
Abstract
Background
Body weight and length are economically important traits in foodfish species influenced by quantitative trait loci (QTL) and environmental factors. It is usually difficult to dissect the genetic and environmental effects. Asian seabass (Lates calcarifer) is an important marine foodfish species with a compact genome (~700 Mb). The recent construction of a first generation linkage map of Asian seabass with 240 microsatellites provides a good opportunity to determine the number and position of QTL, and the magnitude of QTL effects with a genome scan.
Results
We conducted a genome scan for QTL affecting body weight, standard length and condition factors in an F1 family containing 380 full-sib individuals from a breeding stock by using 97 microsatellites evenly covering 24 chromosomes. Interval mapping and multiple QTL model mapping detected five significant and 27 suggestive QTL on ten linkage groups (LGs). Among the five significant QTL detected, three (qBW2-a, qTL2-a and qSL2-a) controlling body weight, total and standard length respectively, were mapped on the same region near Lca287 on LG2, and explained 28.8, 58.9 and 59.7% of the phenotypic variance. The other two QTL affecting body weight, qBW2-b and qBW3, were located on LG2 and 3, and accounted for 6.4 and 8.8% of the phenotypic variance. Suggestive QTL associated with condition factors are located on six different LGs.
Conclusion
This study presents the first example of QTL detection for growth-related traits in an F1 family of a marine foodfish species. The results presented here will enable further fine-mapping of these QTL for marker-assisted selection of the Asian seabass, eventually identifying individual genes responsible for growth-related traits.
Similar content being viewed by others


Background
Asian seabass (Lates calcarifer), also called Barramundi in Australia, is an important farmed marine foodfish species which has been cultured for more than 20 years in Southeast Asian countries [1]. Current global annual production is nearly 400,000 MT according to FAO statistics in 2003, and over 90% of which originated from Taiwan, Hong Kong, Singapore and Thailand [2]. Australia is also presently becoming a major producer. The production of this species is expected to expand more rapidly in the next few years due to high market demand. Studies using allozyme and mtDNA polymorphism showed genetic diversity of the Asian seabass in Australia was low and genetic differentiation among fish from different regions was large [3, 4], while studies using polymorphic microsatellites showed that both cultured and wild stocks of Asian seabass in Southeast Asia contained high allelic and gene diversity [5, 6]. No detailed selective breeding program has yet been reported.
Growth-related traits such as body weight, total length, standard length and condition factor are quantitative traits influenced by a number of genes with relatively small effects, and environmental effects. These traits could be improved by phenotypic selection, but the genetic gain can be increased more rapidly with marker-assisted selection [7, 8]. Genome scans using molecular markers have proved to be effective in the detection of QTL in organisms such as livestock species in experimental populations created by crossing genetically diverse strains [7]. Studies on linkage and QTL mapping in foodfish species started later than those in economically important livestock [9]. To date, linkage maps have been constructed for only 13 foodfish species including rainbow trout [10], salmon [11], tilapia [12], catfish [13], yellowtail [14], and common carp[15].
The genetic and molecular basis of growth-related traits is poorly understood in fish. Genetic studies in fish species have been limited by cost and the difficulty of raising large numbers of fish under the same conditions and the lack of linkage maps. A few QTL for growth-related traits, disease resistance and stress resistance have been detected in experimental F2 or backcross populations of some foodfish species, such as rainbow trout [16–18], salmon [19, 20] and tilapia [21, 22]. QTL analysis in F1 full-sib families from breeding stocks has not been reported in any fish species, although methods for detecting QTL with F1 populations in outbreed species have been developed [23–25], and have been successfully applied to identify QTL in several plant species [26, 27].
Recently, we established a first generation genetic linkage map of Asian seabass using 240 microsatellite markers covering 24 linkage groups with marker interval of 6.2 and 4.7 cM in the female and male maps respectively [28], thus providing a necessary tool for a whole genome scan for QTL affecting economically important traits. In a previous study, we demonstrated that body weight at 90 day post-hatch (dph) was significantly correlated with body weight at harvest (289 dph) in Asian seabass [29]. In an association study, we detected significant association between grow-traits and polymorphism in one parvalbumin gene (PVALB1) [30]. In the present study, we conducted a genome scan for QTL affecting body weight, total length, standard length and condition factors at 90 dph in an F1 family containing 380 individuals from a breeding population. The objectives of this study are: 1) to test whether an F1 family from a breeding stock can be used for detecting QTL affecting growth-related traits, 2) to locate QTL on linkage groups, 3) to estimate effects of QTL and finally, 4) to understand QTL allele substitution effects. This study is an initial step towards the goal of marker-assisted selection in Asian seabass.
Results
Trait values
The phenotypic values of growth-related traits in the F1 family at 90 dph are presented in Table 1. The average body weight of the 380 F1 individuals was 29.4 ± 13.5 g, ranging from 6.7 to 68.2 g. Total lengths (TL) of these fish ranged from 75.4 to 169.0 mm with an average of 122.6 ± 20.1 mm, whereas the standard lengths (SL) were between 62.8 and 139.9 mm (average 101.3 ± 16.8 mm). The average condition factors (KTL and KSL) were 1.49 ± 0.12 and 2.64 ± 0.22 calculated according to TL and SL respectively (Table 1).
Linkage map
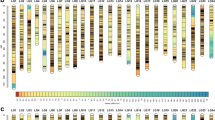
The 97 microsatellite markers selected were mapped into 24 linkage groups with a map length of 690.6 cM. 21 of the 24 linkage groups were covered with at least two markers (Figure 1, 2 and 3). The markers were well-dispersed throughout the genome, and the average distance between markers was 7.12 cM.

A genetic linkage map of Asian seabass. Linkage group (LG) 1–8. The locations of QTL affecting growth-related traits are circled on the map. The names of QTL are shown in italic and bold.

A genetic linkage map of Asian seabass. Linkage group (LG) 9–16. The locations of QTL affecting growth-related traits are circled on the map. The names of QTL are shown in italic and bold.

A genetic linkage map of Asian seabass. Linkage group 17–24. The locations of QTL affecting growth-related traits are circled on the map. The names of QTL are shown in italic and bold.
QTL mapping
QTL affecting body weight, total length and standard length were identified on a genome-wide scale. The genome-wide LOD significance thresholds were 4.5, 6.1 and 6.1 for body weight, total length and standard length, respectively, while the linkage-group-wide LOD significance thresholds varied from 1.7 to 5.4 (Table 2). Five significant (genome-wide significant) and 27 suggestive (linkage-group-wide significant) QTL were detected by the whole genome scan (Table 2 and Figure 1, 2, 3 and 4). Multiple QTL Model (MQM) mapping with initial QTL did not change the results.

Mapping of QTL for growth-related traits. a, QTL for total weight on LG2; b, QTL for body weight on LG3; c, QTL for total length on LG2; d, QTL for body length on LG2; e, QTL for KTL on LG6; f, QTL for KSL on LG6. The lines were drawn by plotting the LOD scores at each marker as well as at 1.0 cM intervals along the linkage group. The QTL were detected with genome-wide threshold for a, b, c and d and linkage-group-wide threshold for e and f.
Three significant QTL (qBW2-a, qTL2-a and qSL2-a) controlling body weight, total length and standard length, were detected on the same region near the microsatellite marker Lca287 on LG2, and explained 28.8, 58.9 and 59.7% of the observed phenotypic variance, respectively (Table 2). The fourth significant QTL(qBW2-b) affecting body weight was detected near Lca371 on LG2. This QTL accounted for 6.4% of the phenotypic variance. The fifth significant QTL (qBW3) affecting body weight was located near Lca154 on LG3, and explained 8.8% of the phenotypic variance.
The 27 suggestive QTL affecting growth-related traits were located on ten linkage groups (LG1, 2, 3, 6, 7, 11, 12, 16, 20 and 21) (Table 2 and Figure 1, 2 and 3). Three suggestive QTL for body weight (i.e. qBW12, qBW16 and qBW20) were detected on LG12, 16 and 20, respectively. These QTL explained relatively low phenotypic variance. Six suggestive QTL for standard and total length were detected on LG1, 2, 3, 12 and 16, respectively. In the regions of Lca148 on LG1, Lca287 and Lca371 on LG2, Lca154 on LG3, Lca436 on LG7, Lca178 on LG12, LcaE22 on LG16, and Lca349 on LG21 as well, QTL controlling more than one growth-related traits were detected.
Suggestive QTL associated with the condition factors were located on six different LGs (LG6, 7, 11, 12, 16 and 21). Some suggestive QTL for KTL and KSL (i.e. qKTL6-a and qKSL6-a, qKTL6-b and qKSL6-b, qKTL7 and qKSL7; qKTL16 and qKSL16, qKTL21-a and qKSL21-a), were located on the same locations of LG6 7, 16 and 21, respectively, but some (e.g. qKTL11-a, qKTL11-b, qKTL12 and qKSL21-a) were mapped on different locations.
Effects of QTL allele substitution
A two-way ANOVA was performed on the 380 progeny using four allelic combinations (m1f1, m1f2, m2f1 and m2f2) from markers nearest to the five significant QTL in order to investigate associations between phenotypic traits and genotypes. The phenotype values of each allelic combination are listed in Table 2. Out of the five significant QTL, progeny with only one m2f1 genotype at any of the four QTL, qBW2-a, qTL2-a, qSL2-a and qBW3, showed higher phenotype values than those without m2f1, while individuals with genotype m2f2 at QTL qBW2-b displayed heavier body weight than individuals without this genotype (Table 2 and Figure 5A and 5B). At the extreme, a difference of 15.8 g in BW were observed between individuals with genotypes m2f1 and m1f2 at the QTL qBW2-a. Similarly, difference in total length between individuals with genotypes m2f1 and m1f2 at QTL qTL2-a was 34.3 mm (Table 2 and Figure 5A and 5B). By using a single marker nearest to every suggestive and significant QTL, the effects of alleles within paternal and maternal parents and their interactions were further analyzed (Table 2). Among the five significant QTL, qBW2-a, qBW2-b, qTL2-a and qSL2-a showed significant effects within both the male and female alleles, and qBW3 showed significant effects within female alleles only. At suggestive QTL, QTL allele substitution effects were also observed (Table 2).

The effects of genotypes on body weight (A), total length and standard length (B). The genotypes were derived from the markers near QTL peaks. Significant differences between phenotype means are the estimated effect of alternate alleles inherited from the male parent (m1 or m2), alleles inherited from the female parent (f1 or f2) and/or their interaction. The letters on the top of bars indicate the level of difference in phenotypic value between genotypes. The same letter indicates that the difference is statistically insignificant, whereas different letters represent statistically significant (Bonferroni T tests at P < 0.01) difference.
Discussion
Linkage mapping
As expected, the selected 97 microsatellites were mapped into 24 linkage groups. For the majority of LGs, three or more markers were mapped. Marker order in every LG were almost identical to that in our published linkage map [28]. The total map length was 690.6 cM, quite similar to the average length of male and female maps we published [28]. The average marker interval was 7.12 cM, smaller than the required minimum of 10–20 cM for QTL mapping [31]. Therefore, this linkage map is suitable for a preliminary genome-wide scan for QTL on growth-related traits.
QTL mapping
Accurate QTL analyses have been developed in recent years to detect and estimate QTL effects on economically important livestock and plant species [32, 33]. In most studies employing QTL mapping, two generations of mapping populations were created by crossing genetically different lines or strains, followed by producing second generation progeny from mating F1 males and females or back-crossing F1 progeny to its parents in order to increase the power for QTL detection [7, 8, 34]. However, for species such as cattle, trees and most marine fish species, long generation interval meant that creating F2 generations or backcross families will take a long time, and therefore, is not easily feasible in practice. Methods for detection of QTL in F1 (full-sib or half-sib) families were developed [23–25] and successfully applied to plants [26, 27], cattle [35] and sheep [36]. The power to detect QTL depends on heritability of traits, average allelic substitution effect of the alleles involved, recombination distance between the QTL and associated markers, and sample size of the progeny [37]. In most marine fish species, as in the Asian seabass, which are broadcast spawners with a long generation interval (3–4 years), they are prolific spawners and are highly fecund. Furthermore, most cultured marine foodfish are progeny of wild-collected brooders having not been selected for any traits, as such, possess high levels of genetic variation. Hence, marine foodfish would be ideal for testing QTL analyses in F1 full-sib families. In this study we conducted a genome scan for QTL for growth-related traits in an F1 family including 380 individuals from a breeding stock. Five significant and 27 suggestive QTL were detected on the genome. Our study represents the first example of detecting QTL for growth-related traits by using a single F1 family with large number of offspring on the whole genome in marine foodfish. However, using of an F1 full-sib family from a breeding population for QTL analysis has its limitation. The power of such QTL analysis is limited. Only the QTL that are segregating in one or both parents can be detected. The detected QTL may not be representative for the genetic architecture of growth of Asian seabass. Thus, the detected QTL should be verified in other Asian seabass families before they are used in selective breeding.
Significant and suggestive QTL for body weight were detected in five LGs in this study, while in rainbow trout, QTL for body weight were detected in seven different LGs using a genome-scan [38]. The proportion of variance explained by the significant QTL for body weight varied from 6.4% to 28.8% in this study, which is similar to the 11–25% that was found in trout [38]. Among the significant QTL detected in this study, qBW2-a, qTL2-a and qSL2-a located on LG2, explained high percentages of the observed phenotypic variance, indicating that growth-related traits of Asian seabass might be controlled by a few QTL with large effects, together with some QTL with minor effects. Our results in this vertebrate system are consistent with a number of genetic studies in plants and insects suggesting that a large proportion of quantitative variation can be explained by the segregation of a few major QTL [39].
At some chromosomal regions, QTL controlling more than one growth-related traits were detected, suggesting either the linkage of two or more QTL (one for each trait), or the presences of a single QTL on each LG with pleiotropic effects. However, distinguishing between a single gene versus a gene cluster with individual QTL proved to be difficult, given the current marker density in the first generation linkage map[28]. More markers are needed to identify the full spectrum of genetic changes, which contribute to growth differences and to narrow down the location of a gene or genes within each chromosome interval with contribution to growth divergence. Isolation of additional microsatellites, SNPs, and microsatellites in ESTs and genes are still underway, to allow for the construction of a high resolution linkage map for fine mapping of QTL and gene of interest.
Suggestive QTL for condition (K) factors were located on six LGs, and their effects were relatively small. In rainbow trout, only a QTL for K factor was mapped in one LG [40], whereas in salmon four significant QTL for K factor were mapped to four LGs [19]. In Asian seabass, some QTL for the traits KTL and KSL were located on the same positions, while others were located in different places, suggesting that the two K factors are controlled by some genes in common and some different genes with small effects. Since the QTL were detected in only an F1 full-sib family and the effects of these QTL are small, confirmation in other families is required.
We have noticed that the total PVE of six significant and suggestive QTL for the trait standard length was almost 100%, which is theoretically impossible. However in practice, similar results have been reported in previous QTL analyses. For example, in the three published papers [19, 41, 42] on QTL mapping in fish, the total PVE of QTL for a certain trait was over 100%. There might be several reasons for this phenomenon. Firstly, interaction among QTL was not included in QTL analysis due to the limitation of the software. If the correlation between QTL is large, they share a certain amount of variation and are not completely independent. Secondly, the proportion of the variation explained by these QTL is likely an overestimate due to the Beavis effect [43, 44]. More makers on the linkage map, larger population size and more improved statistic models to detect QTL interactions are expected to solve this problem.
A two-way ANOVA revealed that allelic substitution at the microsatellite markers located near the five significant QTL showed large effects on growth-related traits. Certain allelic combinations showed significantly improved performances of the traits analyzed. The genotypes at these marker loci may be useful for growth improvement through maker-assisted selection in this family. At four of the five significant QTL, both alleles from the father and the mother showed significant effects, suggesting that QTL alleles of both parents are segregating. Whereas, at one significant QTL (qBW3), alleles from the father showed significant effects, but alleles from mother did not show effects, indicating the segregation of QTL alleles of the father, and fixation of QTL alleles from the mother. QTL allele substitution effects were also seen at suggestive QTL, suggesting the segregation of QTL alleles in both parents or one of the two parents.
Candidate gene approaches have been used for identification of QTL [45]. In a previous study, we demonstrated that the polymorphism in the gene PVALB1 located on LG23 was significantly associated with traits body weight and length in Asian seabass [30]. However, this gene was not polymorphic in the full-sib family for QTL analysis, and no significant QTL for traits was detected on LG23. Another potential candidate gene is the growth hormone (GH) gene, which was mapped near the microsatellite marker Lca226 on the LG3 in the first generation linkage map of Asian seabass [28]. This gene plays an important role in the growth of organisms. Previous studies in pig, chicken and cattle indicated that polymorphisms in the GH gene were associated with growth-related traits and other economically important traits [46–48]. Unfortunately, the microsatellite marker Lca003 located in the promotor of the GH gene was not informative in our mapping family. A significant QTL affecting body weight, explaining 8.8% of the phenotypic variance was mapped on LG3, but the position was about 13.9 cM away from the GH gene, suggesting the GH gene is not associated with growth-related traits in this family. To examine the effects of the GH and PVALB1 genes on economically important traits, more polymorphisms in these two genes will be detected.
Conclusion
We detected five significant and 27 suggestive QTL for growth-related traits in an F1 family from a breeding stock of Asian seabass. This study presents the first example of QTL detection in an F1 family of a marine foodfish. The results presented will enable fine-mapping of the identified QTL for marker-assisted selection and eventually identifying individual genes responsible for these traits for direct selection
Methods
Source of fish
A broodstock consisting of 94 brooders from the Marine Aquaculture Center, Singapore were genotyped with nine polymorphic microsatellites (Lca08, Lca20, Lca21, Lca58, Lca64, Lca69, Lca70, Lca74 and Lca98) as described previously [6, 28]. Genetic relationships among individuals were estimated by using genetic similarity according to Nei and Li [49]. Crosses were performed by selecting distantly related males and females to produce the largest genetic variation in the next generation. All brooders were tagged with passive integrated transponder (PIT) tags (IER, BP, France). From a mass-spawning of ten male and ten female brooders, which showed high genetic difference at the nine loci, about 20,000 fertilized eggs were brought to hatchery and were cultured in a one-ton tank following standard culture protocols [50] with modification until 30 dph. As Asian seabass juveniles are highly carnivorous, grading is necessary. From 30 dph on, these larvae were subsequently graded into three distinct sizes (large, medium and small) every week up to 65 dph, and transferred into cylindrical three-ton capacity tanks with continuous flow-through of optimum quantity of sand-filtered seawater. Juveniles were raised communally in their respective tanks with same stocking density, and were fed to satiation three times (9:30 am, 1:30 pm and 5:30 pm) daily on stringently sized pellets (INVE, PHICHIT, Thailand) until the experiment ended at 90 dph. Other conditions (i.e. air flow, water flow and light intensity) in the tanks were maintained as same as possible.
Traits and DNA samples
At 90 dph, body weight (BW), total length (TL) and standard length (SL) of 600 progeny randomly collected from tanks with the largest and smallest fish were measured. Fulton's coefficients of condition (KTL and KSL) were calculated for each fish according to BW with TL or SL as described [51]. Fin clips of the parents and their progeny were collected and kept in absolute ethanol. DNA was isolated from fin clips using a new method developed by us [52]. The quality of DNA was checked on 1% agarose gel, and the quantity measured using Nanodrop (NanoDrop, DE, USA). DNA concentration of each fish was adjusted to 2.5 ng/μl and arrayed into 96-well PCR plates for later use. By genotyping the fish with nine microsatellite markers [6], pedigree was reconstructed. One parental pair producing the most offspring, 380 of them were used for linkage and QTL analysis.
Microsatellites and genotyping
Ninety-seven microsatellite markers almost evenly covering the 24 LGs were selected from a first-generation linkage map of Asian seabass [28]. Primers were designed for each unique microsatellite using PrimerSelect (Dnastar, WI, USA). One primer of each pair was labeled with FAM or HEX fluorescent dyes at the 5'end. The PCR program for microsatellite amplifications on PTC-100 PCR machines (MJ Research, CA, USA) consisted of the following steps: 94°C for 2 min followed by 37 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 45 s, then a final step of 72°C for 5 min. Each PCR reaction consisted of 1× PCR buffer (Finnzymes, Espoo, Finland) with 1.5 mM MgCl2, 200 nM of each PCR primer, 50 μM of each dNTP, 10 ng genomic DNA and one unit of DNA-polymerase (Finnzymes, Espoo, Finland). Products were analyzed using a DNA sequencer ABI3730xl, and fragment sizes were determined against the size standard ROX-500 (Applied Biosystems, CA, USA) with software GeneMapper V3.5 (Applied Biosystems, CA, USA) as described previously[28].
Linkage mapping
Linkage analyses were conducted through a series of pairwise comparisons between loci using software LINKMFEX [53]. The analyses were executed using segregation data for 97 microsatellite markers in 380 progeny. Pairwise recombination estimates obtained with module LINKMFEX were used as input into module MAPORD to determine linear assignments of markers within a linkage group. A minimum LOD score of 3.0 was used to assign markers to LGs. Map distances were calculated using the Kosambi function with module MAPDIS. Using module MERGE, new LG orders were built from pre-existing LG orders that were obtained from the male and female parents. Linkage map was constructed by averaging recombination rates across the sexes. Map graphics were drawn with MapMaker software [54].
QTL analysis
With genotype data of the 97 markers and phenotypic data of the 380 progeny, QTL analysis was carried out using the program MapQTL 4.0 [25]. Interval mapping and multiple QTL model (MQM) mapping were utilized to detect any significance associating growth-related traits and marker loci in the data sets. The LOD score significance thresholds were calculated by permutation tests in MapQTL 4.0, with a genome-wide significance level of α < 0.05, n = 1000 for significant linkages and a linkage-group-wide significance level of α < 0.05, n = 1000 for suggestive linkages.
To determine the differences among the genotypes of markers nearest to each QTL, an analysis of variance (ANOVA), based on the genotypes (m1f1, m1f2, m2f1 and m2f2) of the marker locus lying closest to the peak in each of the QTL-containing genomic regions were also carried out to characterize the QTL allele substitution effects. A simple Bonferroni correction, to reduce the chance of type 1 error to 0.01 across all tests, gave a significance threshold of 0.0025, since we consider that tests were performed among four genotypes of each marker. This was conducted by using the general linear model (GLM) procedure of SAS (SAS Institute) and the Bonferroni method of multiple comparisons with α < 0.01. For the marker nearest to each QTL, effects of alleles within two parents and the interaction between paternal and maternal alleles were analyzed through ANOVA using GLM of SAS at α < 0.05.
Abbreviations
- QTL:
-
quantitative trait loci
- LG:
-
linkage group
- BW:
-
body weight
- TL:
-
total length
- SL:
-
standard length
- qBW2-a :
-
QTL for body weight located on position a on linkage group 2
- qTL2-a :
-
QTL for total length located on position a on linkage group 2
- qBL2-a :
-
QTL for body length located on position a on linkage group 2
- KTL and KSL:
-
condition factors calculated based on the total length and standard length
- cM:
-
centimorgan
- GH :
-
growth hormone gene
- ANOVA:
-
Analysis of variance
- PVE:
-
percentage of the phenotypic variance explained.
References
Chou R, Lee HB: Commercial marine fish farming in Singapore. Aquac Res. 1997, 28: 767-776. 10.1046/j.1365-2109.1997.00941.x.
FAO: Databases and Statistics. 2003, http://www.fao.org/fi/statist/statist.asp-
Chenoweth SF, Hughes JM, Keenan CP, Lavery S: Concordance between dispersal and mitochondrial gene flow: isolation by distance in a tropical teleost, Lates calcarifer (Australian barramundi). Heredity. 1998, 80: 187-197. 10.1046/j.1365-2540.1998.00292.x.
Keenan CP: Recent evolution of population structure in Australian barramundi, Lates calcarifer (Bloch): an example of isolation by distance in one dimension. Australian Journal of Marine and Freshwater Research. 1994, 45: 1123-1148. 10.1071/MF9941123.
Yue GH, Li Y, Chao TM, Chou R, Orban L: Novel microsatellites from Asian sea bass (Lates calcarifer) and their application to broodstock analysis. Marine Biotechnology. 2002, 4: 503-511.
Zhu ZY, Lin G, Lo LC, Xu YX, Renee C, You GH: Genetic analyses of Asian seabass stocks using novel polymorphic microsatellites. Aquaculture. 2006, 256: 167-173. 10.1016/j.aquaculture.2006.02.033.
Dekkers JCM, Hospital F: Multifactoral genetics: The use of molecular genetics in the improvement of agricultural populations. Nature Reviews Genetics. 2002, 3: 22-32. 10.1038/nrg701.
Andersson L, Georges M: Domestic-animal genomics: deciphering the genetics of complex traits. Nature Reviews Genetics. 2004, 5: 202-212. 10.1038/nrg1294.
Liu ZJ, Cordes JF: DNA marker technologies and their applications in aquaculture genetics. Aquaculture. 2004, 238: 1-37. 10.1016/j.aquaculture.2004.05.027.
Young WP, Wheeler PA, Coryell VH, Keim P, Thorgaard GH: A detailed linkage map of rainbow trout produced using doubled haploids. Genetics. 1998, 148: 839-850.
Gilbey J, Verspoor E, McLay A, Houlihan D: A microsatellite linkage map for Atlantic salmon (Salmo salar). Animal Genetics. 2004, 35: 98-105. 10.1111/j.1365-2052.2004.01091.x.
Kocher TD, Lee WJ, Sobolewska H, Penman D, McAndrew B: A genetic linkage map of a cichlid fish, the tilapia (Oreochromis niloticus). Genetics. 1998, 148: 1225-1232.
Liu Z, Karsi A, Li P, Cao D, Dunham R: An AFLP-based genetic linkage map of channel catfish (Ictalurus punctatus) constructed by using an interspecific hybrid resource family. Genetics. 2003, 165: 687-694.
Ohara E, Nishimura T, Nagakura Y, Sakamoto T, Mushiake K, Okamoto N: Genetic linkage maps of two yellowtails (Seriola quinqueradiata and Seriola lalandi). Aquaculture. 2005, 244: 41-48. 10.1016/j.aquaculture.2004.10.022.
Sun XW, Liang LQ: A genetic linkage map of common carp (Cyprinus carpio L.) And mapping of a locus associated with cold tolerance. Aquaculture. 2004, 238: 165-172. 10.1016/S0044-8486(03)00445-9.
Somorjai IM, Danzmann RG, Ferguson MM: Distribution of temperature tolerance quantitative trait loci in Arctic charr (Salvelinus alpinus) and inferred homologies in rainbow trout (Oncorhynchus mykiss). Genetics. 2003, 165: 1443-1456.
Jackson TR, Ferguson MM, Danzmann RG, Fishback AG, Ihssen PE, O'Connell M, Crease TJ: Identification of two QTL influencing upper temperature tolerance in three rainbow trout (Oncorhynchus mykiss) half-sib families. Heredity. 1998, 80: 143-151. 10.1046/j.1365-2540.1998.00289.x.
Robison BD, Wheeler PA, Sundin K, Sikka P, Thorgaard GH: Composite interval mapping reveals a major locus influencing embryonic development rate in rainbow trout (Oncorhynchus mykiss). J Hered. 2001, 92: 16-22. 10.1093/jhered/92.1.16.
Reid DP, Szanto A, Glebe B, Danzmann RG, Ferguson MM: QTL for body weight and condition factor in Atlantic salmon (Salmo salar): comparative analysis with rainbow trout (Oncorhynchus mykiss) and Arctic charr (Salvelinus alpinus). Heredity. 2005, 94: 166-172. 10.1038/sj.hdy.6800590.
Moen T, Fjalestad KT, Munck H, Gomez-Raya L: A multistage testing strategy for detection of quantitative trait loci affecting disease resistance in Atlantic salmon. Genetics. 2004, 167: 851-858. 10.1534/genetics.103.013227.
Cnaani A, Zilberman N, Tinman S, Hulata G, Ron M: Genome-scan analysis for quantitative trait loci in an F-2 tilapia hybrid. Molecular Genetics and Genomics. 2004, 272: 162-172. 10.1007/s00438-004-1045-1.
Cnaani A, Hallerman EM, Ron M, Weller JI, Indelman M, Kashi Y, Gall GAE, Hulata G: Detection of a chromosomal region with two quantitative trait loci, affecting cold tolerance and fish size, in an F-2 tilapia hybrid. Aquaculture. 2003, 223: 117-128. 10.1016/S0044-8486(03)00163-7.
Knott SA, Haley CS: Maximum likelihood mapping of quantitative trait loci using full-sib families. Genetics. 1992, 132: 1211-1222.
Cervantes-Martinez C, Brown JS: A haplotype-based method for QTL mapping of F1 populations in outbred plant species. Crop Science. 2004, 44:
Van Ooijen JW, Boer MP, Jansen RC, C. M: MapQTL 4.0: Software for the calculation of QTL positions on genetic maps. 2002, , Wageningen (the Netherlands), Plant Research International
Bink MCAM, Sandbrink JM, Jansen RC: Linkage and QTL mapping in related full sib families–a case study in potato. Journal of Agricultual Genomics. 2001, 2: 1-14.
Okogbenin E, Fregene M: Genetic mapping of QTLs affecting productivity and plant architecture in a full-sib cross from non-inbred parents in Cassava (Manihot esculenta Crantz). Theoretical and Applied Genetics. 2003, 8: 1452 -11462.
Wang CM, Zhu ZY, Lo LC, Feng F, Lin G, Li J, Yang WT, Yue GH: A Microsatellite linkage map for Barramundi (Lates calcarifer). Genetics. 2006, submitted:
Wang CM, Zhu ZY, Lo LC, Lin G, Feng F, Li J, Yang WT, Orban L, Renee C, Yue GH: Genetic analysis of growth-related traits using microsatellite-inferred relationships in Asian seabass (Lates calcarifer). Aquaculture. 2006, in revision:
Xu YX, Zhu ZY, Lo LC, Wang CM, Lin G, Feng F, Yue GH: Characterization of two parvalbumin genes and their association with growth traits in Asian seabass (Lates calcarifer). Animal Genetics. 2006, 37: 266-268. 10.1111/j.1365-2052.2006.01423.x.
Beckmann JS, Soller M: Detection of linkage between marker loci and loci affecting quantitative traits in crosses between segregating populations. Theoretical and Applied Genetics. 1988, 76: 228-236. 10.1007/BF00257850.
Haley CS, Knott SA, Elsen JM: Mapping quantitative trait loci in crosses between outbred lines using least-squares. Genetics. 1994, 136: 1195-1207.
Jansen RC, Stam P: High-resolution of quantitative traits into multiple loci via interval mapping. Genetics. 1994, 136: 1447-1455.
Minvielle F, Kayang BB, Inoue-Murayama M, Miwa M, Vignal A, Gourichon D, Neau A, Monvoisin JL, Ito S: Microsatellite mapping of QTL affecting growth, feed consumption, egg production, tonic immobility and body temperature of Japanese quail. BMC Genomics. 2005, 6:
Velmala RJ, Vilkki HJ, Elo KT, de Koning DJ, Maki-Tanila AV: A search for quantitative trait loci for milk production traits on chromosome 6 in Finnish Ayrshire cattle. Animal Genetics. 1999, 30: 136-143. 10.1046/j.1365-2052.1999.00435.x.
Diez-Tascon C, Bayon Y, Arranz JJ, De La Fuente F, San Primitivo F: Mapping quantitative trait loci for milk production traits on ovine chromosome 6. Journal of Dairy Research. 2001, 68: 389-397. 10.1017/S0022029901004988.
Mackay TFC: The nature of quantitative genetic variation revisited: lessons from Drosophila bristles. BioEssays. 1996, 18: 113-121. 10.1002/bies.950180207.
O'Malley KG, Sakamoto T, Danzmann RG, Ferguson MM: Quantitative trait loci for spawning date and body weight in rainbow trout: Testing for conserved effects across ancestrally duplicated chromosomes. Journal of Heredity. 2003, 94: 273-284. 10.1093/jhered/esg067.
Tanksley SD: Mapping polygenes. Ann Rev Genet. 1993, 27: 205-233. 10.1146/annurev.ge.27.120193.001225.
Martyniuk CJ, Perry GML, Mogahadam HK, Ferguson MM, Danzmann RG: The genetic architecture of correlations among growth-related traits and male age at maturation in rainbow trout. Journal of Fish Biology. 2003, 63: 746-764. 10.1046/j.1095-8649.2003.00188.x.
Leder EH, Danzmann RG, Ferguson MM: The candidate gene, Clock, localizes to a strong spawning time quantitative trait locus region in rainbow trout. J Hered. 2006, 97: 74-80. 10.1093/jhered/esj004.
Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jonsson B, Schluter D, Kingsley DM: Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004, 428: 717-723. 10.1038/nature02415.
Xu S: Theoretical basis of the Beavis effect. Genetics. 2003, 165: 2259-2268.
Weller JI, Shlezinger M, Ron M: Correcting for bias in estimation of quantitative trait loci effects. Genet Sel Evol. 2005, 37: 501-522. 10.1051/gse:2005013.
Rothschild M, Jacobson C, Vaske D, Tuggle C, Wang LZ, Short T, Eckardt G, Sasaki S, Vincent A, McLaren D, Southwood O, vanderSteen H, Mileham A, Plastow G: The estrogen receptor locus is associated with a major gene influencing litter size in pigs. Proc Natl Acad Sci U S A. 1996, 93: 201-205. 10.1073/pnas.93.1.201.
Lagziel A, Lipkin E, Ezra E, Soller M, Weller JI: An MspI polymorphism at the bovine growth hormone (bGH) gene is linked to a locus affecting milk protein percentage. Anim Genet. 1999, 30: 296-299. 10.1046/j.1365-2052.1999.00472.x.
Nie Q, Sun B, Zhang D, Luo C, Ishag NA, Lei M, Yang G, Zhang X: High diversity of the chicken growth hormone gene and effects on growth and carcass traits. J Hered. 2005, 96: 698-703. 10.1093/jhered/esi114.
Knorr C, Moser G, Muller E, Geldermann H: Associations of GH gene variants with performance traits in F-2 generations of European wild boar, Pietrain and Meishan pigs. Anim Genet. 1997, 28: 124-128. 10.1111/j.1365-2052.1997.00093.x.
Nei M, Li WH: Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA. 1979, 76: 5269-5273. 10.1073/pnas.76.10.5269.
Pillay TVR: Aquaculture principles and practices. 1990, Oxford, Fishing News Books, 398-407.
Murphy B.R.: Fisheries Techniques. 1996, Bethesda, Maryland, American Fisheries Society
Yue GH, Orban L: A simple and affordable method for high throughput DNA extraction from animal tissues for PCR. Electrophoresis. 2005, 26: 3081-3083. 10.1002/elps.200410411.
Danzmann RG: Linkage analysis package for outcrossed families with male or female exchange of the mapping parent, version 2.3. http://www.uoguelph.ca/~rdanzman/ software/LINKMFEX. 2006
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L: MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987, 1: 174-181. 10.1016/0888-7543(87)90010-3.
Acknowledgements
This study is part of the project "Selective Breeding of Marine Foodfish" funded by AVA, Singapore and the internal research fund from Temasek Life Sciences Laboratory, Singapore. The authors would like to thank F. Feng, G. Lin, J. Li, W.T. Yang, R. Chou, H.S. Lim, J. Tan, H.Y. Pang and H.M. Liu for technical supports. Dr L. Orban was thanked for his comments on the manuscript before submission. We are grateful to Dr L. Zhou, Jiangsu Academy of Agricultural Sciences, China for his help in data analysis with MapQTL 4.0. The members of AnGenMap community are thanked for their very suggestive discussion on the explanation of PVE of QTL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
GHY designed and started the project, and determined the final version of the manuscript. CMW designed the experiment, selected markers, conducted genotyping and QTL analysis, as well as drafted the manuscript. LCL measured and recoded the trait values and conducted most of genotyping. ZYZ helped in data collection, DNA extraction and genotyping. All authors have read and approved the final version of the manuscript.
Chun Ming Wang, Loong Chueng Lo contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, C.M., Lo, L.C., Zhu, Z.Y. et al. A genome scan for quantitative trait loci affecting growth-related traits in an F1 family of Asian seabass (Lates calcarifer). BMC Genomics 7, 274 (2006). https://doi.org/10.1186/1471-2164-7-274
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-7-274