
Abstract
Background
The phylogenetic position of Bryozoa is one of the most controversial issues in metazoan phylogeny. In an attempt to address this issue, the first bryozoan mitochondrial genome from Flustrellidra hispida (Gymnolaemata, Ctenostomata) was recently sequenced and characterized. Unfortunately, it has extensive gene translocation and extremely reduced size. In addition, the phylogenies obtained from the result were conflicting, so they failed to assign a reliable phylogenetic position to Bryozoa or to clarify lophophorate phylogeny. Thus, it is necessary to characterize further mitochondrial genomes from slowly-evolving bryozoans to obtain a more credible lophophorate phylogeny.
Results
The complete mitochondrial genome (15,433 bp) of Bugula neritina (Bryozoa, Gymnolaemata, Cheilostomata), one of the most widely distributed cheliostome bryozoans, is sequenced. This second bryozoan mitochondrial genome contains the set of 37 components generally observed in other metazoans, differing from that of F. hispida (Bryozoa, Gymnolaemata, Ctenostomata), which has only 36 components with loss of tRNAser(ucn) genes. The B. neritina mitochondrial genome possesses 27 multiple noncoding regions. The gene order is more similar to those of the two remaining lophophorate phyla (Brachiopoda and Phoronida) and a chiton Katharina tunicate than to that of F. hispida. Phylogenetic analyses based on the nucleotide sequences or amino acid residues of 12 protein-coding genes showed consistently that, within the Lophotrochozoa, the monophyly of the bryozoan class Gymnolaemata (B. neritina and F. hispida) was strongly supported and the bryozoan clade was grouped with brachiopods. Echiura appeared as a subtaxon of Annelida, and Entoprocta as a sister taxon of Phoronida. The clade of Bryozoa + Brachiopoda was clustered with either the clade of Annelida-Echiura or that of Phoronida + Entoprocta.
Conclusion
This study presents the complete mitochondrial genome of a cheliostome bryozoan, B. neritina. The phylogenetic analyses suggest a close relationship between Bryozoa and Brachiopoda within the Lophotrochozoa. However, the sister group of Bryozoa + Brachiopoda is still ambiguous, although it has some attractions with Annelida-Echiura or Phoronida + Entoprocta. If the latter is a true phylogeny, lophophorate monophyly including Entoprocta is supported. Consequently, the present results imply that Brachiozoa (= Brachiopoda + Phoronida) and the recently-resurrected Bryozoa concept comprising Ectoprocta and Entoprocta may be refuted.
Similar content being viewed by others

Background
Bryozoans (ectoprocts), also known as "moss animals", are aquatic organisms that mostly live in colonies of interconnected individuals. They usually encrust rocky surfaces, shells or algae. They are an ecologically important group, with the marine species forming a dominant component of benthic subtidal marine communities. This group is also economically important because it is a major component of both marine and freshwater biofouling, and evolutionarily important as a long-living phylum with a good fossil record [1]. The phylum is currently reported to contain 4000 extant species. However, it is likely that more than twice that number are currently in existence [2, 3], with new taxa being described annually.
Together with the Brachiopoda and Phoronida, Bryozoa have been classified as "Lophophorata" because they possess a similar suspension feeding apparatus, the lophophore, which is a horseshoe-shaped structure that surrounds the mouth and has ciliated tentacles [4–8]. However, lophophorate phylogeny remains one of the most controversial issues in metazoan animal phylogeny because they display an amalgam of deuterostome and protostome features. The "Lophophorata" have been classified as deuterostomes on the basis of morphological and larval features [9–13]. On the other hand, molecular phylogenetic analyses suggest that the lophophorates have some affinities with mollusks and annelids within the protostomes [14–21].
Lophophorate phylogenies that have been reconstructed with mitochondrial protein-coding genes and nuclear ribosomal DNAs have failed to resolve the detailed relationships among the lophophorates and other related metazoan phyla [15, 17, 22–24]. Most studies of complete mitochondrial genomes have focused on chordate and arthropod phylogenies because only a few mitochondrial genomes from lophotrochozoan phyla have been determined to date. So far, complete lophotrochozoan mitochondrial genome sequences have been published for 94 species from 12 phyla, including 45 mollusks, 8 annelids, 3 brachiopods, 1 bryozoan, 1 phoronid (nearly complete), 2 entoprocts, 28 platyheminths, 1 nemertean (nearly complete), 1 rotifer, 2 chaetognaths, 1 acanthocephalan and 1 echiuran. If the mollusk data are excluded, only 49 mitochondrial genomes have been sequenced from the huge protostome group (the Lophotrochozoa) so far.
Complete mitochondrial genomes have been characterized from a variety of metazoan phyla so that nucleotide, amino acid and gene order data can be used to resolve their phylogenetic relationships. Mitochondrial genomes are generally conserved in terms of gene components (usually 13 protein-coding genes, 2 ribosomal RNA genes and 22 transfer RNA genes) [25], and a number of studies have taken advantage of the various levels of phylogenetic information offered by mitochondrial genomes to solve systematic and evolutionary questions over a broad taxonomic range [26, 27].
Mitochondrial protein-coding genes have recently been used to resolve the phylogenetic relationships of lophophorates [28]. The results show that the phylum Brachiopoda (an articulate brachiopod, Terebratulina retusa) belongs to the lophotrochozoan protostomes and that Brachiopoda have a close relationship with Molluska and Annelida within the monophyletic clade, Lophotrochozoa. The second lophophorate phylum, Phoronida (Phoronis architecta), has also been placed within the Lophotrochozoa. Phoronis has the almost same gene arrangement as the chiton, Katharina tunicata (Molluska, Polyplacophora) [29]. Phylogenies based on most of the molecular data strongly suggest that two lophophorate phyla, Brachiopoda and Phoronida, are closely related to each other (called Phoronizoa or Brachiozoa), and they appear to be sister groups of mollusks and annelids within the Lophotrochozoa [11, 30].
In an attempt to address the phylogenetic position of bryozoans in metazoan phylogeny, the first mitochondrial genome from a ctenostome bryozoan, Flustrellidra hispida (Flustrellidridae), was recently sequenced and characterized. However, F. hispida exhibits a number of peculiar features, such as extensive translocation of gene components including protein-coding and tRNA genes, and extremely reduced size. Phylogenetic trees inferred from the nucleotide and amino acid sequences of its protein-coding genes were mutually conflicting, so the phylogenetic position of F. hispida was not assigned. Thus, it is necessary to sequence additional mitochondrial genomes from more representative and widely-distributed bryozoans in order to address the issue of the phylogenetic position of bryozoans on the basis of mitochondrial genome information.
In this paper, to address whether or not lophophorates are a monophyletic group and to examine the exact phylogenetic position of Bryozoa, we describe the complete mitochondrial genome sequence of Bugula neritina (Bryozoa, Gymnolaemata, Cheilostomata), one of the most widely-distributed cheliostome bryozoans. The result is compared with the F. hispida sequence. We also explore the following: the monophyly of the class Gymnolaemata, the phylogenetic implication of the gene orders in lophophorate mitochondrial genomes, the secondary structures of extremely multiplied noncoding regions, etc.
Results and discussion
Genome organization
The mitochondrial genome sequence of Bugula neritina is 15,433 bp long and consists of 13 protein-coding genes (cox1-3, nad1-nad6, nad4L, atp6, atp8 and cob), two rRNA genes for the small and large subunits (rrnS and rrnL), and 22 tRNA genes, as is typical of the animal mitochondrial genomes published so far (Fig. 1). The A+T content of the entire mitochondrial genome of B. neritina is 70.0%. Interestingly, we found 27 multiplied noncoding regions (NC1-27). All the protein-coding and rRNA genes and 17 of the tRNA genes are transcribed in the same strand in B. neritina; the other five tRNAs are [trnL(cun), trnA, trnE, trnY and trnV], (Fig. 1). The first bryozoan mitochondrial genome reported from F. hispida [31] has only 36 gene components because trnS(ucn) is absent, it is relatively short (13,026 bp), and the A+T content is lower (59.4%). In contrast, B. neritina has features that are more typical of metazoan mitochondrial genomes in general in terms of the number of gene components, whole genome size and A+T content.

A circular map of the complete mitochondrial genome of a bryozoan Bugula neritina (GenBank accession number AY690838). Protein and rRNA genes are abbreviated as follows: atp6 and atp8 (genes for ATPase subunits 6 and 8), cox1-cox3 (genes for cytochrome C oxidase subunits I-III), cob (gene for apocytochrome b), nad1-nad6 and nad4L (genes for NADH dehydrogenase subunits 1–6 and 4L), and rrnS and rrnL (genes for 12S and 16S rRNAs). All 22 tRNA genes are located among protein- and/or tRNA-coding genes. The tRNA genes are named using single-letter amino acid abbreviations, with the exception of those coding for leucine and serine, which are named L1 for the tRNALeu(CUN) (anticodon TAG) gene, L2 for the tRNALeu(UUR) (anticodon TAA) gene, S1 for the tRNASer(AGN) (anticodon GCT) gene and S2 for the tRNASer(UCN) (anticodon TGA) gene. The arrows indicate the orientations of the gene components. The three slashed regions corresponding to NC3, NC4 and NC10 may be related to the mode of regulation of mitochondrial replication and transcription.
Extreme multiplication of noncoding region
Strikingly, the B. neritina mitochondrial genome contains 27 multiplied noncoding regions: 16 noncoding regions (NC1-NC16) larger than 10 bp (Table 1 and Fig. 2A) and 11 smaller (Table 1). The total length of the 16 noncoding regions larger than 10 bp is 864 bp. Three of them – NC3 (271 bp) between trnA and trnK, NC4 (246 bp) between trnK and rrnS and NC10 (68 bp) between trnY and cox1 – could be candidate origins of replication. trnK, one of the five tRNA genes transcribed on the light strand, is located between NC3 and NC4. The placement of trnK between these two possible control regions is likely to have occurred very recently and independently only in the specific evolutionary lineage of B. neritina, since it has never been found in any other metazoan. The remaining 13 noncoding regions (NC1-NC2, NC5-NC9, NC11-NC16) total 279 bp in length and are dispersed throughout the whole genome, ranging from 12 to 36 bp in size (Table 1 and Fig. 2A). In addition, 11 small intergenic gaps (< 10 bp) were identified between some gene components (Table 1).

Multiple noncoding regions of the mitochondrial genome of a bryozoan, Bugula neritina , putative secondary structures of NC3, NC4 and NC10, and "CTT" repeat motif observed in NC10. A) Fifteen (NC1-NC16) larger than 10 bp of the 27 multiple noncoding regions of the Bugula neritina mitochondrial genome (black boxes). The circular genome is linearized. Genes encoded on the opposite strand are shown in gray boxes. NC3, NC4 and NC10 may be related to the mode of regulation of mitochondrial replication and transcription. B) Plausible helix structures predicted from NC3, NC4 and NC10, and 9 "CTT" repeats observed in NC10. The secondary structures and repeats may play important roles in the regulation of mitochondrial replication and transcription. Arabic numbers inside the encircled loop regions of each helix and in parentheses between helices indicate the number of nucleotides in each region.
Most metazoan mitochondrial genomes reported so far possess only a single major noncoding region, which is thought to be involved in the regulation of transcription and the control of DNA replication [32, 33]. In general, possible control regions possess characteristic features such as high A+T contents, hairpin-loop structures, repeat motifs, etc. [25, 34]. In B. neritina, there are three possible control regions (NC3, NC4 and NC10). Their A+T contents are 78.6% in NC3, 78.1% in NC4 and 79.4% in NC10, all of which are much higher than the 70.0% of the mitochondrial genome as a whole. In NC3, NC4 and NC10, we found some hairpin-loop structures that might be related to the mode of regulation of replication and transcription (Fig. 2B). NC3 and NC4 possess no characteristic repeat motifs but have extensive poly "A" and poly "C" tracts (136 "A" and 12 "C" in NC3 and 122 "A" and 36 "C" in NC4), as often observed in mitochondrial control regions in other metazoans [25, 34]. Intriguingly, NC10 (12 A, 15 C, 2 G and 37 T) includes at least nine "CTT" repeats with a short helix consisting of a 5-base-pair stem and a 3-nt loop (Fig. 2B). Despite its short length (68 bp), the existence of "CTT" repeats and a hairpin-loop may suggest that NC10 is important in regulating mitochondrial replication and transcription. In addition to these, NC1 between trnW and trnL(cun) has a helix with a 5-bp stem [additional file 1].
Such multiple noncoding regions are rare in metazoan mitochondrial genomes. The other bryozoan sequenced, F. hispida, has 17 noncoding regions, ranging in size from 1 to 195 bp (506 bp in total). Among these, two possible control regions were observed between trnC and trnG (195 bp) and between cox2 and trnD (114 bp), which are separated by cox2-trnG [31]. The mollusk Loligo bleekeri (Cephalopoda; [35]) has 19 noncoding regions longer than 10 bp. Three of these 19 are 515 bp, 507 bp and 509 bp long, and their sequences are nearly identical, suggesting that all three originated from a single, large, ancestral noncoding region. In Lampsilis ornata (Bivalvia; [36]), 28 noncoding regions were found, ranging from 2 to 282 bp in size. Of these, only one large noncoding region (136 bp long) has an increased A+T content (76.8%), so it is a possible control region. Since no such extreme multiplication of noncoding regions has been observed in any other bivalve or cephalopod mollusk including Katharina tunicata, it is likely that the extreme multiplication of noncoding regions is a homoplasious characteristic, occurring independently in the lineages of L. bleekeri, L. ornata and B. neritina.
Comparative analysis of gene arrangements
Unlike other metazoan mitochondrial genomes in which genes are encoded on both strands, all the protein-coding and rRNA genes and 17 of the tRNA genes – the exceptions being the five tRNA genes trnL(cun), trnA, trnE, trnY and trnV – are transcribed from the same strand in B. neritina (Fig. 1 and Table 1). In F. hispida, one protein-coding gene (cox2), one ribosomal RNA gene (rrnL) and four tRNA genes (trnG, trnC, trnL(uur), trnV and trnV) are reversed. Such a single-strand-dependent transcription tendency has been reported for 137 among the 1428 metazoan species in 23 phyla for which complete or nearly complete mitochondrial genome sequences have been determined to date (Dec. 17, 2008). Except for six tunicates (Deuterostomia, Urochordata), all the remaining 131 cases were from protostomes or primitive metazoan groups: 83 protostomes including 62 lophotrochozoans and 17 nematodes, and 48 primitive metazoans including 29 cnidarians and 19 poriferans, the most primitive metazoan groups (Table 2). The single-strand dependence of transcription might be a plesiomorphic, ancestral characteristic because such a tendency appears in 48 out of 59 primitive metazoans (81.4%) such as Cnidaria and Porifera (Table 2).
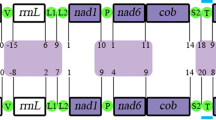
The arrangements of the protein-coding and rRNA genes were compared among two bryozoans (B. neritina and F. hispidia), a brachiopod (T. retusa), a phoronid (P. architecta) and a polyplachophoran (K. tunicata) (Fig. 3). The overall gene arrangement in B. neritina was quite different from those in other metazoans published so far. Compared to the F. hispida sequence, B. neritina needed 6 local translocations and 1 inversion to have the same gene order. On the other hand, only 5 translocations from a brachiopod, T. retusa, and 6 translocations with 1 inversion from a phoronid, P. architecta, would produce the gene arrangement of B. neritina; therefore, the gene arrangement in T. retusa is most similar to that of B. neritina. The B. neritina gene arrangement could be obtained from that of T. retusa by only five translocation events (rrnS/rrnL, nad3/nad2, cox2, nad1 and nad6) with no inversions. The phoronid gene arrangement was identical to that of Katharina with only one exception, a difference in the position of atp6.

Comparison of arrangement of the mitochondrial protein-coding and ribosomal RNA genes for 2 bryozoans, 1 brachiopod, 1 phoronid and 1 polyplacophoran. Protein-coding and ribosomal RNA genes are designated by their abbreviations as shown in Fig. 1. Each gene map commences from cox3 and is oriented so that the gene is transcribed from left to right. The rearrangements that are needed to inter-convert the pair of maps are shown, disregarding tRNA genes in which shared gene arrangements are indicated. A circular arrow indicates inversion of a single gene or a block consisting of more than two genes. Dramatic differences were found in tRNA gene positions, but they are not depicted because they are highly complex.
Nucleotide composition and codon usage
As shown in Table 3, the overall A+T content of the B. neritina mitochondrial genome is 70.0% (+ strand: A = 37.7%; C = 17.6%; G = 12.4%; T = 32.3%), which is typical of the base compositions of metazoan mitochondrial genomes. However, it is unusual in comparison to those of other bryozoans and brachiopods; it is much higher than those of F. hispida (59.4%) and of three brachiopods, T. retusa (57.2%), T. transversa (59.1%) and L. rubellus (58.3%).
Table 3 shows the AT- and CG-skews of each of the 13 protein-coding and 2 ribosomal RNA genes and of the whole genome (total) in B. neritina mitochondria. The results show no marked bias in nucleotide composition. The AT-skew is positive for 11 genes and negative for five on the (+) strand. The CG-skew for all 15 genes on the (+) strand is positive. This means that the B. neritina mitochondrial genome has no biased nucleotide composition. As shown in [additional files 2 and 3], the other bryozoan, F. hispida, has no biased nucleotide composition either. In contrast, the AT-skews of 12 genes in T. transversa and L. rubellus and the CG-skews of nine genes in all three brachiopods seem clearly biased.
The codon usage pattern of the B. neritina mitochondrial protein-coding genes is shown in Table 4. There is a clear preference for A+T-rich codons; the five most frequently used codons are UUA (300 times) for leucine, AUA (281) for methionine, AUU (237) for isoleucine, UUU (178) for phenylalanine and AAA (144) for lysine. Compared to other lophotrochozoans, the B. neritina mitochondrial genome showed a strong bias to A+T codons with dramatically lower G+C content. The anticodon nucleotides in B. neritina were completely identical to those of the brachiopod Laqueus rubellus [37] and the annelid Lumbricus terrestris [38] except for trnL(cun) and trnY. However, two anticodons – UUU in trnK and UCU in trnS(agn) – in B. neritina were different from those used in most other metazoans. The tRNA anticodon corresponding to the codon AGN for serine is UCU, as in nematode mitochondrial genomes, but in most other metazoan mitochondrial genomes such as those of platyhelminthes, mollusks, Drosophila and echinoderms, the serine tRNA anticodon is GCU rather than UCU [25, 38].
Transfer RNA genes
The B. neritina mitochondrial genome contains 22 typical tRNA genes interspersed between the 2 rRNA and 13 protein-coding genes. This result differs from that of F. hispidia, which has only 21 tRNA genes because of the two serine tRNA genes, trnS(agn) and trnS(ucn), trnS(ucn) is absent [31]. If we obtain more bryozoan mitochondrial genome data, it might be possible to provide reasonable evolutionary interpretations through further comparative analyses with respect to the absence/presence of trnS(ucn). Thirteen of the 22 inferred B. neritina mitochondrial tRNAs have uniform features that are invariant in typical cloverleaf-shaped secondary structures with a 7-bp amino-acyl arm, 5-bp anticodon stem and 4-bp variable loop (Fig. 4). Two tRNAs [tRNACys, and tRNATyr] have no DHU arm or TψC arm. The TψC arm and variable loop are replaced by a single TV loop. In four tRNAs [tRNAGln, tRNALeu(uur), tRNASer(agn) and tRNASer(ucn)], the DHU arms are replaced by a loop. The unpaired DHU arm in tRNASer(agn) has been considered a typical feature of animal mitochondrial genomes [25]. tRNASer(ucn) with an unpaired DHU arm has also been reported for some protostomes: 2 nematodes (Caenorhabditis elegans and Ascaris suum [39]), 3 mollusks (1 chiton K. tunicata [40], 2 pulmonates Cepaea nemoralis and Euhadra herklotsi [41]), 2 brachiopods (T. transversa and L. rubellus [37, 42]) and 1 annelid (Lumbricus terrestris [38]). We also found loss of the DHU arm from tRNACys in the brachiopod L. rubellus, as in B. neritina.

Putative secondary structures of the 22 tRNAs identified in the mitochondrial genome of Bugula neritina. Bars indicate Watson-Click base pairings, and dots between G and U pairs mark canonical base pairings appearing in RNA.
Regardless of formation of a stable DHU arm, the first of 2 nts separating the amino-acyl stem from the DHU arm region is "T" in 14 tRNAs and the second is "A" in 19 tRNAs, and 1 nt separating the DHU arm region from the anticodon stem is "A" in 13 tRNAs. The 2 bp preceding the anticodon are always pyrimidines, with two exceptions – 'GU' in tRNALeu(cun) and 'AA' in tRNATyr – and the 1 nt nearest the anticodon is "T" in 21 cases, the exception being 'A' in tRNATyr. The nt immediately after the anticodon is always a purine ["A" in 20 tRNAs] with two exceptions – tRNAGlu and tRNATyr have "U" in the same position. Among the 18 tRNAs that form a stable TψC arm, 4-nt variable arms typical of metazoan mitochondrial tRNAs were observed in 15 tRNAs, 5-nt variable arms in 2 tRNAs, tRNAAsp and tRNASer(agn), and 6-nt variable arms in tRNAGlu. The inferred anticodons for 20 tRNAs in B. neritina were the same as those in the other bryozoan, F. hispida (Fig. 4), but anomalies were detected in two tRNAs: tRNATyr with AUA instead of GUA, and tRNALeu(cun) with GAG instead of UAG. The former has been reported for a few metazoans such as the predatory mite Metaseiulus occidentalis [43] and a onychophoran, Epiperipatus biolleyi [44], but the latter has never previously been reported for any metazoan. The tRNALeu(cun) with GAG may be considered an interesting feature unique to B. neritina. However, further experimental studies are needed to determine whether if it is a truly unique characteristic of B. neritina, or whether it results from a simple error in deducing the anticodon of tRNALeu(cun) from the nucleotide sequence of trnL(cun).
Ribosomal RNA genes
The two rRNA genes are generally separated by at least one gene (trnV in most of cases). In B. neritina, rrnS and rrnL are separated by trnN instead of trnV; trnV is located between trnR and trnQ. Assuming that the rRNA genes occupy all the available space between the adjacent genes, rrnS and rrnL are approximately 840 bp and 1,327 bp in length, respectively. The A+T contents of rrnS (69.1%) and rrnL (69.2%) are similar to the 70.0% of the whole mitochondrial genome. The total size (2,176 bp) of the B. neritina mitochondrial rRNAs was greater than those of the bryozoan F. hispida (1323 bp), 3 brachiopods (T. transversa, 1876 bp; L. rubellus, 1910 bp; T. retusa, 2057 bp), 2 annelids (P. dumerilii, 1962 bp; L. terrestris, 2030 bp) and a polyplacophoran mollusk K. tunicata (2101 bp), but less than those of a bivalve, Mytilis edulis (2189 bp), and a cephalopod, L. bleekeri (2312 bp).
Phylogenetic position of bryozoans and lophophorate phylogeny
As shown in Fig. 5 and [additional files 4, 5, 6], the first step of phylogenetic analysis (ML and BI) was performed on the basis of the nucleotide and amino acid sequences of 12 protein-coding genes in 42 metazoa (Table 5), in order to explore the phylogenetic position of bryozoans and lophophorate phylogeny within the Lophotrochozoa. All four trees showed that the two bryozoans (B. neritina and F. hispida) formed a strong monophyletic group (BP 100% in MLaa (Fig. 5) and MLnt [additional file 4], and BPP 1.0 in BIaa [additional file 5] and BInt [additional file 6]). No tree supported lophophorate monophyly, except for the MLaa tree in Fig. 5, in which lophophorates including Entoprocta are grouped together with a weak node confidence value (BP 40%). The sister group of the bryozoan clade appeared to be brachiopods (BP 88 in MLaa, BP 48 in MLnt and BPP 0.86 in BInt), except that the BIaa tree clustered Bryozoa with Phoronida [additional file 5]. As shown in Fig. 5 and [additional files 4, 5, 6], owing to possibly long-branch attraction artifacts (in particular, Nematoda and Platyhelminthes), all resultant ML and BI trees regardless of the data types employed showed unexpected groupings with extremely low node confidence values. In addition, phylogenetic trees inferred from nucleotide sequence data [additional files 4 and 6] had relatively lower node confidence values especially in deep branches. Amino acid-based trees (Fig. 5 and [additional file 5]) showed relatively higher node confidences in deep branches than the nucleotide-based trees [additional files 4 and 6].

Maximum likelihood tree inferred from amino acid sequences of 12 protein-coding genes of 42 metazoan mitochondrial genomes, showing weak support of the monophyly of lophophorates including Bryozoa, Brachiopoda, Phoronida and Entoprocta and a sister group relationship of Bryozoa and Brachiopoda. The numbers above/below the branches indicate bootstrapping values (BP) that show node confidence values. Gray boxes indicate lophophorate members. Metridium senile and Acropora tenuis were used as outgroups. Refer to Table 5 for more detailed information and classification of the species used. "M" in parenthesis is an abbreviation of the phylum Molluska. The log likelihood value of the best tree is -66427.37.
To resolve the problem of long-branch attraction, 2 nematodes and 3 platyhelminths were excluded from the first data set for the second-round phylogenetic analyses. The ML and BI trees newly obtained with the reduced data set, including 37 taxa comprising 35 protostomes (20 lophotrochzoans and 10 ecdysozoans), 5 deuterostomes and 2 primitive metazoans (outgroup taxa) were improved, robust and reliable with higher nodal support values. Within the Lophotrochozoa, all four trees (Fig. 6) showed that the monophylies of the two bryozoans (B. neritina and F. hispida) and the three brachiopods (T. transversa, L. rubellus, T. retusa) were strongly supported with strong nodal supports (BP 100% in MLaa and MLnt and BPP 1.0 in BIaa and BInt). In all four trees shown in Fig. 6, the strong monophyletic bryozoan clade, within the Lophotrochozoa, was grouped with a monophyletic brachiopod clade (BP 88% and 59% in MLaa and MLnt and BPP 1.0 and 0.98 in BIaa and BInt, respectively). The clade of Bryozoa + Brachiopoda was grouped with the clade of Annelida including Echiura as a subtaxon (BP 90% and 49% in MLaa and MLnt and BPP 0.99 and 0.98 in BIaa and BInt, respectively). P. psamophila (Phoronida) was clustered with Entoprocta in MLaa (BP 77%) and BIaa(BPP 0.90), which is consistent with the result of Yokobori et al. [45] based on mitochondrial protein-coding genes. In contrast, P. psamophila was grouped with a chiton, K. tunicate, in MLaa (BP 51%) and BIaa (BPP 0.97). This indicates that the phylogenetic positions of Phoronida, Entoprocta and K. tunicata are still ambiguous. No tree in Fig. 6 supports lophophorate monophyly.

Maximum likelihood trees inferred from amino acid (A) or nucleotide (B) sequences of 12 protein-coding genes in 37 metazoan mitochondrial genomes, showing a monoclade of Bryozoa and Brachipoda, a sister group relationship of Bryozoa + Brachiopoda and Annelida-Echiura, non-monophyly of lophophorates, and a close relationship of Phoronida and Entoprocta (or Katharina tunicate ). The numbers above and below the branches indicate bootstrapping values in percentage (BP) and Bayesian posterior probabilities (BPP) in order, which show node confidence values. Because the BI tree was very similar to the ML tree, only the ML tree is presented here and the BPP values of the BI tree are shown with BP values of the ML tree on each node. Gray boxes indicate lophophorate members. Metridium senile and Acropora tenuis were used as outgroups. Refer to Table 5 for more detailed information and classification of the species used. M in parenthesis is an abbreviation of the phylum Molluska. The log likelihood values of the best trees are -72906.37 in (A) and -106791.00 in (B).
The results of the present phylogenetic analyses revealed that lophophorates are placed with mollusks and annelids as members of a monophyletic lophotrochozoan group. This is consistent with evidence from 18S rRNA [15, 17, 46], Hox genes [20], Na/K ATPase α-subunit [47] and molecular data [14–18, 29]. Therefore, it strongly suggests that the long-held view inferred from morphological data [10] that deuterostomes have affinity with Bryozoa and the other two lophophorates should be refuted. Recent reports on lophophorate phylogeny based on SSU rRNA gene sequences [24, 48] coincide with the present result in that lophophorates are unambiguously affiliated with protostomes rather than deuterostomes.
Contrary to the present findings, which cluster Bryozoa with Brachiopoda, some previous SSU rRNA-based results have shown that brachiopods and phoronids (called the subphylum 'Phoroniformea', 'Brachiozoa' or 'Poronozoa') form a separate clade from the bryozoans and even suggest that phoronids may be members of the inarticulate brachiopods [11, 15, 17, 19, 21, 23, 30, 48, 49]. However, the present trees did not show the Brachiozoa grouping at all.
To clarify the statistical support for each grouping such as the monophylies of Brachiozoa, Lophophorata, the old-concept Bryozoa (comprising Entoprocta and Ectoprocta) [50, 51] and the sister group Bryozoa + Brachiopoda, we performed tree topology tests (Table 6). The results indicate that on the basis of statistical probability, the sister group of Bryozoa + Brachiopoda could be the Annelida-Echiura or the Phoronida + Entoprocta clade. If the latter is a true phylogeny, lophophorate monophyly including Entoprocta may be supported. The tree topology test is likely to indicate that Brachiozoa (= Brachiopoda + Phoronida) and the recently reinstated old-concept Bryozoa may be refuted, but according to the present data the sister group of Bryozoa is Brachiopoda (Table 6).
Despite intensive phylogenetic analyses, phylogenetic relationships among lophotrochozoan members including lophophorates and others unfortunately remain unclear because there are conflicts among the phylogenetic trees reconstructed by different tree-making methods, with different data types and with different taxon samplings (Figs. 5 and 6 and [additional files 4, 5, 6]). The phylogeny signal of mitochondrial genome nucleotides and/or amino acids alone may be unable to resolve what may have been a relatively rapid radiation during the Cambrian [52, 53]. Recently, to overcome such limitations, huge EST data sets from a number of metazoans have been employed to resolve metazoan phylogeny [49]. The results still left the phylogenetic position of bryozoans unclear, and lophophorates did not form a monophyletic group. Further more intensive studies seem to be necessary to resolve the exact phylogenetic position of the bryozoans and to examine the question of lophophorate monophyly.
Conclusion
This study presents the complete mitochondrial genome of a cheliostome bryozoan, B. neritina. Comparison of the orders of the protein-coding genes showed the possibility that three lophophorates are closely related, including K. tunicata. The present phylogenetic analyses suggest the probable relationships ((Bryozoa, Brachiopoda), Annelida-Echiura), or ((Bryozoa, Brachiopoda), (Phoronida, Entoprocta)), but the phylogenetic position of phoronids is still ambiguous. Consequently, the results seem to imply that the three lophophorate members did not form a monophyletic group in the phylogenetic trees and this possibility was also refuted statistically. However, according to the tree topology test, lophophorate monophyly including Entoprocta – ((Bryozoa, Brachiopoda), (Phoronida, Entoprocta)) – was not refuted. In addition, Brachiozoa (= Brachiopoda + Phoronida) and the recently-reinstated old-concept Bryozoa may be refuted, but according to the present data the sister group of Bryozoa is Brachiopoda (Table 6). However, because only a few samples of lophophorates were used here and there were some conflicts among the resultant trees, it is better to postpone a final decision on the phylogenetic position of bryozoans and on lophophorate phylogeny. Until more mitochondrial genomes become available and until we know more about the evolution of these organelle genomes, we may not come to any conclusion with respect to the monophyly or polyphyly of the lophophorates.
Methods
Specimen collection and DNA extraction
Bugula neritina (Bryozoa) was collected at Cheonsuman, Taean Gun, Chungnam Province, Korea. Total genomic DNA was extracted using a DNeasy tissue kit (QIAGEN Co., Hilden, Germany) following the manufacturer's protocol.
PCR amplification and cloning
The entire Bugula mitochondrial genome was amplified by two kinds of overlapping polymerase chain reactions (PCR). The PCR strategy was as follows: the ca. 2.5 kb fragment from cox1 to rrnL was amplified with previously reported universal primers, 16SA (5'-CGC CTG TTT ATC AAA AAC AT-3'; [54]) and HCO2198 (5'-TAA ACT TCA GGG TGA CCA AA AAA -3'; [55]). From the newly-sequenced ca. 2.5-kb sequences, the following two Bugula-specific primers were designed to amplify the remaining part (ca. 13.5 kb) of the mitochondrial genome: bnCOI (5'-AGC CAT TTT CTC TTT ACA CCT TGC-3') and bn16S (5'-TCA CTA CAA ACT CTA CAG GGT CTT-3').
The 2.5-kb PCR product was directly ligated to the pGEM T-easy vector (Promega), and the 13.5-kb PCR product was digested with Pst I, generating four fragments (approximately 0.9, 2.7, 2.7 and 7 kb). The two internal Pst I-restricted fragments (0.9 kb and 2.7 kb) were ligated into Pst I-digested pUC19 vector and both the end fragments (2.7 kb and 7 kb) with A-tailings were ligated into the modified, Pst I-digested pGEM T-easy vector (Promega Co.). All ligates were cloned with Escherichia coli DH5α strain. Correct recombinants were selected by the blue/white colony selection method using X-gal and IPTG. Plasmid DNAs were purified using an AtmanBio Plasmid Miniprep Kit (Takara Co., Japan).
Sequencing and sequence analysis
The purified plasmid DNA was sequenced using a primer walking method with the ABI PRISM BigDye terminator system and analyzed on an ABI3700 model automatic sequencer (Genotech Co., Korea). DNA sequences were analyzed using GeneJockey II, Version 1.6 (BIOSOFT Inc., Cambridge, UK). Thirteen mitochondrial protein-coding genes were initially identified by a BLAST comparison with other animal mitochondrial genomes, with start codons inferred as eligible in-frame start codons corresponding at least to the extent of alignment that does not overlap the upstream gene. Protein gene termini were inferred to be at the first in-frame stop codon unless this was located within the sequence of a downstream gene. Otherwise, a truncated stop codon (T or TA) adjacent to the beginning of the downstream gene was designated the termination codon, assuming that it could be completed by polyadenylation after transcript cleavage [56]. Ribosomal RNAs were identified by a BLAST search. A preliminary screening for tRNA genes was carried out using tRNAscan-SE, version 1.1 [57]. The tRNA genes that were not identified in this way were visually identified by inspection of anticodon sequences and their proposed cloverleaf secondary structures [58]. The sequence data obtained here are available from DDBJ/EMBL/GenBank under accession number AY690838.
Phylogenetic analysis
For the first step in the present phylogenetic analyses, we employed 40 protostomes and deuterostomes as ingroup taxa and 2 primitive metazoans as outgroup taxa, as listed in Table 5. When we selected the taxa for the present analyses, we tried to include all the lophotrochzoans for which complete mitochondrial genomes had already been sequenced. Some representative and/or slowly-evolving ecdysozoans and deuterostomes were included as reference taxa. All mitochondrial genome sequences obtained from members of the phyla Bryozoa (2 species), Brachiopoda (3), Phoronida (1), Echiura (1) and Entoprocta (2) were used here. However, since complete mitochondrial genome sequences from a number of members of the phyla Molluska (45), Platyhelminthes (28), and Annelida (8) have been determined, we selected only 3 each from Annelida and Platyhelminthes and 8 from Molluska, in order to reduce the calculation time in the present analyses. Those selected are representative and/or slowly-evolving ones in each phylum. Paraspadella gotoi and Spadella cephaloptera (Phyum Chaetognatha) and Cephalothrix rufifrons (Phylum Nemertea) were not included in the present analyses because they do not have atp6 and atp8, or have some genes that are as yet unidentified.
The nucleotide and amino acid sequences of the 12 protein-coding genes were used for the analyses. Only the 12 multiple alignment subsets of these sequences were created using a Clustal X multiple alignment program [59] under the default option. Only well-aligned, conserved alignment sites were extracted from each alignment subset using the Gblock program [60] with the default option. The conserved blocks extracted were subsequently concatenated into a single, unified, large alignment set with the Gblock program. In the second-round phylogenetic analyses, to resolve the problem of long-branch attraction, 5 taxa (2 nematodes and 3 platyhelminths) showing extremely long branches (Fig. 5 and [additional file 4]) were excluded from the original data set used in the first step. In total, the nucleotide and amino acid sequences of the mitochondrial protein-coding genes for 37 taxa were aligned and conserved blocks were extracted as described above.
For the first-round phylogenetic analyses with 42 metazoan mitochondrial genomes, the refined alignments (1735 aa and 4470 nt positions in length) were subjected to two different tree-making algorithms: the maximum likelihood (ML) and Bayesian inference (BI) methods. For phylogenetic analyses based on amino acid sequences, rather than using hierarchical likelihood ratio tests to select the best-fitting model for the evolution of sequences, and to calculate the related parameter values (I and Ã), ProtTest ver. 1.3 was used under the Akaike Information Criterion (AIC) because it has several important advantages [61]. Among the 36 models implemented in this program, the best-fitting model selected was MtArt [62] with among-site substitution-rate heterogeneity described by a gamma distribution (Ã = 0.732) and a fraction of sites constrained to be invariable (I= 0.072). For phylogenetic analyses based on nucleotide sequences, the best-fitting evolutionary model was estimated by Model Test 3.6 [63], from which the GTR+G+I (general time reversible model + among site rate variation + invariable sites) model was selected. Model Test 3.6 was also used to estimate the substitution rate parameters between nucleotides (AC 1.64479, AG 3.36847, AT 1.24161, CG 3.28174, CT 3.48682, and GT 1.00000) for the GTR model, base frequencies (A = 0.244605, C = 0.141275, G = 0.184743, T = 0.429377), assumed proportion of invariable sites (I = 0.126031), and the shape parameter (alpha) of the among-site rate variation (G = 0.665080).
For the second-round phylogenetic analyses with 35 protostomes and deuterostomes and 2 outgroup taxa, the refined alignments (2127 aa and 4965 nt positions in length) were subjected to the two different tree-making algorithms, ML and BI. For phylogenetic analyses based on amino acid sequences, MtArt was selected as the best-fitting model [62] with among-site substitution-rate heterogeneity described by a gamma distribution (Γ = 0.714) and a fraction of sites constrained to be invariable (I = 0.1511). For phylogenetic analyses based on nucleotide sequences, GTR+G+I (general time reversible model + among site rate variation + invariable sites) was selected as the best-fitting model. The substitution rate parameters between nucleotides were AC 1.08325, AG 3.02089, AT 1.20831, CG 2.51010, CT 2.92091, and GT 1.00000 for the GTR model, the base frequencies were A = 0.259281, C = 0.176486, G = 0.176848, T = 0.387385, the invariable site parameter (I) was 0.105884, and the shape parameter (alpha) of the among-site rate variation was G = 0.593221.
All the parameters estimated were then employed for ML and BI analyses in the first and second round phylogenetic analyses, respectively. Four rate categories were used in the present study. The ML analysis was carried out using PHYML v2.4.4 [64] and Treefinder [65]. The bootstrap proportions in percentage (BP) of the ML tree were obtained with 500 replicates by the fast-ML method using PHYML and Treefinder. The BI analysis was carried out using the MrBayes v3.0b4 program [66] with the following options: 1,000,000 generations, 4 chains (1 hot and 3 cold) and a burn-in step of the first 10,000. The node confidence values of the BI tree were presented with Bayesian posterior probabilities (BPP).
Statistical confidence values for possible groupings of the ML tree based on the amino acid residues of 12 protein-coding genes were computed by applying expected likelihood weights (ELWs) [67] to all local rearrangements (LR) of tree topology around an edge (1,000 replicates) using the program TREEFINDER.
Abbreviations
- atp6 and atp8:
-
genes for the ATPase subunits 6 and 8
- cox1-cox3 :
-
genes for cytochrome C oxidase subunits I-III
- cob :
-
a gene for apocytochrome b
- nad1-nad6 and nad4L:
-
genes for NADH dehydrogenase subunits 1–6 and 4L
- rrnS and rrnL:
-
genes for 12S and 16S rRNAs
- trnX :
-
where X is replaced by single-letter amino acid abbreviations of the corresponding amino acids
- trnL1 and trnL2:
-
genes for tRNALeu(UUR) (anticodon TAA) and tRNALeu(CUN) (anticodon TAG): respectively
- trnS1 and trnS2:
-
genes for the tRNASer(UCN) (anticodon TGA) and tRNASer(AGN) (anticodon GCT): respectively
- ML:
-
the maximum likelihood method
- BI:
-
Bayesian inference
- BPP:
-
Bayesian posterior probabilities
- BP:
-
bootstrap proportions
- MLnt:
-
the maximum likelihood tree inferred from nucleotide sequences
- MLaa:
-
the maximum likelihood tree inferred from amino acid sequences
- BInt:
-
the Bayesian inference tree inferred from nucleotide sequences
- BIaa:
-
the Bayesian inference tree inferred from amino acid sequences.
References
McKinney ML, Jackson JBC: Bryozoan Evolution. 1989, Chicago: University of Chicago Press
Hayward PJ, Ryland JS: Cheilostomatous Bryozoa Part 1 Aeteoidea-Cribrilinoidea. Edited by: Hayward PJ, Ryland JS. 1998, Brill and Backhuys
Ryland JS: Bryozoa: an introductory overview. Moostiere (Bryozoa), Moss Animals (Bryozoa) Denisia. Edited by: Wöss ER. 2005, 16: 9-20.
Hyman LH: The invertebrates: smaller coelomate groups. 1959, New York; Toronto; London: McGraw-Hill
Emig CC: On the origin of the lophophorata. Z Zool Syst Evolut-Forsch. 1984, 22: 91-94.
Brusca RC, Brusca GJ: Invertebrates. 1990, Sunderland, MA: Sinauer Associates Inc
Willmer P: Invertebrate Relationships: Patterns in animal evolution. 1990, New York: Cambrige University Press
Eming CC: Les lophophorates constituent-ils un embranchement?. Bull Soc Zool France. 1997, 122: 279-288.
Eernisse DJ, Albert JS, FE A: Annelida and arthropoda are not sister taxa: a phylogenetic analysis of spiralian metazoan phylogeny. Syst Biol. 1992, 41: 305-330.
Nielsen C, Scharff N, Eibye-Jacobsen D: Cladistic analyses of the animal kingdom. Biol J Linn Soc Lond. 1996, 57: 385-410. 10.1111/j.1095-8312.1996.tb01857.x.
Zrzavy J, Milhulka S, Kepka P, Bezdek A, Tietz DF: Phylogeny of the Metazoa based on morphological and 18S ribosomal DNA evidence. Cladistics. 1998, 14: 249-285.
Nielsen C: Animal evolution: Interrelationships of the living phyla. Edited by: Nielsen C. 2001, USA, Oxford University Press, 232-263. 2
Brusca RC, Brusca GJ: Invertebrates. Edited by: Brusca RC, Brusca GJ. 2003, MA, Sinauer Press, 771-779. 2
Conway-Morris S: Nailing the Lophophorates. Science. 1995, 375: 365-366.
Halanych KM, Bacheller JD, Aguinaldo AM, Liva SM, Hillis DM, Lake JA: Evidence from 18S ribosomal DNA that the lophophorates are protostome animals. Science. 1995, 267: 1641-1643. 10.1126/science.7886451.
Conway-Morris S, Cohen BL, Gawthrop AB, Cavalier-Smith T, Winnepenninckx B: Lophophorate phylogeny. Science. 1996, 272: 282-10.1126/science.272.5259.282.
Mackey LY, Winnepenninckx B, Dewachter R, Backeljau T, Emschermann P, Garey JR: 18S rRNA suggests that Entoprocta are protostomes, unrelated to Ectoprocta. J Mol Evol. 1996, 42: 552-559. 10.1007/BF02352285.
Cohen BL, Gawthrop AB: The brachiopod genome. Treatise on invertebrate paleontology. Edited by: Williams A, Lawrence KS. 1997, Geological Society of America and University of Kansas Press, 189-211.
Giribet G, Distel DL, Polz M, Sterrer W, Wheeler WC: Triploblastic relationships with emphasis on the acoelomates and the position of Gnathostomulida, Cycliophora, Platyhelminthes, and Chaetognatha: a combined approach of 18S rDNA sequences an morphology. Syst Biol. 2000, 49: 539-562. 10.1080/10635159950127385.
Passamaneck YL, Halanych KM: Evidence from Hox genes that bryozoans are lophotrochozoans. Evol Develop. 2004, 6: 275-281. 10.1111/j.1525-142X.2004.04032.x.
Peterson KJ, Eernisse DJ: Animal phylogeny and the ancestry of bilaterians: inferences from morphology and 18S rDNA gene sequences. Evol Develop. 2001, 3: 170-205. 10.1046/j.1525-142x.2001.003003170.x.
Field KG, Olsen GJ, Lane DJ, Giovannoni SJ, Ghiselin MT, Raff EC, Pace MR, Raff RA: Molecular phylogeny of the animal kingdom. Science. 1988, 239: 748-753. 10.1126/science.3277277.
Cohen BL, Gawthrop AB, Cavalier-Smith T: Molecular phylogeny of brachiopods and phoronids based on nuclear-encoded small subunit ribosomal RNA gene sequences. Phil Trans R Soc Lond B. 1998, 353: 2039-2061. 10.1098/rstb.1998.0351.
Cohen BL, Stark S, Gawthrop AB, Burke ME, Thayer CW: Comparison of articulate brachiopod nuclear and mitochondrial gene trees leads to a clade-based redefinition of protostomes Protostomozoa and deuterostomes Deuterostomozoa. Proc R Soc Lond Ser B Biol Sci. 1998, 265: 475-482. 10.1098/rspb.1998.0319.
Wolstenholme DR: Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992, 141: 173-216. 10.1016/S0074-7696(08)62066-5.
Hassanin A, Léger N, Deutsch J: Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol. 2005, 54: 277-298. 10.1080/10635150590947843.
Swire J, Judson O, Burt A: Mitochondrial genetic codes evolve to match amino acid requirements of proteins. J Mol Evol. 2005, 60: 128-139. 10.1007/s00239-004-0077-9.
Stechmann A, Schlegel M: Analysis of the complete mitochondrial DNA sequence of the brachiopod Tereburatulina retusa places Brachiopoda within the protosmes. P Roy Soc B Biol Sci. 1999, 266: 2043-2052. 10.1098/rspb.1999.0885.
Helfenbein KG, Boore JL: The mitochondrial genome of Phoronis architecta comparisons demonstrate that phoronids are lophotrochozoan protosmes. Mol Biol Evol. 2004, 21: 153-157. 10.1093/molbev/msh011.
Cavalier-Smith T: A revised six-kingdom system of life. Biol Rev. 1998, 73: 203-266. 10.1017/S0006323198005167.
Waeschenbach A, Telford MJ, Porter JS, Littlewood TJ: The complete mitochondrial genome of Flustrellidra hispida and the phylogenetic position of Bryozoa among the Metazoa. Mol Phylogenet Evol. 2006, 40: 195-207. 10.1016/j.ympev.2006.03.007.
Clayton DA: Replication of animal mitochondrial DNA. Cell. 1982, 28: 693-705. 10.1016/0092-8674(82)90049-6.
Clayton DA: Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol. 1999, 7: 453-478. 10.1146/annurev.cb.07.110191.002321.
Jacobs HT, Elliott DJ, Math VB, Farquharson A: Nucleotide sequence and gene organization of sea urchin mitochondrial DNA. J Mol Biol. 1988, 202: 185-217. 10.1016/0022-2836(88)90452-4.
Tomita K, Yokobori SI, Oshima T, Ueda T, Watanabae K: The Cephalopod Loligo bleekeri mitochondrial genome: multipled noncoding regions and transposition of tRNA genes. J Mol Biol. 2002, 54: 486-500.
Serb JM, Lydeard C: Complete mtDNA sequence of the North American freshwater mussel, Lampsilis ornata (Unionidae): an examination of the evolution and phylogenetic utility of mitochondrial genome organization in bivalvia (Molluska). Mol Biol Evol. 2003, 20: 1854-1866. 10.1093/molbev/msg218.
Noguchi Y, Endo K, Tajima F, Ueshima R: The mitochondrial genome of the brachiopod Laqueus rubellus. Genetics. 2000, 155: 245-259.
Boore JL, Brown WM: Complete sequence of the mitocondrial DNA of the annelid worm Lumburicus terrestris. Genetics. 1995, 141: 305-319.
Wolstenholme DR, Okimoto R, Macfarlane JL: Nucleotide correlations that suggest tertiary interactions in the TV-replacement loop-containing tochondrial tRNAs of the nematodes, Caenorhabditis elegans and Ascaris suum. Nucleic Acids Res. 1994, 22: 4300-4306. 10.1093/nar/22.20.4300.
Boore JL, Brown WM: Complete DNA sequence of the mitochondrial genome of the black chiton, Katharina tunicate. Genetics. 1994, 138: 423-443.
Yamazaki N, Ueshima R, Terrett JA, Thomas RH: Evolution of pulmonate gastropod mitochondrial genomes: comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics. 1997, 145: 749-758.
Helfenbein KG, Brown WM, Boore JL: The complete mitochondrial genome of the articulate brachiopoda Terabratalia transversa. Mol Biol Evol. 2001, 18: 1734-1744.
Jeyaprakash A, Hoy M: The mitochondrial genome of the predatory mite Metaseiulus occidentalis (Arthropoda: Chelicerata: Acari: Phytoseiidae) is unexpectedly large and contains several novel features. Gene. 2007, 391: 264-274. 10.1016/j.gene.2007.01.012.
Lars P, Anke B, Georg M: The complete mitochondrial genome of the Onychophoran Epiperipatus biolleyi reveals a unique transfer RNA set and provides further support for the ecdysozoa hypothesis. Mol Biol Evol. 2008, 25: 42-51.
Yokobori S, Iseto T, Asakawa S, Sasaki T, Shimizu N, Yamagishi A, Oshima T, Hirose E: Complete nucleotide sequences of mitochondrial genomes of two solitary entoprocts, Loxocorone allax and Loxosomella aloxiata: Implications for ophotrochozoan phylogeny. Mol Phylogenet Evol. 2008, 47 (2): 612-628. 10.1016/j.ympev.2008.02.013.
Eernisse DJ: Arthropod and annelid relationships re-examined. 1997, London: Chapman & Hall
Anderson FE, Córdoba AJ, Thollesson M: Bilaterian phylogeny based on analyses of a region of the sodium-potassium ATPase α-subunit gene. J Mol Evol. 2004, 58: 252-268. 10.1007/s00239-003-2548-9.
Cohen BL: Monophyly of brachiopods and phoronids: reconciliation of molecular evidence with Linnaean classification (the subphylum Phoronifirmea nov.). Proc R Soc Lond B. 2000, 267: 225-231. 10.1098/rspb.2000.0991.
Dunn C, Hejnol a, Matus D, Pang K, Browne W, Smith S, Seaver E, Rouse G, Obst M, Edgecombe G, et al: Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008, 452: 745-749. 10.1038/nature06614.
Hausdorf B, Helmkampf M, Meyer A, Witek A, Herlyn H, Bruchhaus I, Hankeln T, Struck T, Lieb B: Spiralian Phylogenomics Supports the Resurrection of Bryozoa Comprising Ectoprocta and Entoprocta. Mol Biol Evol. 2007, 24: 2723-2729. 10.1093/molbev/msm214.
Helmkampf M, Bruchhaus I, Hausdorf B: Phylogenomic analyses of lophophorates (brachiopods, phoronids and bryozoans) confirm the Lophotrochozoa concept. Proc R Soc B. 2008, 275: 1927-1933. 10.1098/rspb.2008.0372.
Adoutte A, Balavoine G, Lartillot N, Lespinet O, Prud'homme B, De Rosa R: The new animal phylogeny: reliability and implications. Proc Natl Acad Sci U S A. 2000, 97 (9): 4453-4456. 10.1073/pnas.97.9.4453.
Halanych K: The new view of animal phylogeny. Ann Rev Ecol Evol Syst. 2004, 35: 229-256. 10.1146/annurev.ecolsys.35.112202.130124.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P: Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a comilation of conserved polymerase chain reaction primers. Ann Entomol Soc Amer. 1994, 87: 651-701.
Folmer O, Black M, Hoeh R, Lutz RA, Vrijenhoek R: DNA primers for amplification of mitochondrial cytchrome C oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994, 3: 294-299.
Ojala D, Montoya J, Attardi G: tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981, 290: 470-474. 10.1038/290470a0.
Lowe TM, Eddy SR: tRNA-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-964. 10.1093/nar/25.5.955.
Kumazawa Y, Nishida M: Variations in mitochondrial tRNA gene organization of reptiles as phylogenetic markers. Mol Biol Evol. 1995, 12: 759-772.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24: 4876-4882. 10.1093/nar/25.24.4876.
Castresana J: Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000, 17: 540-552.
Posada D, Buckley T: Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst Biol. 2004, 53: 793-808. 10.1080/10635150490522304.
Adachi J, Hasegawa M: Model of amino acid substitution in proteins encoded by mitochondrial DNA. J Mol Evol. 1996, 42: 459-468. 10.1007/BF02498640.
Posada D, Crandall KA: Modeltest: testing the model of DNA substitution. Bioinformaitcs. 1998, 14: 817-818. 10.1093/bioinformatics/14.9.817.
Guindon S, Gascuel O: A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003, 52: 696-704. 10.1080/10635150390235520.
Jobb G: TREEFINDER version of may 2006. Munich, Germany: Distributed by the author. [http://www.treefinder.de]
Huelsenbeck JP, Ronquist F: MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001, 17: 754-755. 10.1093/bioinformatics/17.8.754.
Strimmer k, Rambaut A: Inferring confidence sets of possibly misspecified gene trees. Proc R Soc B. 2002, 269: 137-142. 10.1098/rspb.2001.1862.
Acknowledgements
The authors greatly appreciate Prof. Ji Eun Seo (Dept. of Social Welfare with addiction rehabilitation, Woosuk University, Jeonbuk, Korea) for providing the Bugula neritina sample for us, and also heartily thank the laboratory staffs of UWH (Mr. Yong Seok Lee, Ms. Shin Ju Park, and Mr. Jong Tae Lim) for their help with the experiments and sequence analysis. We also appreciate two anonymous reviewers for critical and valuable comments on the manuscript. This study was supported by both a grant from the Korea Science and Engineering Foundation (KOSEF; R01-2008-000-21028-0) and a year-2008 grant from the National Institute of Biological Resources, Korean Government (Origin of Biological Diversity of Korea: Molecular Phylogenetic Analyses of Major Korean Taxa) awarded to UWH.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
KHJ and UWH made substantial contributions to the conception and design of the study, acquisition of the data, and analysis and interpretation of the data. KHJ wrote the early draft of this manuscript, and UWH revised and rewrote all parts of the manuscript. Both authors read and approved the final version of the manuscript. UWH gave final approval of the version to be published.
Electronic supplementary material
12864_2008_2051_MOESM1_ESM.eps
Additional file 1:A hairpin-loop structure of a noncoding region NC1 in the mitochondrial genome of a bryozoan, Bugula neritina.(EPS 417 KB)
12864_2008_2051_MOESM2_ESM.docx
Additional file 2:AT-skew of mitochondrial protein-coding and ribosomal RNA genes of 14 lophotrochozoan species.(DOCX 17 KB)
12864_2008_2051_MOESM3_ESM.docx
Additional file 3:CG-skew of mitochondrial protein-coding and ribosomal RNA genes of 14 lophotrochozoan species.(DOCX 16 KB)
12864_2008_2051_MOESM4_ESM.eps
Additional file 4: Maximum likelihood tree inferred from nucleotide sequences of 12 protein-coding genes of 42 metazoan mitochondrial genomes, showing non-monophyly of lophophorates and a sister group relationship of Bryozoa and Brachiopoda. The numbers above/below the branches indicate bootstrapping values (BP) that show node confidence values. Gray boxes indicate lophophorate members. Metridium senile and Acropora tenuis were used as outgroups. Refer to Table 5 for more detailed information and classification of the species used. M in a parenthesis is an abbreviation of the phylum Molluska. The log likelihood value of the best tree is -112314.88. (EPS 2 MB)
12864_2008_2051_MOESM5_ESM.eps
Additional file 5: Bayesian Inference tree inferred from amino acid residues of 12 protein-coding genes of 42 metazoan mitochondrial genomes. The numbers above/below the branches indicate Bayesian posterior probabilities (BPP) that show node confidence values. Metridium senile and Acropora tenuis were used as outgroups. The log likelihood value of the best tree is -68516.902. Refer to Table 5 for more detailed information. (EPS 2 MB)
12864_2008_2051_MOESM6_ESM.eps
Additional file 6: Bayesian Inference tree inferred from nucleotide sequences of 12 protein-coding genes of 42 metazoan mitochondrial genomes. The numbers above/below the branches indicate Bayesian posterior probabilities (BPP) that show node confidence values. Metridium senile and Acropora tenuis were used as outgroups. The log likelihood value of the best tree is -112068.205. Refer to Table 5 for more detailed information. (EPS 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jang, K.H., Hwang, U.W. Complete mitochondrial genome of Bugula neritina (Bryozoa, Gymnolaemata, Cheilostomata): phylogenetic position of Bryozoa and phylogeny of lophophorates within the Lophotrochozoa. BMC Genomics 10, 167 (2009). https://doi.org/10.1186/1471-2164-10-167
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-167