
Abstract
Background
GSL1 and GSL2, Gibberellin Stimulated-Like proteins (also known as Snakin-1 and Snakin-2), are cysteine-rich peptides from potato (Solanum tuberosum L.) with antimicrobial properties. Similar peptides in other species have been implicated in diverse biological processes and are hypothesised to play a role in several aspects of plant development, plant responses to biotic or abiotic stress through their participation in hormone crosstalk, and redox homeostasis. To help resolve the biological roles of GSL1 and GSL2 peptides we have undertaken an in depth analysis of the structure and expression of these genes in potato.
Results
We have characterised the full length genes for both GSL1 (chromosome 4) and GSL2 (chromosome 1) from diploid and tetraploid potato using the reference genome sequence of potato, coupled with further next generation sequencing of four highly heterozygous tetraploid cultivars. The frequency of SNPs in GSL1 and GSL2 were very low with only one SNP every 67 and 53 nucleotides in exon regions of GSL1 and GSL2, respectively. Analysis of comprehensive RNA-seq data substantiated the role of specific promoter motifs in transcriptional control of gene expression. Expression analysis based on the frequency of next generation sequence reads established that GSL2 was expressed at a higher level than GSL1 in 30 out of 32 tissue and treatment libraries. Furthermore, both the GSL1 and GSL2 genes exhibited constitutive expression that was not up regulated in response to biotic or abiotic stresses, hormone treatments or wounding. Potato transformation with antisense knock-down expression cassettes failed to recover viable plants.
Conclusions
The potato GSL1 and GSL2 genes are very highly conserved suggesting they contribute to an important biological function. The known antimicrobial activity of the GSL proteins, coupled with the FPKM analysis from RNA-seq data, implies that both genes contribute to the constitutive defence barriers in potatoes. The lethality of antisense knock-down expression of GSL1 and GSL2, coupled with the rare incidence of SNPs in these genes, suggests an essential role for this gene family. These features are consistent with the GSL protein family playing a role in several aspects of plant development in addition to plant defence against biotic stresses.
Similar content being viewed by others


Background
The gibberellin stimulated-like proteins GSL1 (also known as Snakin-1) and GSL2 (also known as Snakin-2) are cysteine-rich peptides from potato (Solanum tuberosum L.) with in vitro antimicrobial activity against a wide range of bacteria and fungi [1–5], as well as nematodes [6]. The spectrum of antimicrobial activity is almost identical for GSL1 and GSL2 [2, 3]. GSL1 and GSL2 induce rapid aggregation of both Gram-negative and Gram-positive bacteria, and although this response does not correlate with antimicrobial activity, it is still considered that these proteins may play an in vivo role in controlling pathogen migration [1–3, 5].
Amino acid sequence alignment of GSL1 and GSL2 show similarity with the GAST1 (gibberellic acid stimulated transcript) from tomato [7] and the GASA family (gibberellic acid stimulated in arabidopsis) from arabidopsis [8] and similar members from a wide range of dicotyledonous and monocotyledonous species [9–14]. Based on a limited similarity in amino acid sequence to the hemotoxic, desintegrin-like snake venoms, GSL1 and GSL2 were formerly referred to as Snakin-1 and Snakin-2 [2, 3]. However, the term Snakin is inappropriate for these plant-based proteins since GSL1 and GSL2 do not share the RGD residues and functional properties of snake venoms responsible for desintegrin action [15].
Both the StGSL1 and StGSL2 genes encode polypeptides that have similar structural features with an N-terminal putative signal sequence congruent with a sub-cellular location in the plant cell wall and a cysteine-rich C-terminal domain [2, 3]. GSL1 has a signal sequence of 25 amino acid residues, followed by a 63-amino acid mature peptide (6.9 kDa) with 12 highly conserved cysteine residues [2]. GSL2 has a 23 amino acid-residue signal peptide, followed by an intermediate 15-residue acidic peptide, and then a mature peptide (7.0 kDa) of a 66 amino acid basic peptide with the 12 conserved cysteine residues [3]. GSL1 and GSL2 peptides share several features characteristic of all antimicrobial peptides. The cysteine-rich nature of these peptides is critical for the occurrence of disulphide bridges that are important for enhancing the structural stability under diverse stress conditions [16]. A high frequency of charged amino acids appears to play a key role in the activity against microbes [16], along with the amphipathic structure and cationic charge at physiological pH [17–19]. The prediction of GSL1 three-dimensional structure and disulfide bonding pattern revealed two long α-helices stabilized and maintained by six knotted disulfide bonds between specific cysteine residues [20].
Northern analysis in potatoes established that transcripts of StGSL1 exhibited highest accumulation in stems, shoot apices, young floral buds and petals, with expression also detected in tubers and carpels, but not in roots, stolons, leaves, sepals or stamens [2, 3]. Transcripts in leaves were not induced by either abiotic or biotic stress, or chemical treatments such as jasmonic acid, salicylic acid, isonicotinic acid, abscisic acid, gibberellic acid, and indolacetic acid, leading to the conclusion that GSL1 is a component of the constitutive defence barriers, especially of the storage and reproductive organs [2]. Similar studies on StGSL2 expression detected the highest accumulation of transcripts in tubers, petals and carpels, with expression also in stems, shoot apices, leaves, flower buds and stamens, but not in roots, stolons and sepals [3]. In contrast to StGSL1, the StGSL2 gene was locally up-regulated in leaves by wounding and abscisic acid treatments, responded weakly to salinity stress, while drought stress or treatments with gibberellic acid, chitosan, jasmonic acid, ethylene, benzo(1,2,3)thiadiazole-7-carbothioic acid or S-methyl ester had no effect [3]. StGSL2 expression was also up-regulated upon infection of tubers with the compatible fungus Botrytis cinerea, but down-regulated by the bacterial pathogens Ralstonia solanacearum and Dickeya chrysanthemi (formerly known as Erwinia chrysanthemi), resulting in the overall hypothesis that GSL2 is a component of both constitutive and inducible defence barriers to pathogens [3].
Over-expression of the StGSL1 gene in transgenic potato plants enhances resistance against two important potato pathogens Pectobacterium carotovorum subspecies carotovorum (formerly known as Erwinia carotovora) and Rhizoctonia solani[21]. Transgenic wheat plants over-expressing the Solanum chacoense GSL1 gene exhibited improved resistance to Blumeria graminis f.sp. tritici[22]. Likewise, over-expression of the tomato GSL2 gene in tomato enhanced tolerance to Clavibacter michiganensis subsp. michiganensis that causes bacterial canker and wilt disease [23]. Viral-induced gene silencing of GSL2 in Nicotiana benthamiana increased susceptibility to wilt disease development induced by C. michiganensis subsp. michiganensis[24]. Similarly, virus-induced gene silencing of GSL2 in Capsicum annuum increased susceptibility to root-knot nematodes (Meloidogyne spp.) [6]. A defence role for GSL1 was also suggested from the observation of decreased virulence of GSL1-sensitive mutants of Dickeya chrysanthemi (formerly known as Erwinia chrysanthemi) to potato tubers [25]. The antimicrobial mechanism of action for GSL peptides is not known, but in contrast to other antimicrobial peptides from plants, GSL1 and GSL2 do not interact with artificial lipid membranes [1]. A cysteine-rich 6.8 kDa orthologue of GSL2 from French bean (Phaseolus vulgaris) was demonstrated to tightly bind to a 25 kDa polypeptide of a proline-rich protein family from legumes and thought to function as a two-component chitin-receptor involved in plant-pathogen interactions through antimicrobial activity and/or signalling [26].
There is no consensus on the biological roles of GSL proteins. Given their in vitro antimicrobial activity they are often considered to play important roles in the innate defence against invading microorganisms [2, 3, 6] and/or to be a key determinant during the interaction between plants and pathogens [25, 26]. Similar genes in other species have been implicated in diverse biological processes, including: cell division, cell elongation, cell growth, transition to flowering, somatic embryogenesis and signalling pathways [10–12, 27–30]. Despite the highly conserved nature of GSL/GASA amino acid sequences, including 12 cysteine residues at the C-terminus that are probably responsible for the protein structure and biochemical activity, the functions and mode of action of GSL/GASA proteins are not completely elucidated [31]. The prevailing view is that GSL/GASA proteins play a role in several aspects of plant development, plant responses to biotic or abiotic stress through their participation in hormone crosstalk, and redox homeostasis [31]. This is supported by partial silencing of GSL1 in potato using antisense approaches that resulted in plants with reduced height and smaller leaves resulting from reduced cell division, altered leaf metabolism and cell wall composition [32].
To help resolve the biological roles of GSL1 and GSL2 peptides we have undertaken a thorough analysis of the structure and expression of these genes in potato. We have characterised the full length genes for both GSL1 and GSL2 from diploid and tetraploid potato using the genome sequence of potato [33], coupled with further next generation sequencing of highly heterozygous tetraploid cultivars. Specific promoter motifs and exon regions are highly conserved among multiple alleles, suggesting their importance for biological function. Analysis of comprehensive transcriptome data substantiates the role of specific promoter motifs in transcriptional control of gene expression. The lethality of antisense knock-down expression suggests the essential role of this gene family in potatoes.
Results and discussion
Allelic polymorphism of GSL genes in potato
PCR isolation, cloning and sequencing of the coding region from the autotetraploid potato cultivar ‘Iwa’ revealed two alleles (a1 and a2) for the GSL1 gene (GenBank accessions FJ195646 and FJ195647) and two alleles (b1 and b2) for the GSL2 gene (GenBank accessions EU848497 and EU848498). From the frequency of clones with the GSL1 alleles, it is estimated that Iwa has three copies of the a1 allele (15 of 16 clones) and one copy of the a2 allele (1 of 16 clones). Similarly for the GSL2 gene, Iwa has three copies of the b1 allele (10 of 12 clones) and one copy of the b2 allele (2 of 12 clones). The a1 and a2 alleles of the GSL1 gene differed by 18 Single Nucleotide Polymorphisms (SNPs) and four indels of 1–7 nucleotides. All of these variant nucleotides were in the introns, except for only three synonymous SNPs in the exons. Similarly, the b1 and b2 alleles of the GSL2 gene were polymorphic for 19 SNPs and five indels of 1–18 nucleotides, with only four synonymous SNPs all occurring in the third exon of GSL2.
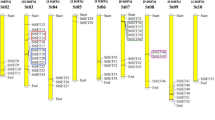
PCR isolation and direct sequencing of cDNA products determined the sequence of cDNA from mature transcripts for both the GSL1 (GenBank accession GU137307) and GSL2 (GenBank accession JF683606) genes. Alignment of the genomic and cDNA sequences confirmed the exon and intron regions in both the GSL1 and GSL2 genes (Figure 1). The GSL1 gene consists of two exons of 82 and 187 nucleotides interrupted by a single intron of 525 nucleotides. The GSL2 gene is composed of three exons of 87, 46 and 182 nucleotides respectively, interspersed with two introns of 268 and 172 nucleotides. The exon/intron boundaries were identical in all alleles of both the GSL1 and GSL2 genes, with the splice sites possessing the conserved 5’GT and 3’AG dinucleotides (Additional file 1: Figure S1 and Additional file 2: Figure S2), consistent with the consensus sequences of the intron at both donor and acceptor sites [34].

Schematic representation of the GSL1 gene (A) and GSL2 gene (B). The 5’UTR and 3’UTR, exon and intron regions are indicated in red, orange and yellow, respectively. The vertical blue lines indicate positions at which SNPs were observed, the horizontal blue bars are positions where indels occur and the green ovals mark the positions of promoter motifs with the encased numbers indicative of the specific motifs noted in Tables 2 and 3 for the GSL1 and GSL2 genes, respectively. This representation is based on a consensus sequence of all genotypes analysed. Due to the presence of polymorphic indels, the exact nucleotide positions do not necessarily match the information for the genotype DM presented in Tables 2 and 3, or Additional file 1: Figure S1 and Additional file 2: Figure S2.
Structure of full length GSL genes
The sequence of the coding regions of the GSL1 and GSL2 genes described above were used for interrogation of the potato genome sequence [33]. Firstly, GSL1 and GSL2 nucleotide sequences were used to BLAST search the reference CDS and genomic sequences to identify gene locations on superscaffold assemblies. Secondly, annotated protein sequences from potato genome assembly version 3.4 of the genotype DM1-3 516 R44 (DM) were also searched for the presence of the GASA pfam motif PF02704 (http://pfam.sanger.ac.uk/).
Results showed single copy genes for each of GSL1 and GSL2. GSL1 is located on superscaffold PGSC0003DMB000000381 which has been mapped to chromosome 4, whereas GSL2 is located on superscaffold PGSC0003DMB000000290 which has been mapped to chromosome 1 (Additional file 3: Table S1). Several other genes similar to GSL and GASA genes were also identified; results of these searches are shown in Additional file 3: Table S1.
The general structure of the GSL1 and GSL2 genes was analysed based on the DM sequence of the potato genome [33]. For the GSL1 gene, up to 1967 nucleotides were recovered upstream from the putative transcription start site. We were only able to confirm up to 616 nucleotides upstream of the putative transcription start site for the GSL2 gene due to a gap in the assembly of the reference potato genome in the upstream region of the GSL2 promoter. However, we documented at least 1000 nucleotides downstream of the stop codon for both GSL genes. A putative transcription start site was predicted based on a plant dimer motif YR Rule [35] at 33 nucleotides and 38 nucleotides upstream from the first base of the translation start site (ATG) for the GSL1 and GSL2 genes, respectively. Putative cis-elements were identified for the GSL1 gene (Additional file 1: Figure S1). These are TATA-box (nucleotides −32 to −27), a pyrimidine patch (Y Patch, nucleotides −26 to −20) and CAAT-box (nucleotides −48 to −44). In GSL2, a putative TATA-box and Y Patch were located from nucleotides −50 to −45 and nucleotides −59 to −51, respectively. Since we were unable to locate a satisfactory CAAT-box, we identified a ‘hypothetical’ CAAT-box (nucleotides −65 to −61) in the GSL2 promoter (Additional file 2: Figure S2).
Next generation sequencing and SNP discovery
Genomic regions for GSL1 and GSL2 were further analyzed for sequence variation by aligning re-sequence data generated from Illumina reads of the diploid RH89-039-16 (RH) [33] and data generated from four tetraploid lines using the Illumina GAIIx platform. Illumina short insert read pair data were generated for each line. Reads were aligned to the reference genome using BWA [36]. Alignments were further analysed using SAMtools [37], polySNP (an in-house developed tool for SNP calling; https://github.com/mfiers/polysnp) and visualized using Geneious [38].
For the diploid RH and the four tetraploid potato genotypes the structure of the GSL1 and GSL2 genes and sequence polymorphisms for the various alleles were annotated manually and compared to the reference DM potato genome. The locations of SNPs and indels (insertion/deletions) identified across all alleles from all genotypes is illustrated in Figure 1 and summarized in Table 1. In GSL1 multiple SNPs and indels were identified within the non-coding regions, with the greatest frequency occurring within the intron and the region from −2000 to −500 nucleotide positions relative to the 5’UTR. The 5’UTR contains only a single SNP that is found in one re-sequenced genotype. The exon regions have no indels and only very rare synonymous SNPs, with an overall SNP frequency in exons of one SNP/67 nucleotides (Table 1, Additional file 4: Table S2). In contrast, the single intron contains 24 SNPs and 16 different indels. The GSL2 gene also exhibits the highest SNP frequency within the introns (Table 1). A deletion of 21 nucleotides was found within the 5’UTR of DM, but not in the other genotypes. Other indels were found in all gene components except the exons. Consistent with the GSL1 gene, the exons of GSL2 exhibit only very rare synonymous SNPs among the alleles from all genotypes, with an overall SNP frequency in exons of one SNP/53 nucleotides (Table 1, Additional file 4: Table S2).
The rare frequency of SNPs observed in GSL1 and GSL2 within and between the diploid homozygous DM, the diploid heterozygous RH, and the four tetraploid potato genotypes was comparable to other GSL and GASA-like genes, as well as other highly conserved housekeeping genes (Additional file 4: Table S2). The rare SNP incidence in these genes across these 19 haplotypes is substantially lower than the one SNP every 29 nucleotides observed in a 6.6 Mb region analyzed for only two RH haplotypes associated with the potato genome sequence [33], and the one SNP every 24 nucleotides (exons) and one SNP every 15 nucleotides (non-coding/introns) reported by Uitdewilligen et al. [39] based on targeted resequencing of 83 tetraploid cultivars. This confirms the highly conserved nature of the GSL1 and GSL2 genes, and therefore suggests that they play an essential role in biological function.
Expression profiles of GSL genes
FPKM (fragments per kilobase per million mapped reads) as expression values for each transcript were extracted from previous data sets [33] representing a range of different potato tissues and treatments (Figure 2). FPKM levels from a total of 32 tissue and treatment libraries from the genotype DM were analyzed for GSL1 and GSL2 expression. Of the 32 samples analyzed, GSL2 was expressed at a higher level in 30 samples compared with GSL1, often by over an order of magnitude in FPKM values.

FPKM values as a representation of transcriptional expression for the potato GSL1 and GSL2 genes. Where FPKM values were zero, no data point is graphed. GSL1 data points are represented by squares and GSL2 data points are represented by diamonds. (A) Different organs of potato DM plant material. (B) Stress-related conditions and plant growth regulator treatments using in vitro grown potato DM plant material. FPKM values of the controls are represented by an open data point and the treatment values have solid colour.
For GSL1 expression, Segura et al. [2] showed by northern analysis that transcripts of StGSL1 exhibited highest accumulation in stems, shoot apices, young floral buds and petals. Similarly, FPKM reads showed highest levels in mature flowers, immature fruit, shoots, petals and carpels (Figure 2A). Northern analysis also detected expression in tubers and carpels, but not in roots, stolons, leaves, sepals or stamens [2]. In contrast to the northern analysis, FPKM analysis showed that GSL1 was expressed in roots, stolons, leaves and sepals. Northern analysis and FPKM data are in agreement for the absence of GSL1 expression in stamens; with the FPKM data also showing no expression in mature fruit and callus tissue. Northern analysis also established GSL1 expression is not induced by biotic or abiotic stresses [2], while the FPKM data show an absence of GSL1 transcripts in response to heat stress or BABA treatments and a slight increase in transcripts in response to BTH treatment (Figure 2B). Analysis of Arabidopsis thaliana plants transgenic for GUS fusions to the potato GSL1 promoter revealed GUS expression in root vascular tissue, cotyledons, young leaves and floral organs [40]. This analysis of transcriptional control by the GSL1 promoter is more consistent with the FPKM data (Figure 2) than the previously published northern analysis [2].
FPKM values for the GSL2 gene (Figure 2) and northern analysis [3] are in agreement with highest levels of expression being in carpels and petals, and generally high expression in all tissues examined. However, northern analysis did not detect GSL2 expression in stolons or roots, which is in contrast to the high levels of expression seen in FPKM data (Figure 2A). In addition, FPKM analysis indicates that GSL2 expression is not induced by biotic or abiotic stresses, plant growth regulator treatments or wounding (Figure 2B), although slight reductions are observed in GSL2 transcripts in response to stress induced by heat, salt, mannitol and BABA treatments. This is in contrast with northern analysis, where GSL2 expression responded to biotic stress, was up-regulated by wounding and ABA treatments, down regulated in response to GA3 and showed no response to salinity or drought treatments [3].
The differences between previously published northern analysis for GSL1[2] and GSL2[3] and the FPKM values in this study (Figure 2) may reflect differences in cultivar/ploidy level and growth/treatment conditions. Overall, the FPKM data support the conclusion that GSL1 is a component of the constitutive defense barriers, especially of the storage and reproductive organs [2]. The FPKM analysis supports the same conclusion for GSL2, which is in contrast to the previous view that GSL2 is a component of both constitutive and inducible defense barriers [3].
Analysis of GSL promoters
Since the GSL1 and GSL2 genes differed markedly in their magnitude and specificity of transcript accumulation (Figure 2), the promoter regions were analysed for motifs using Genomatix-MatInspector [41] based on PLACE [42]. A total of 58 and 28 different motifs, previously characterised in other studies, were identified in the GSL1 and GSL2 promoter regions, respectively (Additional file 5: Table S3 and Additional file 6: Table S4). Based on the known transcriptional expression of the GSL genes (Figure 2; [2, 3]), the putative roles of GSL proteins [31, 32], the repeated occurrence of motifs, their presence in all potato genotypes, and their relative position in the promoter region, key motifs with potential functional significance were identified for GSL1 (Table 2) and GSL2 (Table 3).
Eight different key motifs were identified in the GSL1 promoter, which are repeated up to eleven times resulting in a total of 42 motifs (Table 2). These involve motifs associated with roles for response to disease and biotic stress, abiotic stresses, light induction, and plant development and were found in the genome sequence of DM as well as all four tetraploid genotypes. However, allelic polymorphisms involving disruptions of these motifs were occasionally observed in the tetraploid genotypes. Polymorphic SNPs were observed in seven of the 42 motifs, with polymorphic indels also observed for nine motifs (Table 2).
In the GSL2 promoter one key motif was identified that occurs five times and ten other key motifs were identified that occur only once (Table 3). Similar to the promoter of the GSL1 gene, these have known roles associated with biotic stress, abiotic stress, and development. Additional motifs present in the GSL2 promoter are associated with sugar signaling and hormone responses. These fifteen motifs were all observed in the four tetraploid genotypes, although two were observed to be polymorphic for SNPs and one was polymorphic for an insertion into at least one allele of the genotype VTn62-33-3 (Table 3).
The conservation of these motifs across the genome of DM and all four re-sequenced tetraploid genotypes substantiates their importance. Their presence in the GSL1 and GSL2 promoter regions aligns with the transcriptional expression of the respective genes observed by previous northern analysis [2, 3] and/or the FPKM data (Figure 2). The presence of allelic polymorphisms involving sequence disruptions in some of these motifs could be representative of alleles with potentially altered transcriptional expression of the GSL1 and GSL2 genes.
Antisense knockdown of GSL expression
Using our standard Agrobacterium-mediated transformation protocol for potato [43], we failed to recover any transformants of potato cultivar Iwa with antisense constructs for either the GSL1 or GSL2 genes. Over 100 leaf explants were subjected to Agrobacterium-mediated transformation in each of three experiments for both GSL genes using our well established protocol. We would normally expect to recover at least one regenerated transformant per leaf explant for the potato cultivar Iwa when selecting for the kanamycin resistance marker gene used on the binary vector. This expected frequency was achieved in concurrent related experiments using the GSL sense constructs [44]. However, a total of only 33 and 49 putative transformed cell colonies were recovered from all three co-cultivation experiments with Agrobacterium containing the GSL1 and GSL2 antisense constructs, respectively (Additional file 7: Table S5). All potato cell colonies transformed with the antisense constructs failed to continue growth and eventually senesced and died before complete shoots were regenerated (Additional file 8: Figure S3). The senescing cultures were sub-cultured onto medium without Timentin™ and DNA was extracted from those exhibiting no Agrobacterium growth. PCR using primers that bridged the Lhca3 promoter and the antisense GSL coding regions confirmed that these cell colonies were transformed with the intended construct prior to their death (Additional file 9: Figure S4). The same DNA samples failed to amplify PCR products using primers specific to the Agrobacterium virG gene. This confirms the absence of Agrobacterium cells in the plant tissue that would otherwise compromise the PCR testing of the transformed potato cell colonies.
The lethality of antisense knock-down expression of GSL1 and GSL2 suggests an essential role of the GSL gene family for potato development. A previous study achieved partial silencing of GSL1 in potato by expressing an antisense RNA under the control of the 35S promoter. This resulted in plants with reduced height and smaller leaves resulting from reduced cell division, changed leaf metabolism and cell wall composition [32]. The Lhca 3.St.1 promoter used in the present study is known to confer higher and more stable transgene expression than the 35S promoter [45]. Consequently, the lethality of GSL1 and GSL2 antisense knock-down under the control of the Lhca3 promoter is not unexpected given the dramatic phenotypes observed with the partial silencing from the use of the 35S promoter [32]. It is plausible that these GSL1 or GSL2 knock-down impacts, resulting from antisense expression driven by either the 35S or the Lhca3 promoters, could also arise by interference in expression of other closely related GSL and GASA genes. The three most closely related genes to GSL1 show 68-78% identity in exon regions, whereas the identity with all the other related genes was only 42-56% (Additional file 3: Table S1). For GSL2, the related GSL and GASA genes have only 44-60% identity in exon regions (Additional file 3: Table S1). Although this level of identity may be sufficient to trigger a partial knock-down of these related genes, it is unlikely to result in complete knock-down necessary for lethality. Therefore, the lethality of GSL1 and GSL2 antisense expression under the control of the Lhca3 promoter can be attributed to knock-down of the GSL1 and GSL2 genes.
Conclusions
GSL1 and GSL2, Gibberellin Stimulated-Like proteins (also known as Snakin-1 and Snakin-2), are cysteine-rich peptides from potato (Solanum tuberosum L.) with antimicrobial properties [2, 3]. Given their in vitro antimicrobial activity, the GSL1 and GSL2 genes are often considered to play important roles in the innate defence against invading microorganisms [2, 3, 6] and/or to be a key determinant during the interaction between plants and pathogens [25, 26]. In other species similar GSL/GASA proteins are hypothesised to play diverse biological roles in several aspects of plant development, plant responses to biotic or abiotic stress through their participation in hormone crosstalk, and redox homeostasis [31]. To further the understanding of the biological roles of GSL proteins, we undertook a thorough analysis of the structure and expression of these genes in potato.
We isolated and sequenced the coding regions and cDNAs for both GSL1 and GSL2 genes from the potato cultivar Iwa. This revealed two alleles (a1 and a2) for the GSL1 gene (GenBank accessions FJ195646 and FJ195647) and two alleles (b1 and b2) for the GSL2 gene (GenBank accessions EU848497 and EU848498). Alignment of the genomic and cDNA sequences confirmed the exon and intron regions in both the GSL1 and GSL2 genes (Figure 1). The GSL1 gene consists of two exons of 82 and 187 nucleotides interrupted by a single intron of 525 nucleotides. The GSL2 gene is composed of three exons of 87, 46 and 182 nucleotides respectively, alternating with two introns of 268 and 172 nucleotides.
We have also characterised the full length genes for both GSL1 (chromosome 4) and GSL2 (chromosome 1) using the genome sequence of diploid potato [33], coupled with further next generation sequencing of four highly heterozygous tetraploid potato genotypes; cultivars Summer Delight and Karaka, and breeding lines 1021/1 and VTn62-33-3. The frequency of SNPs in GSL1 and GSL2 was very low with only one SNP every 67 and 53 nucleotides in exon regions of GSL1 and GSL2, respectively (Table 1, Additional file 4: Table S2), similar to other highly conserved housekeeping genes in potato (Additional file 4: Table S2).
Specific promoter motifs were also highly conserved among multiple alleles representing the 17 haplotypes from DM and the four re-sequenced tetraploid genotypes (Tables 2 and 3), suggesting their importance for biological function. Analysis of comprehensive RNA-seq data substantiated the role of specific promoter motifs in transcriptional control of gene expression (Figure 2). FPKM analysis established that GSL2 was expressed at a higher level than GSL1 in 30 out of 32 libraries, often by an order of magnitude. Furthermore, both the GSL1 and GSL2 genes exhibited constitutive expression that was not up-regulated in response to biotic or abiotic stresses, hormone treatments or wounding. The FPKM analysis did not always agree with previous northern analysis [2, 3], although closely matched conclusions from the analysis of Arabidopsis thaliana plants transgenic for GUS fusions to the potato GSL1 promoter [40].
The GSL1 and GSL2 genes from potato are very highly conserved suggesting they contribute to an important biological function. The known antimicrobial activity of the GSL proteins, coupled with the FPKM analysis from RNA-seq data, suggests that both genes contribute to the constitutive defence barriers in potatoes. The lethality of antisense knock-down expression of GSL1 and GSL2, coupled with the rare incidence of SNPs in these genes, suggests an essential role for this gene family. These features are consistent with the GSL protein family playing a role in several aspects of plant development and plant defence responses.
Methods
Extraction of potato DNA and RNA for analysis of GSL genes
For cloning and sequencing of the GSL genes, genomic DNA was isolated from in vitro shoots of potato, Solanum tuberosum L., cv Iwa based on the method described by Bernatzky and Tanksley [46]. Total RNA was isolated from the youngest, fully expanded leaves of 2 month old greenhouse-grown Iwa potato plants using the Illustra RNAspin Mini Isolation Kit (GE healthcare, Buckinghamshire, UK), including DNase treatment according to the manufacturer’s instructions. The integrity of the total RNA was checked by electrophoresis in 1% agarose gel in Tris-acetate-EDTA (TAE) buffer and quantity was determined with a NanoVue™ Spectrophotometer (GE healthcare).
PCR isolation of GSL genes
Primers GSL1-F2 (5’-AAATGAAGTTATTTCTATTAACTCTGC-3’) and GSL1-R2 (5’-TGTGAAGACGCAAATATAACCAC-3’) were designed based on the reference gene sequence of StGSL1 (Genbank accession AJ320185) to isolate the GSL1 gene. The reference gene sequence of StGSL2 gene (Genbank accession AJ312424) was used to design the primers GSL2-F (5’-AAATATTTCAAATTCCAATGGC-3’) and GSL2-R (5’-CAATACAATGCAAACCAGAACAA-3’) to isolate the GSL2 gene. PCRs were carried out in a Mastercycler (Eppendorf, Hamburg, Germany). The 50 μl PCR mix contained 1x Expand High FidelityPLUS Reaction Buffer containing 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.4 μM of each primer, 1 μl of DNA (~100 ng) and 2.5 U of Expand High Fidelity Taq DNA polymerase (Roche Applied Science, Mannheim, Germany). The conditions for PCR for the GSL1 gene were: 93°C for 1 min, 35 cycles of 30 s 92°C, 30 s 57°C, 90 s 72°C, followed by 6 min extension at 72°C. For the GSL2 gene, PCR was performed using the same PCR conditions with 58°C annealing temperature. Amplified products were separated by electrophoresis in a 1% agarose gel in 1xTAE buffer and visualized under UV light after staining with ethidium bromide. Additional primers were designed to flank the previously designed primer regions and following PCR the products were sequenced to confirm the authenticity of the sequence over the previous primer regions.
Cloning and sequencing of GSL genes
PCR fragments of the expected size (813 bp for GSL1 and 953 bp for GSL2) were extracted from an agarose gel using a QIAquick gel extraction kit (QIAGEN, Hilden, Germany) and cloned into pGEM®-T Easy vector (Promega, Mannheim, Germany). The resulting plasmids were transformed into Subcloning Efficiency™ DH5α™ Competent Cells (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. Plasmid DNA from white clones was isolated using High Pure Plasmid Isolation Kit (Roche Applied Science) and tested by restriction analysis using FastDigest® Not I enzyme (Fermentas, Hanover, Maryland, USA) to identify whether they contained GSL gene inserts. Plasmid DNA from 16 clones of each GSL gene was sequenced using Applied Biosystems BigDye® Terminator v3.1 kit. Sequencing reactions were analysed using an ABI 3130xl automated sequencer (Applied Biosystems, Foster City, USA). Each fragment was sequenced from both directions individually using 3.2 pmole of primer. M13 forward and M13 reverse primers were used individually as sequencing primer in each sequencing reaction. Vector NTI Advance 10 software package (Invitrogen) was used to analyse the sequences and assemble into contigs.
Sequencing the coding regions of GSL genes
First-strand cDNA was synthesised from isolated RNA using the SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, Carlsbad, USA) according to the manufacturer’s instructions. VILO™ Reaction Mix contains random primers, MgCl2 and dNTPs in a buffer formulation. Single-stranded cDNA was then used as a template in the PCR reactions using the Expand High FidelityPLUS PCR system (Roche Applied Science). Approximately 50 ng of cDNA, corresponding to the amount of total RNA isolated from Iwa plants, was used as a template. The PCRs were carried out in a C1000™ Thermal Cycler (Bio-Rad). The 50 μl PCR mix contained 1x Expand High FidelityPLUS Reaction Buffer containing 1.5 mM MgCl2, 0.2 mM of each dNTP, 0.4 μM of each primer and, 2.5 U of Expand High Fidelity Taq DNA polymerase (Roche Applied Science). For the GSL1 coding region, the primers were GSL1-exonF2 (5’-ATGAAGTTATTTCTATTAACTCTGCTTT-3’) and GSL1-exonR2 (5’-TCAAGGGCATTTAGACTTGC-3’). For the GSL2 coding region, the nucleotide sequences of the primers were GSL2-exonF (5’-ATGGCCATTTCGAAAGC-3’) and GSL2-exonR (5’-TTAAGGGCATTTACGTTTGTT-3’).
The PCR conditions for the GSL1 coding region were: 94°C for 2 min, 34 cycles of 30 s 94°C, 30 s 59°C, 30 s 72°C, followed by 7 min extension at 72°C. The PCR for the GSL2 coding region was performed using the same PCR conditions with 57°C annealing temperature. PCR products were separated by electrophoresis in a 2% agarose gel in TAE buffer and visualized under UV light after staining with ethidium bromide.
PCR products were purified using the Illustra GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare) according to the manufacturer’s recommendations and sequenced directly using Applied Biosystems BigDye® Terminator v3.1 kit. Sequencing reactions were analysed using an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA). Each fragment was sequenced from both directions individually using 3.2 pmole of primer. The primers described above were used individually as sequencing primer in each sequencing reaction. Sequences of GSL1 and GSL2 coding regions were analysed and aligned with their reference sequences by MUSCLE alignment method [47] using Geneious software [38].
Next generation sequencing
Genomic DNA was isolated from young shoots of greenhouse grown potato plants of cultivars Summer Delight and Karaka, and breeding lines 1021/1 and VTn62-33-3, based on the method described by Bernatzky and Tanksley [46]. Illumina short insert read pair data were generated for each line using the Illumina GAIIX platform (Illumina, San Diego).
Transcript profiling
Transcriptome analyses were performed using data sets produced previously by The Potato Genome Sequencing Consortium [33]. Transcriptome sequences were generated from 32 libraries of genotype DM using RNA-seq with the Illumina Genome Analyser II platform. These 32 libraries represent a wide range of tissues/organs as well as abiotic, biotic and plant growth regulator treatments. Full experimental details are described in the Supplementary Material and Table S4 of The Potato Genome Sequencing Consortium [33]. The number of expressed genes and RNA-seq reads for each of the libraries is presented in Massa et al. [48]. To provide a normalized unit to allow comparisons within and between samples, the abundance of transcripts was expressed in fragments per kilobase of exon model per million mapped reads (FPKM) as implemented in Cufflinks [49].
Bioinformatic analysis
Illumina short insert paired-end reads of the four tetraploid potato lines were aligned to the reference genome using BWA [36]. For 1021/1, VTn62-33-3, Karaka and Summer Delight, 60.6 × 106 (71%), 53.4 × 106 (69%), 49.4 × 106 (72%) and 37.1 × 106 (72%) read pairs were mapped to the reference genome, respectively. This resulted in approximately 9 to 15 fold coverage of the ~840 MB potato genome for these four genotypes. Alignments were further analysed using SAMtools [37]. Single nucleotide polymorphisms were detected using an in-house tool, polySNP (https://github.com/mfiers/polysnp). PolySNP calls SNPs based on Samtools mpileup mapping quality scores. Only high confidence SNPs for uniquely mapped reads with sequence scores above phred 15 were considered. SNPs had to be present in at least three reads to be counted. Output from polySNP was validated by manual confirmation using the software package Geneious [38].
The analysis of promoter regions for motifs predicted to be involved in transcription factor binding sites was performed using the Genomatix MatInspector software [41] with the selection of Plant IUPAC Library based on PLACE [42].
Construction of the expression cassette
Sequence information derived from the StLhca3 promoter and terminator regions was used to design a potato expression cassette into which coding regions of other potato genes can be cloned. A region consisting of nucleotides 1–600 from the StLhca3 promoter (GenBank accession EU234502) and a region consisting of nucleotides 101–487 from the StLhca3 terminator (GenBank accession EU293853) were adjoined in silico to generate a unique Psi I restriction site at their junction. These sequences were synthesized as a single 988 bp fragment (Genscript Corporation, Piscataway, NJ, USA) and cloned into pUC57 to produce pStLhca3cas (Additional file 10: Figure S5A).
Construction of antisense vectors
PCR was performed to isolate blunt-end GSL1 and GSL2 sequences from the pGEM®-T Easy plasmids harbouring specific GSL alleles described above. VentR DNA polymerase (New England BioLabs, Massachusetts, USA) was used to isolate blunt-end fragments from the a1 allele of GSL1 (GenBank accession FJ195646) using the primers GSL1-F2 and GSL1-R2 and the b1 allele of GSL2 (GenBank accession EU848497) using the primers GSL2-F and GSL2-R. PCR products of the expected size (813 bp for the a1 allele of GSL1 gene and 955 bp for the b1 allele of GSL2 gene) were gel-purified using QIAquick Gel Extraction Kit (QIAGEN). Quick Blunting Kit (New England BioLabs) was used for phosphorylation of the 5’ ends of the blunt-ended DNA fragments. The fragments were ligated into the Psi I site of the expression cassette (pStLhca3cas) using T4 DNA Ligase (New England BioLabs). One Shot® TOP10 Electrocomp™ E. coli Cells (Invitrogen) were transformed with DNA from blunt-end ligation reactions. Plasmid DNA from clones was isolated using High Pure Plasmid Isolation Kit (Roche Applied Science) and tested by restriction analysis using Hin dIII enzyme. The recombinant plasmid was sequenced using Applied Biosystems BigDye® Terminator v3.1 kit to confirm the orientation of the expression cassette. The primers Cab-Fa (5’-TTCTAGTGGAGCTAAGTGTTCA-3’) and Cab-Ra (5’-TGTTACATTACACATAAGAGAAGG-3’) were used individually as sequencing primers in each sequencing reaction. Sequencing reactions were analysed using an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA). Plasmids containing StLhca3 expression cassettes with the GSL genes in the sense orientation and those with inserts in the opposite orientation (antisense expression cassettes) were identified by sequencing.
Plasmids of antisense expression cassettes were digested with Hin dIII and the resulting fragments, Lhca3-StGSL1 (1631 bp) and Lhca3-StGSL2 (1770 bp) were blunt-ended using Quick Blunting Kit (New England BioLabs) and ligated individually into the blunt-ended Not I site of the binary vector pMOA33 [50] using T4 DNA Ligase (New England BioLabs). Ligation products were transformed into MAX Efficiency® DH5αTM Competent Cells (Invitrogen). Colonies were screened using colony PCR with Cab-Fa and Cab-Ra primers to identify intact clones. Individual colonies were picked using a sterile pipette tip and resuspended in 10 μl of PCR mix. Each 10 μl PCR mix contained 1xThermoPol Reaction Buffer, 0.2 mM of each dNTP, 0.2 μM of each primer and, 0.4 U of Taq DNA polymerase (New England BioLabs). The PCRs were carried out in a Mastercycler (Eppendorf). The conditions for PCR were: 94°C for 4 min, 34 cycles of 15 s 93°C, 30 s 55°C, 90 s 72°C followed by 10 min extension at 72°C.
Plasmid DNA from clones selected by colony PCR was isolated using the QIAprep Spin Miniprep kit (QIAGEN). The orientation of the expression cassettes within the T-DNA in the binary vectors, pMOA33-Lhca3-antiGSL1 and pMOA33-Lhca3-antiGSL2, was tested by restriction analysis (Eco RV and Sca I for pMOA33-Lhca3-antiGSL1; Eco RV and Xho I for pMOA33-Lhca3-antiGSL2) to select a binary vector that contains the Lhca3 promoter adjacent to the right border within the T-DNA.
Potato transformation
The resulting binary vectors (Additional file 10: Figures S5B and S5C) were transferred to the disarmed Agrobacterium tumefaciens strain EHA105 [51] using the freeze-thaw method [52]. Agrobacterium cultures harbouring the binary vectors were cultured overnight on a shaking table at 28°C in LB broth supplemented with 300 mg l-1 spectinomycin. Leaf segments from virus-free plants of potato (cultivar Iwa) were transformed using our well established protocol [43] with 100 mg l-1 kanamycin to select for transformed potato cells and 200 mg l-1 Timentin™ to prevent Agrobacterium overgrowth.
Molecular confirmation of transformation
Independently-derived putative transformed potato cell colonies were sub-cultured onto culture medium without Timentin™. Genomic DNA was extracted from those exhibiting no Agrobacterium growth using a modified CTAB method [53]. To confirm the presence of the antisense-GSL constructs in the cell colonies, primers specific to the Lhca3 promoter region and the GSL genes were used to avoid endogenous gene amplification. The PCR for the Lhca3-antiGSL1 gene used the primers Cab-Fa (5’-TTCTAGTGGAGCTAAGTGTTCA-3’) and GSL1-F1 (5’-ACCCTTCTCTCATTCAAACT-3’) with a predicted amplicon of 840 bp. The PCR for the Lhca3-antiGSL2 gene used the primers Cab-Fa and GSL2-bF1 (5’-TCAGACCGATCAAGTGGTGA-3’) with a predicted amplicon of 940 bp. The following PCR conditions were used: 1 cycle at 94°C for 1 min, 34 cycles of 20 s 93°C, 20 s 55°C, 80 s 72°C, followed by a 3 min extension at 72°C. Finally, primers specific to the Agrobacterium virG gene, GMT24virGF (5’-GCGGTAGCCGACAG-3’) and GMT25virGR (5’-GCGTCAAAGAAATA-3’) producing a predicted amplicon of 692 bp were used to investigate the possible presence of Agrobacterium contamination remaining in the plant tissue. The PCR conditions were 2 min at 94°C, then 34 cycles of 30 s 94°C, 30 s 45°C, 30 s 72°C, followed by a 5 min extension at 72°C. All PCRs were conducted in 10 μL reactions containing 1x ThermoPol Reaction Buffer [20 mM Tris–HCl, 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 0.1% Triton X-100, pH 8.8 at 25°C], 0.2 mM of each dNTP, 0.2 μM of each primer and 0.4 U of Taq DNA Polymerase (New England Biolabs). PCRs were carried out in a Mastercycler (Eppendorf, Hamburg, Germany) and amplified products were separated by electrophoresis in a 1% agarose gel in 1xTAE buffer at 5.5 V/cm for 40 min and visualized under UV light after staining with ethidium bromide (5 mg l-1) for 15 min.
References
Caaveiro JMM, Molina A, González-Mañas JM, Rodríguez-Palenzuela P, García-Olmedo F, Goñi FM: Differential effect of five types of antipathogenic plant peptides on model membranes. FEBS Lett. 1997, 410: 338-342. 10.1016/S0014-5793(97)00613-3.
Segura A, Moreno M, Madueño F, Molina A, Garcia-Olmedo F: Snakin-1, a peptide from potato that is active against plant pathogens. Mol Plant-Microbe Interact. 1999, 12: 16-23. 10.1094/MPMI.1999.12.1.16.
Berrocal-Lobo M, Segura A, Moreno M, Lopez G, Garcia-Olmedo F, Molina A: Snakin-2, an antimicrobial peptide from potato whose gene is locally induced by wounding and responds to pathogen infection. Plant Physiol. 2002, 128: 951-961. 10.1104/pp.010685.
López-Solanilla E, González-Zorn B, Novella S, Vázquez-Boland JA, Rodríguez-Palenzuela P: Susceptibility of Listeria monocytogenes to antimicrobial peptides. FEMS Microbiol Lett. 2003, 226: 101-105. 10.1016/S0378-1097(03)00579-2.
Kovalskaya N, Hammond RW: Expression and functional characterization of the plant antimicrobial snakin-1 and defensin recombinant proteins. Protein Expr Purif. 2009, 63: 12-17. 10.1016/j.pep.2008.08.013.
Mao ZC, Zheng JY, Wang YS, Chen GH, Yang YH, Feng DX, Xie BY: The new CaSn gene belonging to the snakin family induces resistance against root-knot nematode infection in pepper. Phytoparasitica. 2011, 39: 151-164. 10.1007/s12600-011-0149-5.
Shi L, Gast RT, Gopalraj M, Olszewski NE: Characterization of a shoot-specific, GA3- and ABA-regulated gene from tomato. Plant J. 1992, 2: 153-159.
Zhang S, Yang C, Peng J, Sun S, Wan X: GASA5 a regulator of flowering time and stem growth in Arabidopsis thaliana. Plant Mol Biol. 2009, 69: 745-759. 10.1007/s11103-009-9452-7.
Ben-Nissan G, Weiss D: The petunia homologue of the tomato gast1: transcript accumulation coincides with gibberellin-induced corolla cell elongation. Plant Mol Biol. 1996, 32: 1067-1074. 10.1007/BF00041390.
Kotilainen M, Helariutta Y, Mehto M, Pöllänen E, Albert VA, Elomaa P, Teeri TL: GEG participates in the regulation of cell and organ shape during corolla and carpel development in Gerbera hybrida. Plant Cell. 1999, 11: 1093-1104.
Ben-Nissan G, Lee JY, Borohov A, Weiss D: Gip, a Petunia hybrida GA-induced cysteine-rich protein: a possible role in shoot elongation and transition to flowering. Plant J. 2004, 37: 229-238. 10.1046/j.1365-313X.2003.01950.x.
Furukawa T, Sakaguchi N, Shimada H: Two OsGASR genes, rice GAST homologue genes that are abundant in proliferating tissues, show different expression patterns in developing panicles. Genes Genetic Syst. 2006, 81: 171-180. 10.1266/ggs.81.171.
Alleman M, Sidorenko L, McGinnis K, Seshadri V, Dorweiler JE, White J, Sikkink K, Chandler VL: An RNA-dependent RNA polymerase is required for paramutation in maize. Nature. 2006, 442: 295-298. 10.1038/nature04884.
Silverstein KAT, Moskal WA, Wu HC, Underwood BA, Graham MA, Town CD, VandenBosch KA: Small cysteine-rich peptides resembling antimicrobial peptides have been under-predicted in plants. Plant J. 2007, 51: 262-280. 10.1111/j.1365-313X.2007.03136.x.
Adler M, Lazarus RA, Dennis MS, Wagner G: Solution structure of kistrin, a potent platelet aggregation inhibitor and GPIIb-GIIIa antagonist. Science. 1991, 253: 445-448. 10.1126/science.1862345.
Pelegrini PB, del Sarto RP, Silva ON, Franco OL, Grossi-de-Sa MF: Antibacterial peptides from plants: what they are and how they probably work. Biochem Res Internat. 2011, Article ID 250349, 9 pp, doi:10.1155/2011/250349
Epand RM, Vogel HJ: Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999, 1462: 11-28. 10.1016/S0005-2736(99)00198-4.
Sitaram N, Nagaraj R: Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim Biophys Acta. 1999, 1462: 29-54. 10.1016/S0005-2736(99)00199-6.
Shai Y: Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002, 66: 236-248. 10.1002/bip.10260.
Porto WF, Franco OL: Theoretical structural insights into the snakin/GASA family. Peptides. 2013, 44: 163-167.
Almasia NI, Bazzini AA, Hopp HE, Vazquez-Rovere C: Overexpression of Snakin-1 gene enhances resistance to Rhizoctonia solani and Erwinia carotovora in transgenic potato plants. Mol Plant Path. 2008, 9: 329-338. 10.1111/j.1364-3703.2008.00469.x.
Faccio P, Vazquez-Rovere C, Hopp E, González G, Décima-Oneto C, Favret E, Díaz Paleo A, Franzone P: Increased tolerance to wheat powdery mildew by heterologous constitutive expression of the Solanum chacoense snakin-1 gene. Czech J Genetics Plant Breed. 2011, 47: S135-S141.
Balaji V, Smart CD: Over-expression of snakin-2 and extension-like protein genes restricts pathogen invasiveness and enhances tolerance to Clavibacter michiganensis subsp. michiganensis in transgenic tomato (Solanum lycopersicum). Transgenic Res. 2012, 21: 23-37. 10.1007/s11248-011-9506-x.
Balaji V, Sessa G, Smart CD: Silencing of host basal defense response-related gene expression increases susceptibility of Nicotiana benthamiana to Clavibacter michiganensis subsp. michiganensis. Phytopathology. 2011, 101: 349-357. 10.1094/PHYTO-05-10-0132.
López-Solanilla E, García-Olmedo F, Rodríguez-Palenzuela P: Inactivation of the sapA to sapF locus of Erwinia chrysanthemi reveals common features in plant and animal bacterial pathogenesis. Plant Cell. 1998, 10: 917-924.
Bindschedler LV, Whitelegge JP, Millar DJ, Bolwell GP: A two component chitin-binding protein from French bean – association of a proline-rich protein with a cysteine-rich polypeptide. FEBS Lett. 2006, 580: 1541-1546. 10.1016/j.febslet.2006.01.079.
Aubert D, Chevillard M, Dorne AM, Arlaud G, Herzog M: Expression patterns of GASA genes in Arabidopsis thaliana: the GASA4 gene is up-regulated by gibberellins in meristematic regions. Plant Mol Biol. 1998, 36: 871-883. 10.1023/A:1005938624418.
de la Fuente JI, Amaya I, Castillejo C, Sanchez-Sevilla JF, Quesada MA, Botella MA, Valpuesta V: The strawberry gene FaGAST affects plant growth through inhibition of cell elongation. J Exp Bot. 2006, 57: 2401-2411. 10.1093/jxb/erj213.
Roxrud I, Lid SE, Fletcher JC, Schmidt EDL, Opsahl-Sorteberg HG: GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol. 2007, 48: 471-483. 10.1093/pcp/pcm016.
Lucau-Danila A, Laborde L, Legrand S, Huot L, Hot D, Lemoine Y, Hilbert JL, Hawkins S, Quillet MC, Hendriks T, Blervacq AS: Identification of novel genes potentially involved in somatic embryogenesis in chicory (Cichorium intybus L.). BMC Plant Biol. 2010, 10: 122-10.1186/1471-2229-10-122.
Nahirñak V, Almasia N, Hopp H, Vazquez-Rovere C: Snakin/GASA proteins: Involvement in hormone crosstalk and redox homeostasis. Plant Signal Behav. 2012, 7: 1004-1008. 10.4161/psb.20813.
Nahirñak V, Almasia NI, Fernandez PV, Hopp HE, Estevez JM, Carrari F, Vazquez-Rovere C: Potato Snakin-1 gene silencing affects cell division, primary metabolism, and cell wall composition. Plant Physiol. 2012, 158: 252-263. 10.1104/pp.111.186544.
The Potato Genome Sequencing Consortium: Genome sequence and analysis of the tuber crop potato. Nature. 2011, 475: 189-195. 10.1038/nature10158.
Breathnach R, Chambon P: Organization and expression of eukaryotic split genes coding for proteins. Ann Rev Biochem. 1981, 50: 349-383. 10.1146/annurev.bi.50.070181.002025.
Yamamoto YY, Ichida H, Matsui M, Obokata J, Sakurai T, Satou M, Seki M, Shinozaki K, Abe T: Identification of plant promoter constituents by analysis of local distribution of short sequences. BMC Genomics. 2007, 8: 67-10.1186/1471-2164-8-67.
Li H, Durbin R: Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009, 25: 1754-1760. 10.1093/bioinformatics/btp324.
Li H, Handsaker B, Wysoker A, Fennel T, Ruan J, Homer N, Mart G, Abecasi G, Durbin R, 1000 Genome Project Data Processing Subgroup: The sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A: Geneious v5.4.http://www.geneious.com,
Uitdewilligen JGAML, Wolters AMA, D'hoop BB, Borm TJA, Visser RGF, van Eck HJ: A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS One. 2013, 8: e62355-10.1371/journal.pone.0062355.
Almasia NI, Narhirñak V, Hopp HE, Vazquez-Rovere C: Isolation and characterisation of the tissue and development-specific potato snakin-1 promoter inducible by temperature and wounding. Electron J Biotech. 2010, 13: 5-http://dx.doi.org/10.2225/vol13-issue5-fulltext-12,
Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T: MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005, 21: 2933-2942. 10.1093/bioinformatics/bti473.
Higo K, Ugawa Y, Iwamoto M, Korenaga T: Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999, 27: 297-300. 10.1093/nar/27.1.297.
Meiyalaghan S, Jacobs JME, Butler RC, Wratten SD, Conner AJ: Expression of cry1Ac9 and cry9Aa2 genes under a potato light-inducible Lhca3 promoter in transgenic potatoes for tuber moth resistance. Euphytica. 2006, 147: 297-309. 10.1007/s10681-005-9012-4.
Mohan S: Snakin genes from potato: overexpression confers blackleg disease resistance. 2012, Canterbury, New Zealand: PhD Thesis, Lincoln University
Annadana S, Udayakumar M, de Jong J, Nap JP: The potato Lhca3.St.1 promoter confers high and stable transgene expression in chrysanthemum, in contrast to CaMV-based promoters. Mol Breed. 2001, 8: 335-344.
Bernatzky R, Tanksley SD: Genetics of actin-related sequences in tomato. Theor Appl Genet. 1986, 72: 314-339. 10.1007/BF00288567.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32: 1792-1797. 10.1093/nar/gkh340.
Massa AN, Childs KL, Lin H, Bryan GJ, Giuliano G, Buell CR: The transcriptome of the reference potato genome Solanum tuberosum Group Phureja clone DM1-3 516R44. PLoS One. 2011, 6: e26801-10.1371/journal.pone.0026801.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Barens MJ, Salzberg SL, Wold BJ, Pachter L: Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotech. 2010, 28: 511-515. 10.1038/nbt.1621.
Barrell PJ, Conner AJ: Minimal T-DNA vectors suitable for agricultural deployment of transgenic plants. Biotechniques. 2006, 41: 708-710. 10.2144/000112306.
Hood EE, Gelvin SB, Melchers LS, Hoekema A: New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res. 1993, 2: 208-218. 10.1007/BF01977351.
Höfgen R, Willmitzer L: Storage of competent cells for Agrobacterium transformation. Nucleic Acids Res. 1988, 16: 9877-10.1093/nar/16.20.9877.
Doyle JJ, Doyle JL: Isolation of plant DNA from fresh tissue. Focus. 1990, 12: 13-15.
Sharma SK, Bolser D, de Boer J, Sønderkær M, Amoros W, Carboni MF, D’Ambrosio JM, de la Cruz G, Di Genova A, Douches DS, Eguiluz M, Guo X, Guzman F, Hackett CA, Hamilton JP, Li G, Li Y, Lozano R, Maass A, Marshall D, Martinez D, McLean K, Mejía N, Milne L, Munive S, Nagy I, Ponce O, Ramirez M, Simon R, Thomson SJ, Torres Y, et al: Construction of reference chromosome-scale pseudomolecules for potato: integrating the potato genome with genetic and physical maps. G3. 2013, 3: 2031-2047. 2013.
de Boer JM, Borm TJA, Jesse T, Brugmans B, Tang X, Bryan GJ, Bakker J, van Eck HJ, Visser RGF: A hybrid BAC physical map of potato: a framework for sequencing a heterozygous genome. BMC Genomics. 2011, 12: 594-10.1186/1471-2164-12-594.
Acknowledgements
We thank Katrina Monaghan for technical assistance with DNA isolation, Donna Gibson for the illustrations, and Andrew Gleave and Simon Deroles for comments on earlier drafts of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SMe cloned and sequenced cDNA and genomic DNA for the GSL genes, constructed the antisense vectors, annotated the sequences, and analysed promoter motifs. SJT and MWEJF performed bioinformatic analyses and determined SNP frequencies. MWEJF designed the polySNP tool. PJB interpreted the expression profile data of GSL genes and assisted in sequence annotation. JML, SMo and EEJ contributed to the cDNA sequencing, annotation of GSL genes, and carried out the potato transformations. AJC conceived the study, coordinated data analysis and wrote the manuscript. JMEJ conceived the study, undertook the next generation sequencing and data generation, and wrote the manuscript. All authors contributed to, read and approved the final manuscript.
Electronic supplementary material
12864_2013_5646_MOESM1_ESM.pdf
Additional file 1: Figure S1: Nucleotide sequence of the GSL1 gene with 5’upstream regulatory and terminator regions from potato DM; derived from The Potato Genome Sequencing Consortium [33]. Numbering is defined by the putative transcription start site (TSS, +1) predicted at 33 nt from the first base of the translation start site (ATG), based on a plant dimer motif YR Rule (TG, -1/+1). Putative cis-elements TATA-box (−32 to −27, highlighted violet), a pyrimidine patch (Y Patch, -26 to −20, highlighted pink) and CAAT-box (−48 to −44, highlighted red) were also identified. Other nucleotide sequences highlighted are: positions of promoter motifs annotated as numbered ovals on Figure 1A and listed in Table 2 (blue); 5’UTR (grey); exons (yellow); and intron (green). The start and stop codons are marked in red font. (PDF 625 KB)
12864_2013_5646_MOESM2_ESM.pdf
Additional file 2: Figure S2: Nucleotide sequence of the GSL2 gene with 5’upstream regulatory and terminator regions from potato DM; derived from The Potato Genome Sequencing Consortium [33]. Numbering is defined by the putative transcription start site (TSS, +1) predicted at 38 nt from the first base of the translation start site (ATG), based on a plant dimer motif YR Rule (TG, -1/+1). Putative cis-elements TATA-box (−50 to −45, highlighted violet), a pyrimidine patch (Y Patch, -59 to −51, highlighted pink) and hypothetical CAAT-box (−65 to −61, highlighted red) were also identified. Other nucleotide sequences highlighted are: positions of promoter motifs annotated as numbered ovals in Figure 1B and listed in Table 3 (blue); 5’UTR (grey); exons (yellow); and introns (green). The start and stop codons are marked in red font. (PDF 622 KB)
12864_2013_5646_MOESM3_ESM.pdf
Additional file 3: Table S1: List of GSL and GASA genes and their genetic position in potato. The chromosomal location is supported by super-scaffolds anchored via a genetic map generated for DM [54], incorporating information from RH and tomato, or a genetic map of RH generated by whole genome profiling (WGP) [55]. Locations of the GSL1 and GSL2 genes were identified by the superscaffold location on the physical map given in the agp file generated by the Potato Genome Sequencing Consortium (PGSC) (http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml). The alignment of coding regions to determine identity to GSL1 and GSL2 used MUSCLE [47] and was based on allele a1 for GSL1 (FJ195646) and allele b1 for GSL2 (EU848498). (PDF 424 KB)
12864_2013_5646_MOESM4_ESM.pdf
Additional file 4: Table S2: SNP frequency in all GSL and GASA-like genes and a number of housekeeping genes in potato. The next generation sequence data from four tetraploid potato genotypes (‘Karaka’, ‘Summer Delight’, 1021/1, VTn62-33-3), plus the diploid RH [33], were aligned with the genome of DM [33]. Output is from polySNP tool with stringent calling (https://github.com/mfiers/polysnp). SNP frequency is given as nucleotides/SNP; ‘-‘ indicates no SNPs present. (PDF 292 KB)
12864_2013_5646_MOESM7_ESM.pdf
Additional file 7: Table S5: Transformation of potato antisense constructs of the GSL1 and GSL2 genes. Results are presented for three independent Agrobacterium-mediated transformation experiments of potato cultivar Iwa using the binary vectors pMOA33-Lhca3-antiGSL1 (Additional file 10: Figure S4B) and pMOA33-Lhca3-antiGSL2 (Additional file 10: Figure S4C). (PDF 292 KB)
12864_2013_5646_MOESM8_ESM.pdf
Additional file 8: Figure S3: Senescing potato cell colonies transformed with antisense constructs of the GSL1 gene. Identical results were obtained for the antisense construct of the GSL2 gene. (PDF 253 KB)
12864_2013_5646_MOESM9_ESM.pdf
Additional file 9: Figure S4: PCR confirmation of transgenic status of potato cell colonies transformed with antisense constructs of the GSL1 and GSL2 genes. (PDF 440 KB)
12864_2013_5646_MOESM10_ESM.pdf
Additional file 10: Figure S5: Plasmids constructed and used in this study. A. pStLhca3cas; B. pMOA33-Lhca3-antiGSL1; C. pMOA33-Lhca3-antiGSL2. (PDF 466 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Meiyalaghan, S., Thomson, S.J., Fiers, M.W. et al. Structure and expression of GSL1 and GSL2 genes encoding gibberellin stimulated-like proteins in diploid and highly heterozygous tetraploid potato reveals their highly conserved and essential status. BMC Genomics 15, 2 (2014). https://doi.org/10.1186/1471-2164-15-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-2