added when appropriate. E. coli strains DH5α [54] and BL21 (DE3) [55] were used as host for cloning and heterologous expression, respectively.
For growth and amino acid production experiments, exponentially growing cells of LB precultures (50 ml) were harvested by centrifugation (3200 × g, 10 min), and washed in CgXII medium [56] without carbon source. Cultures of 50 ml CgXII media containing 4% (w/v) glucose, 100 μg/ml spectinomycin or 25 μg/ml kanamycin, and 1 mM IPTG were inoculated to a final optical density (OD600) of 1 and incubated in 500 ml baffled shake flasks at 30°C. The OD600 was measured in dilutions resulting in an OD600 between 0.05 and 0.25 using a Shimadzu UV-1202 spectrophotometer (Duisburg, Germany). For enzymatic activity determination in cell-free extracts, cells were grown in LB medium to mid-exponential phase (OD600 of 3.5 to 4), harvested by centrifugation (10 min at 3200 × g, 4°C) and washed in 100 mM TEA-Cl pH 7.4. Cells were stored at -20°C until usage.
Overexpression of pgk in C. glutamicum
For overexpression of pgk, the gene was amplified via PCR from genomic DNA of C. glutamicum WT. The PCR was performed using the oligonucleotide primers listed in Table 5. To allow IPTG inducible expression of pgk in C. glutamicum the PCR-product was ligated into SmaI restricted vector pEKEx3 resulting in pEKEx3-pgk and XbaI restricted vector pVWEx1 resulting pVWEx1-pgk. Sequencing confirmed the integrity of the construct.
Overproduction of PGK in E. coli, protein purification and molecular weight determination
For heterologous expression of the 3-phosphoglycerate-kinase gene pgk (Cg1790) in E. coli BL21 (DE3), pgk was amplified via PCR from genomic DNA of C. glutamicum WT using the following oligonucleotide primers listed in Table 5. The 1208 bp amplification product was cloned into vector pGEM-T (Promega, Mannheim, Germany) resulting in vector pGEM-T-pgk. After restriction with NdeI, the 1208 bp product from pGEM-T-pgk was ligated to NdeI restricted pET16b (Novagen, Madison, WI, USA). The vector, pET16b-pgk, allows IPTG-inducible expression of an N-terminal tenfold His-tagged pgk in E. coli BL21 (DE3). Cultivation of BL21 (DE3) (pET16b-pgk), Cells were grown in an incubator/shaker at 37°C to an absorbance reading (A600) of 0.6–0.8 at which point IPTG (0.5 mM) was added and the flasks were cooled to 22°C and further incubated for 4 hours. Cells were harvested by centrifugation (20 min at 3200 × g) and the cell pellet was washed with 20 mM Tris, 300 mM NaCl, 5 mM imidazole (TNI buffer), 5% (vol/vol) glycerol and stored at -80°C. Prior to lysis by French press, cells were resuspended in TNI buffer, and protease activity was inhibited by addition of 1 mM phenylmethylsulfonyl fluoride (PMSF) and 1 mM diisopropylfluorophosphate (DFP). The extract was cleared by centrifugation for 1 h at 25000 × g. Peak fractions of Ni-nitrilotriacetic acid (Ni-NTA) agarose affinity chromatography eluted with 20 mM Tris, 300 mM NaCl, 100, 200, or 400 mM imidazol, and 5% (vol/vol) glycerol were pooled, and the pooled fractions were desalted using Sephadex G25 gel filtration (Amersham Bioscience, Uppsala) and buffered in 100 mM triethanolamine hydrochloride (TEA-Cl), pH 7.4.
The molecular weight of PGKHis was determined using gel filtration and by cross-linking experiments. For gel filtration, a Bio-Prep SE-1000/17 column (BioRad, Richmond, VA, USA) was used and calibrated with Gel Filtration Standard (BioRad) containing thyroglobulin (670 kDa), bovine γ - globulin (158 kDa), chicken ovalbumin (44 kDa), equine myoglobin (17 kDa), and vitamin B12 (1.35 kDa). The buffer used contained the optimal conditions for activity of PGK plus 100 mM TEA-Cl pH 7.4). To determine the PGK elution volume, two gel filtrations were performed, one in a mix with the indicated protein standards containing 2 mg/ml of PGK and a second with 4 mg/ml of PGK. Protein elution time was measured at 280 nm. Eluting PGKHis was collected in 0.5 ml fractions and confirmed by enzyme activity measurement.
Assay conditions
The phosphoglycerate kinase activity was determined with purified enzyme and cell-free extracts of WT(pEKEx3) and WT(pEKEx3-pgk). Cell-free extracts were prepared according to Stansen et al.[57]. In vitro enzyme assays were carried out spectrophotometrically in a coupled assay in which, product of the first reaction catalyzed by Cg PGK, namely 1, 3 - bisphosphoglyerate is reduced by the second enzyme glyceraldehyde-3-phosphate dehydrogenase, which uses NADH. Assays were conducted in 100 mM triethanolamine-HCl buffer (pH-7.4) containing 1 mM EDTA, 2 mM MgSO4, 1 mM ATP, 10 mM 3-PGA, 0.2 mM NADH, 10 U/ml of GAPDH (rabbit muscle). Approximately 30 ng of PGKHis was used for each assay. The enzyme activity was measured by following the decrease in absorbance at 340 nm due to oxidation of NADH [14]. All spectrophotometric measurements were carried out using a Shimadzu UV-1202 spectrophotometer (Duisburg, Germany) at 30°C, NAD+ formation was followed at λ = 340 nm (ϵ340nm = 6.3 mM-1 cm-1). Kinetic parameters were calculated using Michaelis–Menten kinetics. One unit (U) of enzyme activity is defined as 1 μmol × min-1 × mg-1 of protein. PEPCx was assayed at 30°C in 1 ml of 100 mM Tris–HCl buffer (pH 8.0) containing 10 mM MgSO4, 25 mM NaHCO3, 1 mM dithiothreitol (DTT), 0.2 mM NADH, 10 U of malate dehydrogenase, and 8 mM PEP. One unit (U) of activity is defined as 1 mmol of NADH consumed per min. TPI was assayed at 30°C in 1 ml of 300 mM triethanolamine buffer (pH 7.6), 0.2 mM NADH, 2 U of glycerolphosphate dehydrogenase, 5 mM glyceraldehyde-3-phosphate as the substrate. The decrease of NADH was monitored at 340 nm.
To determine the pH-optimum TEA-Cl was replaced by the following buffers (50 mM): acetate (pH 5.0-6.0), phosphate (pH 6.0-7.0), TEA-Cl (pH 7.0-9.0), and glycine-NaOH (pH 9.0-10.0) under standard conditions. The pH was adjusted at room temperature. The effect of metal ions and EDTA on kinase activity was measured under standard conditions in the presence of Zn2+, Ca2+, Co2+, Cd2+, Cu2+, Mg2, Fe2+, Mn2+, Ni2+, at 0.5 and 1 mM final concentration in the reaction mixture. For determination of optimum temperature, the reaction mix was allowed to equilibrate for 5 min at each temperature point.
Overexpression of pgk in production strains
Cells were removed from the culture samples by centrifugation for 10 min at 14,000 × g and supernatant was analyzed using a high-pressure liquid chromatography system (HPLC, 1200 series, Agilent Technologies). L-lysine, L-ornithine and L-arginine concentrations were determined by automatic precolumn derivatization with ortho-phthaldialdehyde and reversed-phase high-performance liquid chromatography (RP-HPLC) with fluorimetric detection (excitation at 230 nm; emission at 450 nm). The buffer gradient consisted of 0.1 M sodium acetate, pH 7.2 (with 0.03% sodium azide), as the polar phase and methanol as the nonpolar phase. L-asparagine was used as internal standard for L-lysine, L-ornithine and L-arginine respectively.
Computational analysis
Sequence comparisons were carried out with protein sequences obtained from the NCBI database (http://www.ncbi.nlm.nih.gov) using CLUSTAL W [60], the alignment was formatted using BoxShade and phylogenetic trees were constructed using the neighbor-joining method [61] with 1,000 bootstrap replicates, the tree was rooted against phosphoglycerate kinase of E. coli (data not shown). Besides the protein sequence of C. glutamicum PGK (Cg1790), protein sequences of characterized PGKs from the following organisms were used: E. coli (b2926), Methanothermus ferviduds (Mfer_0156), Pyrococcus woesi (Accession ID P61884.1), Sulfolobus solfataricus (CAA 56459), Trypanosoma brucei ( AAA32120.1).
Abbreviations
- PGK:
-
3-phosphoglycerate kinase
- PGKHis:
-
PGK with histidine tag
- 3-PG:
-
3-phosphoglycerate
- EV:
-
Empty vector
- GAP:
-
Glyceraldehyde 3-phosphate
- GAPDH/GapA:
-
Glyceraldehyde-3-phosphate dehydrogenase
- HPLC:
-
High performance liquid chromatography
- IPTG:
-
Isopropyl β-D-1-thiogalactopyranoside
- ATP:
-
Adenosine tri phosphate
- GTP:
-
Guanosine tri phosphate
- AMP:
-
Adenosine mono phosphate
- ADP:
-
Adenosine di phosphate
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TEA-Cl:
-
Triethanolamine hydrochloride.
References
Noor E, Eden E, Milo R, Alon U: Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell. 2010, 39: 809-820.
Smith CD, Chattopadhyay D, Pal B: Crystal structure of Plasmodium falciparum phosphoglycerate kinase: evidence for anion binding in the basic patch. Biochem Biophys Res Commun. 2011, 412: 203-206.
Gao B, Gupta RS: Phylogenetic framework and molecular signatures for the main clades of the phylum actinobacteria. Microbiol Mol Biol Rev. 2012, 76: 66-112.
Stabler N, Oikawa T, Bott M, Eggeling L: Corynebacterium glutamicum as a host for synthesis and export of D-Amino Acids. J Bacteriol. 2011, 193: 1702-1709.
Wendisch VF, Bott M, Eikmanns BJ: Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for biotechnological production of organic acids and amino acids. Curr Opin Microbiol. 2006, 9: 268-274.
Schneider J, Niermann K, Wendisch VF: Production of the amino acids L-glutamate, -lysine, -ornithine and l-arginine from arabinose by recombinant Corynebacterium glutamicum. J Biotechnol. 2011, 154: 191-198.
Blombach B, Hans S, Bathe B, Eikmanns BJ: Acetohydroxyacid synthase, a novel target for improvement of L-lysine production by Corynebacterium glutamicum. Appl Environ Microbiol. 2009, 75: 419-427.
Wieschalka S, Blombach B, Bott M, Eikmanns BJ: Bio-based production of organic acids with Corynebacterium glutamicum. Microb Biotechnol. 2013, 6: 87-102.
Buschke N, Schroder H, Wittmann C: Metabolic engineering of Corynebacterium glutamicum for production of 1,5-diaminopentane from hemicellulose. Biotechnol J. 2011, 6: 306-317.
Meiswinkel TM, Rittmann D, Lindner SN, Wendisch VF: Crude glycerol-based production of amino acids and putrescine by Corynebacterium glutamicum. Bioresour Technol. 2013, 145: 254-258.
Jensen JV, Wendisch VF: Ornithine cyclodeaminase-based proline production by Corynebacterium glutamicum. Microb Cell Fact. 2013, 12: 63-
Xu J, Zhang J, Guo Y, Zai Y, Zhang W: Improvement of cell growth and L-lysine production by genetically modified Corynebacterium glutamicum during growth on molasses. J Ind Microbiol Biotechnol. 2013, 40: 1423-1432. doi:10.1007/s10295-013-1329-8. Epub 2013 Sep 13
Wendisch VF: Genetic regulation of Corynebacterium glutamicum metabolism. J Microbiol Biotechnol. 2006, 16 (7): 999-1009.
Eikmanns BJ: Identification, sequence analysis, and expression of a Corynebacterium glutamicum gene cluster encoding the three glycolytic enzymes glyceraldehyde-3-phosphate dehydrogenase, 3-phosphoglycerate kinase, and triosephosphate isomerase. J Bacteriol. 1992, 174: 6076-6086.
Schwinde JW, Thum-Schmitz N, Eikmanns BJ, Sahm H: Transcriptional analysis of the gap-pgk-tpi-ppc gene cluster of Corynebacterium glutamicum. J Bacteriol. 1993, 175: 3905-3908.
Toyoda K, Teramoto H, Gunji W, Inui M, Yukawa H: Involvement of regulatory interactions among global regulators GlxR, SugR, and RamA in expression of ramA in Corynebacterium glutamicum. J Bacteriol. 2013, 195: 1718-1726.
Engels V, Lindner SN, Wendisch VF: The global repressor SugR controls expression of genes of glycolysis and of the L-lactate dehydrogenase LdhA in Corynebacterium glutamicum. J Bacteriol. 2008, 190: 8033-8044.
Toyoda K, Teramoto H, Inui M, Yukawa H: Expression of the gapA gene encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum is regulated by the global regulator SugR. Appl Microbiol Biotechnol. 2008, 81: 291-301.
Hensel R, Schramm A, Hess D, Russell RJM: 3-Phosphoglycerate kinase and triose phosphate isomerase from hyperthermophilic Archaea: features of biochemical thermoadaptation. Thermophiles: the key to molecular evolution and the origin of life?. Edited by: Wiegel A. 1998, London, UK: CRC Press, Taylor & Francis, 311-323.
Misset O, Bos OJ, Opperdoes FR: Glycolytic enzymes of Trypanosoma brucei. Simultaneous purification, intraglycosomal concentrations and physical properties. Eur J Biochem. 1986, 157: 441-453.
Bernstein BE, Hol WG: Crystal structures of substrates and products bound to the phosphoglycerate kinase active site reveal the catalytic mechanism. Biochemistry. 1998, 37: 4429-4436.
Hess D, Kruger K, Knappik A, Palm P, Hensel R: Dimeric 3-phosphoglycerate kinases from hyperthermophilic Archaea. Cloning, sequencing and expression of the 3-phosphoglycerate kinase gene of Pyrococcus woesei in Escherichia coli and characterization of the protein. Structural and functional comparison. Eur J Biochem. 1995, 233: 227-237.
Zomer AW, Allert S, Chevalier N, Callens M, Opperdoes FR, Michels PA: Purification and characterisation of the phosphoglycerate kinase isoenzymes of Trypanosoma brucei expressed in Escherichia coli. Biochim Biophys Acta. 1998, 1386: 179-188.
D’Alession G, Josse J: Phosphoglycerate kinase and phosphoglyceromutase from Escherichia coli. Methods Enzym. 1975, 42: 139-144.
Fifis T, Scopes RK: Purification of 3-phosphoglycerate kinase from diverse sources by affinity elution chromatography. Biochem J. 1978, 175: 311-319.
Jones CE, Fleming TM, Cowan DA, Littlechild JA, Piper PW: The phosphoglycerate kinase and glyceraldehyde-3-phosphate dehydrogenase genes from the thermophilic archaeon Sulfolobus solfataricus overlap by 8-bp. Isolation, sequencing of the genes and expression in Escherichia coli. Eur J Biochem. 1995, 233: 800-808.
Pegoraro B, Lee CY: Purification and characterization of two isozymes of 3-phosphoglycerate kinase from the mouse. Biochim Biophys Acta. 1978, 522: 423-433.
Ijeoma O, Hollowell HN, Bodnar MA, Britt BM: Thermodynamic analysis of the nondenaturational conformational change of baker’s yeast phosphoglycerate kinase at 24 degrees C. Arch Biochem Biophys. 2008, 478: 206-211.
Kirino H, Aoki M, Aoshima M, Hayashi Y, Ohba M, Yamagishi A, Wakagi T, Oshima T: Hydrophobic interaction at the subunit interface contributes to the thermostability of 3-isopropylmalate dehydrogenase from an extreme thermophile, Thermus thermophilus. Eur J Biochem. 1994, 220: 275-281.
McHarg J, Kelly SM, Price NC, Cooper A, Littlechild JA: Site-directed mutagenesis of proline 204 in the “hinge” region of yeast phosphoglycerate kinase. Eur J Biochem. 1999, 259: 939-945.
Szabo J, Varga A, Flachner B, Konarev PV, Svergun DI, Zavodszky P, Vas M: Communication between the nucleotide site and the main molecular hinge of 3-phosphoglycerate kinase. Biochemistry. 2008, 47: 6735-6744.
Mukherjee K, Ghosh S, Ray M, Ray S: Purification and characterization of 3-phosphoglycerate kinase from Ehrlich ascites carcinoma cells. Indian J Biochem Biophys. 2002, 39: 332-341.
Lee CY: 3-Phosphoglycerate kinase isozymes and genetic variants from mouse. Methods Enzym. 1982, 90 Pt E: 121-126.
Mensonides FIC, Bakker BM, Cremazy F, Messiha HL, Mendes P, Boogerd FC, Westerhoff HV: A new regulatory principle for in vivo biochemistry: Pleiotropic low affinity regulation by the adenine nucleotides - Illustrated for the glycolytic enzymes of Saccharomyces cerevisiae. FEBS Lett. 2013, 587: 2860-2867.
Bentahir M, Feller G, Aittaleb M, Lamotte-Brasseur J, Himri T, Chessa JP, Gerday C: Structural, kinetic, and calorimetric characterization of the cold-active phosphoglycerate kinase from the antarctic Pseudomonas sp. TACII18. J Biol Chem. 2000, 275: 11147-11153.
Moritz B, Striegel K, de Graaf AA, Sahm H: Changes of pentose phosphate pathway flux in vivo in Corynebacterium glutamicum during leucine-limited batch cultivation as determined from intracellular metabolite concentration measurements. Metab Eng. 2002, 4: 295-305.
Larsson-Raznikiewicz M, Arvidsson L: Inhibition of phosphoglycerate kinase by products and product homologues. Eur J Biochem. 1971, 22: 506-512.
Stolzenberger J, Lindner SN, Wendisch VF: The methylotrophic Bacillus methanolicus MGA3 possesses two distinct fructose 1,6-bisphosphate aldolases. Microbiology. 2013, 159: 1770-1781.
Netzer R: Untersuchungen zur Glykolyse und zum L-Serin-Stoffwechsel in Corynebacterium glutamicum. Berichte des Forschungszentrums Jülich. 2004, 4130: 1-141. ISSN 0944-2952
Omumasaba CA, Okai N, Inui M, Yukawa H: Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J Mol Microbiol Biotechnol. 2004, 8: 91-103.
Riedel C, Rittmann D, Dangel P, Möckel B, Petersen S, Sahm H, Eikmanns BJ: Characterization of the phosphoenolpyruvate carboxykinase gene from Corynebacterium glutamicum and significance of the enzyme for growth and amino acid production. J Mol Microbiol Biotechnol. 2001, 3: 573-583.
Rittmann D, Schaffer S, Wendisch VF, Sahm H: Fructose-1,6-bisphosphatase from Corynebacterium glutamicum: expression and deletion of the fbp gene and biochemical characterization of the enzyme. Arch Microbiol. 2003, 180: 285-292.
Netzer R, Krause M, Rittmann D, Peters-Wendisch PG, Eggeling L, Wendisch VF, Sahm H: Roles of pyruvate kinase and malic enzyme in Corynebacterium glutamicum for growth on carbon sources requiring gluconeogenesis. Arch Microbiol. 2004, 182: 354-363.
Siedler S, Lindner SN, Bringer S, Wendisch VF, Bott M: Reductive whole-cell biotransformation with Corynebacterium glutamicum: improvement of NADPH generation from glucose by a cyclized pentose phosphate pathway using pfkA and gapA deletion mutants. Appl Microbiol Biotechnol. 2012, 97: 143-152.
Bartek T, Blombach B, Zonnchen E, Makus P, Lang S, Eikmanns BJ, Oldiges M: Importance of NADPH supply for improved L-valine formation in Corynebacterium glutamicum. Biotechnol Prog. 2010, 26: 361-371.
Becker J, Klopprogge C, Herold A, Zelder O, Bolten CJ, Wittmann C: Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum–over expression and modification of G6P dehydrogenase. J Biotechnol. 2007, 132: 99-109.
Georgi T, Rittmann D, Wendisch VF: Lysine and glutamate production by Corynebacterium glutamicum on glucose, fructose and sucrose: roles of malic enzyme and fructose-1,6-bisphosphatase. Metab Eng. 2005, 7: 291-301.
Tsuge Y, Yamamoto S, Suda M, Inui M, Yukawa H: Reactions upstream of glycerate-1,3-bisphosphate drive Corynebacterium glutamicum D-lactate productivity under oxygen deprivation. Appl Microbiol Biotechnol. 2013, 97: 6693-6703.
Litsanov B, Brocker M, Bott M: Toward homosuccinate fermentation: metabolic engineering of glucose and formate. Appl Environ Microbiol. 2012, 78 (9): 3325-3337.
Yamamoto S, Gunji W, Suzuki H, Toda H, Suda M, Jojima T, Inui M, Yukawa H: Overexpression of genes encoding glycolytic enzymes in Corynebacterium glutamicum enhances glucose metabolism and alanine production under oxygen deprivation conditions. Appl Environ Microbiol. 2012, 78 (12): 4447-4457.
Abe Takayarna K, Kinoshita SS: Taxonomical studies on glutamic acid producing bacteria. J Gen Appl Microbiol. 1967, 13: 279-301.
Schneider J, Wendisch VF: Putrescine production by engineered Corynebacterium glutamicum. Appl Microbiol Biotechnol. 2010, 88: 859-868.
Sambrook J, Fitsch EF, Maniatis T: Molecular cloning: a laboratory manual. 1989, Cold Spring Harbor: Cold Spring Harbor Press
Hanahan D: Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983, 166: 557-580.
Studier FW, Moffatt BA: Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986, 189: 113-130.
Handbook of Corynebacterium glutamicum. Edited by: Eggeling LBM. 2005, Boca Raton: CRC Press LLC
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF: Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Env Microbiol. 2005, 71: 5920-5928.
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ: Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol. 2001, 3: 295-300.
Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A: Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994, 145: 69-73.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Acknowledgements
This work was funded by the German Federal Ministry of Education and Research through project “SysEnCor” (0315598E). The authors would like to thank Dr. Sonja Siwiora Brenke for providing the facilities, equipment and technical assistance to perform size exclusion chromatography and molecular mass estimation of the purified protein. Acknowledge support of the publication fee by Deutsche Forschungsgemeinschaft and the Open Access Publication Funds of Bielefeld University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors do not declare competing interests.
Authors’ contributions
VFW and GKR designed the experiments. GKR conducted the experiments, analyzed the results, and wrote the manuscript. VFW reviewed and revised the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Reddy, G.K., Wendisch, V.F. Characterization of 3-phosphoglycerate kinase from Corynebacterium glutamicum and its impact on amino acid production. BMC Microbiol 14, 54 (2014). https://doi.org/10.1186/1471-2180-14-54
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2180-14-54