
Abstract
Background
The mitochondrial DNA (mtDNA) of most animals evolves more rapidly than nuclear DNA, and often shows higher levels of intraspecific polymorphism and population subdivision. The mtDNA of anthozoans (corals, sea fans, and their kin), by contrast, appears to evolve slowly. Slow mtDNA evolution has been reported for several anthozoans, however this slow pace has been difficult to put in phylogenetic context without parallel surveys of nuclear variation or calibrated rates of synonymous substitution that could permit quantitative rate comparisons across taxa. Here, I survey variation in the coding region of a mitochondrial gene from a coral species (Balanophyllia elegans) known to possess high levels of nuclear gene variation, and estimate synonymous rates of mtDNA substitution by comparison to another coral (Tubastrea coccinea).
Results
The mtDNA surveyed (630 bp of cytochrome oxidase subunit I) was invariant among individuals sampled from 18 populations spanning 3000 km of the range of B. elegans, despite high levels of variation and population subdivision for allozymes over these same populations. The synonymous substitution rate between B. elegans and T. coccinea (0.05%/site/106 years) is similar to that in most plants, but 50–100 times lower than rates typical for most animals. In addition, while substitutions to mtDNA in most animals exhibit a strong bias toward transitions, mtDNA from these corals does not.
Conclusion
Slow rates of mitochondrial nucleotide substitution result in low levels of intraspecific mtDNA variation in corals, even when nuclear loci vary. Slow mtDNA evolution appears to be the basal condition among eukaryotes. mtDNA substitution rates switch from slow to fast abruptly and unidirectionally. This switch may stem from the loss of just one or a few mitochondrion-specific DNA repair or replication genes.
Similar content being viewed by others

Background
Rates of nucleotide substitution for mitochondrial DNA (mtDNA) are several times higher than those for nuclear DNA (nDNA) for most animals. Several reasons for this difference in rates have been proposed. Because mtDNA is haploid and usually maternally inherited, the effective population size of mitochondrial genes is one quarter that for nuclear genes, which should speed neutral divergence [1]. Unlike nDNA, mitochondria lack histones, which leaves them exposed to mutagens. Furthermore, because mitochondria are centers of oxidative metabolism, mtDNA faces more of the free radicals responsible for many mutations than does nDNA [2]. As a result, intraspecific variation for mtDNA may exceed that for nDNA [3]. This variation is often partitioned among populations, such that surveys of mtDNA may reveal population subdivision when other markers such as allozymes do not [4–7].
Yet not all mtDNA evolves rapidly. Rates of nucleotide substitution are slow in the mtDNA of plants, both in absolute terms and relative to nDNA [8]. Rates of mtDNA substitution appear to be slow in some basal animals as well, including sponges [9, 10] and anthozoans (corals, anemones, and their kin, [11–15]). This is especially surprising for corals. Corals and other anthozoans do not sequester their germ cells [16], yet single coral colonies may live many hundreds of years [17]. All else being equal, this combination of a nonsequestered germline and great longevity should lead to high rates of mitochondrial mutation, as any mutations accumulated over a long life could be passed on to offspring.
Contrary to this expectation, mtDNA divergence among closely related anthozoans is low [14, 18, 19], in fact lower than that for nDNA from the same taxa [20, 21]. Among the few intraspecific studies to survey mitochondrial variation from many (>20) individuals, most have focused on non-coding regions [22] or rRNA genes [23]. Variation in these regions has generally been less than or equal to that for nuclear genes [24, 25].
Such non-coding substitution rates are difficult to compare across taxa due to the effects of variation in evolutionary constraints on substitution rates and to difficulties in aligning homologous sites. Synonymous (silent) substitutions within coding regions, however, can often be aligned unambiguously among distant taxa. Synonymous sites show low variation in rates among different loci in the same genome [26], consistent with their neutral evolution. To date, surveys of mitochondrial coding regions in corals [25, 27, 28] have found little or no variation, but these studies have included just a few individuals (≤ 8) and a few localities (≤ 3), and have not been accompanied by surveys of variation at single-copy nuclear markers.
Here, I survey intraspecific variation and estimate the interspecific divergence rate for two species of coral, Balanophyllia elegans and Tubastrea coccinea, using a widely sequenced mitochondrial coding region from cytochrome c oxidase subunit I (coxI). Allozyme surveys in B. elegans have previously established that single copy nuclear markers are both variable and subdivided among populations [29, 30]. Divergence between the two species surveyed allows me to calculate fossil-calibrated rates of synonymous substitution and to place these rates of mtDNA substitution in the phylogenetic context of synonymous rates in other eukaryotic lineages
Results
coxI was invariant among all 67 B. elegans (GenBank Accession DQ445805) sampled from 18 populations spanning 3000 km of its geographic range, including 16 individuals from the site (Bodega Bay) that was most polymorphic for allozymes [29]. The seven T. coccinea samples from the Caribbean and the Eastern Pacific were likewise identical at coxI (Accession DQ445806). Rate calculations were based on this Caribbean/Eastern Pacific consensus sequence. The five sequences from Hawaiian T. coccinea were identical to each other, but differed from the Caribbean/Eastern Pacific consensus sequence by a single nonsynonymous substitution. Reading frames for all sequences remain open for all of these sequences when translated using the cnidarianmitochondrial genetic code [31].
The fossil record indicates that Balanophyllia and Tubastrea diverged at least 50 MY ago [32], yet raw coxI divergence between B. elegans and T. coccinea is only 2.7%. The synonymous substitution rate is 0.00055 substitutions per site per MY (Table 1). This rate is the same as that for two angiosperms (rice and maize) that diverged at about the same time as Balanophyllia and Tubastrea [26]. For comparison, diverse animal lineages sundered by the Isthmus of Panama three MY ago show rates of synonymous substitution roughly 100 times greater than for angiosperms and corals (Table 1); even the notably slow mtDNA of sharks [33] is 50 times faster. Nonsynonymous (amino acid altering) substitution rates for corals and plants are similar as well, but are only slightly slower than those bilateral animals (Table 1).
Patterns of nucleotide substitution between the two corals are also more similar to angiosperms [8] than to other animals. About half of coral mtDNA substitutions are transversions (9/17). In contrast, the mtDNA of other animals typically shows a strong transition bias [34, 35]. For example, the Tegula species in Table 1 show a 15-fold excess of transitions compared to transversions between closely-related species [36].
Discussion
Low levels of mitochondrial variation in corals
This study revealed little intraspecific variation within either of two corals, Balanophyllia elegans or Tubastrea coccinea. B. elegans was sampled over much (> 3000 km) of its broad geographic range, where high levels of allozyme variation have been found previously [29, 30]. The single substitution (a nonsynonymous one) observed within T. coccinea mtDNA occurred between populations separated by over 4000 km of uninhabitable ocean. Previous surveys of genetic variation in coding regions of coral mtDNA have found similar patterns: either no variation [27] or very little variation restricted to nonsynonymous sites [28].
Such low levels of genetic variation are not characteristic of anthozoan nuclear genes. Indeed, a comparison of allozyme polymorphism and heterozygosity found variation in cnidarians and sponges to be higher than those for all other animals [37]. High allozyme heterozygosity in B. elegans [29, 30] shows it is no exception to this trend. Intraspecific surveys of nDNA sequence variation from coding regions are lacking for corals, but intron sequences are quite variable [20, 24, 38], and microsatellites [39] and AFLPs [25] have revealed both high heterozygosities and population subdivision. While more extensive surveys of nucleotide variation from coding nDNA are needed, low variation appears to be restricted to the mitochondrial genome of corals.
Relatively low levels of mtDNA variation can result from range expansions, where the smaller effective population size of mtDNA genes can enhances founder effects. This may account for the lack of variation between Eastern Pacific and Caribbean T. coccinea, if the latter were indeed recently introduced as suggested by Cairns [40]. This does not appear to be the case for B. elegans, however. Subdivision within this species [29] does not suggest any anthropogenic range changes. Natural poleward range expansions following climatic cooling events can homogenize the mtDNA of newly founded populations, but more equatorial populations continue to harbor variation [41]. Sampled populations of B. elegans include its southern range limit, however, but still reveal no variation. Selective sweeps can also homogenize mtDNA within species. However, mtDNA regions are often identical among different species, genera, and even families [21, 42], a pattern that would require very strong stabilizing selection to maintain homogeneity (even at silent sites) over millions of years. The most likely explanation of low levels of mtDNA variation within coral species, then, is a low rate of nucleotide substitution.
Slow rates of synonymous substitution in corals
A growing body of evidence suggests that the mtDNA of anthozoans evolves slowly [11–15, 19]. The very low divergence found here for two genera with independent fossil records extending back over 50 MY provides an estimate of just how slowly: 0.055% per MY. This rate of synonymous substitution is 50–100 times slower than those reported previously for an array of animals (Table 1), including hydrozoans [43].
These low rates have practical consequences. First, the dearth of variation in anthozoan mtDNA makes routine phylogeographic surveys impossible. Alternative approaches employing microsatellite variation have revealed genetically isolated regions within coral species [39], but primers for single-copy nuclear gene regions that both amplify across diverse taxa and consistently reveal variation within species remain to be developed. Second, low variation means that mtDNA sequences cannot be counted on to reveal differences between closely related species. Indeed, the mtDNA region nominated for such DNA barcoding [44] is the very same coxI used here. Hebert et al. [45] stated previously that barcoding fails in cnidarians due to low variation; Figure 1 suggests that this limitation applies only to a subset of the Cnidaria, the anthozoans. Still, low rates of coxI evolution in corals, sea fans, and sponges mean that the very bricks and mortar of tropical reefs are not amenable to barcoding diversity surveys (although cox1 should still prove useful for resolving deeper phylogenetic relationships, e.g. [21]).

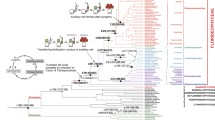
Phylogenetic correlation of tempo and mode of nucleotide substitution in mitochondrial DNA for flowering plants (Angio = Angiosperms), Fungi, Porifera (sponges), Cnidarians (Anthozoans and Scyphozoans), and bilateral animals [Platy = Platyzoa (flatworms), Lopho = Lophotrochozoa (molluscs, annelid worms, bryozoans), Ecdysozoa (arthropods, nematodes), and Deutero = Deuterostoma (echinoderms, tunicates, vertebrates)]. Supporting data: 1 [49] *see text for exceptions; 2 [8]; 3 [43,56]; 4 [59]; 5 [79,83]; 6 [36]; 7 [35]; 8 [80]; 9 [84]; 10 [34]; 11 [81,85].
Multiple losses of mtDNA repair function
As with corals, the available data for fungi [46] and sponges [47] suggest that rates of synonymous substitution in mtDNA are slower than nDNA in these taxa. Taken together with rates for plants and animals, these data suggest that mtDNA evolves in two distinct modes: one slow relative to nDNA and with little bias toward transitions, the other fast relative to nDNA and often (but not always, [48]) transition-biased (Figure 1). Phylogenetic analysis suggests that the switch from the slow mode to the fast mode occurs abruptly (without any apparent intermediate state) and always in the direction toward the fast mode. This switch has occurred at least four times: twice in flowering plants (in geraniums and plantains, [49]), and twice in animals (Figure 1).
One change in animals occurred within the phylum Cnidaria between the basal Anthozoa [50, 51] and the derived Medusozoa (hydroids and jellyfish). The independently derived fast mtDNA rates in the Medusozoa are similar to those in bilateral animals. In fact, 16S divergence rates in hydroids of the genus Hydractinia [52] are greater that of their hermit crab hosts [53], with whom they have probably co-speciated [54]). Phylogenetic analyses reveal interspecific variation typical for most animals among recent radiations of scyphozoans [55]. Scyphozoans also have high levels of intraspecific variation [56, 57] and show a transition:transversion bias > 10:1 (e. g. Cassiopea coxI data from [58]). A second switch from slow to fast mtDNA occurred at the base of the Bilateria (which includes the vertebrates); even flatworms show high intraspecific mtDNA variation and a strong transition bias [59].
This qualitative switch in the mode of mtDNA evolution is not a simple extension of the quantitative variation in mtDNA rates noted previously [33]. Within vertebrates, relative substitution rates in mtDNA and nDNA are correlated within taxa, despite variation in absolute rates among taxa [60]. Even in bilateral animals with notably slow mutation rates (e.g. the ancient asexual ostracod Darwinula stevensoni, [61]), the rate of mitochondrial substitution remains faster than that for nuclear loci. In contrast, the relative rates of substitution in mtDNA and nDNA are reversed in angiosperms, fungi, anthozoans, and probably sponges (nDNA evolves an order of magnitude faster than mtDNA) when compared to bilateral animals (where nDNA evolves an order of magnitude slower than mtDNA, [34, 35]).
No attribute of the mitochondrial genome itself correlates obviously with slow, unbiased evolution. For example, anthozoan mtDNA has group I introns [31, 62] like some plants and fungi, but is compact in size (ca. 16–18 kb) like most other animals [63]. Instead, the abrupt and unidirectional switch from slow to fast modes of mtDNA substitution is consistent with the sudden loss of one or a few mitochondrion-specific DNA repair or replication genes. A possible candidate gene has been proposed previously to account for low variation in anthozoans [11, 12]: an ortholog of the mismatch repair gene MSH1 (but see [64]). MSH1 is known to be mitochondrion-specific in yeast [65], and is present in the mitochondrial genome of octocorallian anthozoans [66] but missing from the bilateral animals whose genomes have been sequenced. Loss of any of the many genes involved in repair [67] could potentially speed synonymous substitution rates. Note also that the mitochondrial location of the putative MSH1 homolog in octocorallians is exceptional; MSH1 is not present in the mitochondrial genome of corals [19, 62], and mitochondrion-specific repair genes are generally encoded in the nuclear genome.
Regardless of the particular genes responsible, a loss of mtDNA-specific repair function (or crippling of mtDNA replication genes) could explain observed low levels of mtDNA variation and divergence compared to nuclear genes. Differences in the fidelity of mtDNA repair and replication could have broad implications. If mutations to mtDNA caused by oxidative stress promote cellular aging [68] and organismal senescence [69–71], then the loss of mtDNA repair abilities may place a physiological ceiling on longevity. Exploring this possibility will require phylogenetic comparisons of covariation in synonymous rates of substitution in mtDNA and patterns of senescence, as well as closer examination of the molecular mechanisms of mtDNA replication and repair across slow and fast mitochondrial lineages.
Conclusion
Both of the corals surveyed here showed low variation in mitochondrial coding gene sequence, despite the demonstration of extensive nuclear gene variation at allozymes in one of them previously. Combined with other types of nuclear variation (AFLPs, ITS, microsatellites) and phylogenetic studies reported previously, these results suggest that the lack of variation in coral mtDNA results from mechanisms specific to the mitochondrial genome. Synonymous substitution rates suggest coral mtDNA evolves at rates typical for plants, but about 100 times slower than for most animals.
Methods
Balanophyllia elegans and Tubastrea coccinea
Balanophyllia elegans is a small solitary coral common at shallow depths in temperate waters ranging between northern Baja California and southeastern Alaska. Average longevity for B. elegans has been estimated at about 8.5 years [72]. Dislodgment and overgrowth by algae appear to be the principle causes of death in B. elegans; adults do not appear to senesce nor do the growth rates of large individuals slow [72].
The larvae of B. elegans crawl along the seafloor during their brief dispersal [73]. As expected given such limited larval dispersal, genetic subdivision (inferred using allozymes) is high between localities separated by hundreds or thousands of kilometers (FST = 0.28, [29]). The allozyme markers employed in these genetic surveys were highly variable, both in terms of their mean heterozygosity (0.3, ranging above 0.5 for some loci at some locations) and number of alleles per locus (2.5). These markers have been demonstrated as single copy and Mendelian using controlled crosses [74].
Like B. elegans, Tubastrea coccinea belongs to the family Dendrophylliidae and broods its larvae. T. coccinea is currently recognized as a single species with a circum-tropical distribution [32], however this species has a long history of taxonomic splitting and synonymization. The Atlantic form is morphologically indistinguishable from the Pacific T. coccinea and does not appear in the Caribbean fossil record, suggesting this species may have been recently reduced to the Atlantic [40, 75]. However these populations appear to be differentiated at the allozyme level (E. Weil, pers. comm.); this Atlantic form has been called T. aurea.
Population sampling
Samples of B. elegans were collected from 18 localities spanning over 3000 km of the Pacific coast of North America, namely: Moresby Island, McInnes Island, Stubb's Island, Nanaimo and Bamfield from British Columbia; Tatoosh Island from Washington; Cape Arago from Oregon; Trinidad Harbor, Caspar, Bodega Bay, Santa Cruz, Monterey, San Simeon, Goleta, East Anacapa Island, and Point Loma from California; and Punta Banda and Isla San Geronimo from Baja California.(see [29]). Three individuals were selected for sequencing from each locality, each from a different subpopulation within that locality. In addition, two individuals were sequenced from each of eight subpopulations (a total of 16) from Bodega Bay, where the highest level of allozyme variation occurred (H = 0.42, averaged over seven loci [29]).
T. coccinea were collected from four localities: Kaneohe Bay, Oahu, Hawaii (5 individuals); La Paz and Isla Cerralvo in Baja California Sur, Mexico (2); Margarita Reef, Isla Magueyes, Puerto Rico (1); and four localities along the coast of Curaçao (4). All samples were taken from 5–15 m depth.
PCR amplification, sequencing and analysis
DNA was extracted from coral samples using the QIAamp DNA Mini Kit (Qiagen). A 710-bp fragment of coxI was initially amplified from B. elegans using primers LCO1490 and HCO2198 of Folmer et al. [76]. The resulting sequence was used to design an internal primer (Lc2COI, 5'-CGTTATTTTAGTATTTGGGATTGG-3') that was used in combination with HCO2198 for all subsequent amplification and sequencing.
Amplification products were sequenced directly on an ABI 377 using Big Dye Terminator chemistry, except for six templates (one from an Anacapa B. elegans, and T. coccinea from Puerto Rico and Curaçao plus three from Hawaii), which were cloned before sequencing. Multiple sequences were obtained from these clones to avoid misinterpretation of PCR errors.
Calculation of substitution rates
Genetic distances and transition/transversion ratios were estimated using MEGA3 [77]. Jukes-Cantor estimates were used because all mitochondrial sequences were similar (< 3 % divergence from raw counts) and no strong transition bias was evident. Numbers of synonymous and nonsynonymous sites were estimated using the methods of Nei and Gojobori [78], making appropriate adjustments for taxon-specific variation in the mitochondrial genetic code. Rates for trans-Ismuthian pairs were calculated using GenBank sequences for Tegula ([36]: AF080668, [79]: AF132340), Alpheus ([80]: AF309923, AF309904), Echinometra ([81]: AF255539, AF255502) and Sphyrna ([33]: L08042, L08043). Rates for the trans-Ismuthian teleost pair Abudefduf saxatlis and A. torschelli were taken from the literature [82]; sequences for A. saxatlis have not been deposited in GenBank and so nonsynonymous rates could not be calculated for this pair. Transition/transversion ratios for scyphozoans were calculated using Cassiopea sequences from [58] (AY319448-AY319473).
Abbreviations
- mitochondrial DNA:
-
(mtDNA)
- nuclear DNA:
-
(nDNA)
- cytochrome oxidase subunit I:
-
(coxI)
- MY:
-
(million years)
References
Birky CW, Fuerst P, Maruyama T: Organelle gene diversity under migration, mutation, and drift: equilibrium expectations, approach to equilibrium, effects of heteroplasmic cells, and comparison to nuclear genes. Genetics. 1989, 121: 613-627.
Shigenaga MK, Hagen TM, Ames BN: Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA. 1994, 91: 10771-10778.
Avise JC, Lansman RA: Polymorphism of mitochondrial DNA in populations of higher animals. Evolution of Genes and Proteins. Edited by: Nei M, Koehn RK. 1983, Sinauer: Sunderland, 147-164.
Reeb CA, Avise JC: A genetic discontinuity in a continuously distributed species – mitochondrial DNA in the American oyster, Crassostrea virginica. Genetics. 1989, 124: 397-406.
Burton RS, Lee B-N: Nuclear and mitochondrial gene genealogies and allozyme polymorphism across a major phylogeographic break in the copepod Tigriopus californicus. Proc Natl Acad Sci USA. 1994, 91: 5197-5201.
Buonaccorsi VP, Reece KS, Morgan LW, Graves JE: Geographic distribution of molecular variance within the blue marlin (Makaira nigricans): A hierarchical analysis of allozyme, single-copy nuclear DNA, and mitochondrial DNA markers. Evolution. 1999, 53: 568-579.
Wlasiuk G, Garza JC, Lessa EP: Genetic and geographic differentiation in the Rio Negro tuco-tuco (Ctenomys rionegrensis): Inferring the roles of migration and drift from multiple genetic markers. Evolution. 2003, 57: 913-926.
Wolfe KH, Li W-H, Sharp PM: Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci USA. 1987, 84: 9054-9058.
Duran S, Pascual M, Turon X: Low levels of genetic variation in mtDNA sequences over the western Mediterranean and Atlantic range of the sponge Crambe crambe (Poecilosclerida). Mar Biol. 2004, 144: 131-135. 10.1007/s00227-003-1178-5.
Wörheide G: Low variation in partial cytochrome oxidase subunit I (COI) mitochondrial sequiences in the coralline demosponge Astrosclera willeyana across the Indo-Pacific. Mar Biol. 2005,
France SC, Rosel PE, Agenboard JE, Mullineaux LS, Kocher TD: DNA sequence variation of mitochondrial large-subunit rRNA provides support for a two subclass organization of the Anthozoa (Cnidaria). Mar Mol Biol Biotechnol. 1996, 5: 15-28.
van Oppen MJH, Willis B, Miller DJ: Atypically low rate of cytochrome b evolution in the scleractinian coral genus Acropora. Proc R Soc Lond B. 1999, 266: 179-183. 10.1098/rspb.1999.0619.
Chen CA, Wallace GC, Yu JK, Wei NV: Strategies for amplification by polymerase chain reaction of the complete sequence of the gene encoding nuclear large subunit ribosomal RNA in corals. Mar Biotechnol. 2000, 2: 558-570. 10.1007/s101260000040.
Shearer TL, van Oppen MJH, Romano SL, Worheide G: Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol. 2002, 11: 2475-2487. 10.1046/j.1365-294X.2002.01652.x.
Tseng CC, Wallace CC, Chen CA: Mitogenomic analysis of Montipora cactus and Anacropora matthai (Cnidaria; Scleractinia; Acroporidae) indicates an unequal rate of mitochondrial evolution among Acroporidae corals. Coral Reefs. 2005, 24: 502-508. 10.1007/s00338-005-0499-x.
Buss LW: Evolution, development, and the units of selection. Proc Natl Acad Sci USA. 1983, 80: 1387-1391.
Potts DC, Done TJ, Isdale PJ, Fisk DA: Dominance of a coral community by the genus Porites (Scleractinia). Mar Ecol Prog Ser. 1985, 23: 79-84.
van Oppen MJH, Koolmees EM, Veron JEN: Patterns of evolution in the scleractinian coral genus Montipora (Acroporidae). Marine Biology. 2004, 144: 9-18. 10.1007/s00227-003-1188-3.
Fukami H, Knowlton N: Analysis of complete mitochondrial DNA sequences of three members of the Montastrea annularis coral species complex (Cnidaria, Anthozoa, Scleractinia). Coral Reefs. 2005, 24: 410-417. 10.1007/s00338-005-0023-3.
van Oppen MJH, McDonald BJ, Willis B, Miller DJ: The evolutionary history of the coral genus Acropora (Scleractinia, Cnidaria) based on a mitochondrial and a nuclear marker: Reticulation, incomplete lineage sorting, or morphological convergence?. Mol Biol Evol. 2001, 18: 1315-1329.
Fukami H, Budd AF, Paulay G, Sole-Cava A, Chen CA, Iwao K, Knowlton N: Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nature. 2004, 427: 832-836. 10.1038/nature02339.
Smith PJ, McVeagh SM, Mingoia JT, France SC: Mitochondrial DNA sequence variation in deep-sea bamboo coral (Keratosidinae) species in the southwest and northwest Pacific Ocean. Mar Biol. 2004, 144: 253-261. 10.1007/s00227-003-1206-5.
Watanabe T, Nishida M, Watanabe K, Wewengkang DS, Hidaka M: Polymorphism in nucleotide sequence of mitochondrial intergenic region in scleractinian coral (Galaxea fascicularis). Mar Biotechnol. 2005, 7: 33-38. 10.1007/s10126-004-3200-4.
Vollmer SV, Palumbi SR: Hybridization and the evolution of reef coral diversity. Science. 2002, 296: 2023-2025. 10.1126/science.1069524.
Fukami H, Budd AF, Levitan DR, Jara J, Kersanach R, Knowlton N: Geographic differences in species boundaries among members of the Montastrea annularis complex based on molecular and morphological markers. Evolution. 2004, 58: 324-337.
Li W-H: Molecular evolution. 1997, Sinauer, Sunderland, MA
Snell TL, Foltz DW, Sammarco PW: Variation in morphology vs. conservatism of a mitochondrial gene in Montastrea cavernosa (Cnidaria, Scleractinia). Gulf of Mexico Sci. 1998, 188-195.
Medina M, Weil E, Szmant AM: Examination of the Montastrea annularis species complex (Cnidaria: Scleractinia) using ITS and COI sequences. Mar Biotechnol. 1999, 1: 89-97. 10.1007/PL00011756.
Hellberg ME: Relationships between inferred levels of gene flow and geographic distance in a philopatric coral, Balanophyllia elegans. Evolution. 1994, 48: 1829-1854. [http://www.biology.lsu.edu/webfac/mhellberg/pubs/hellberg1994.pdf]
Hellberg ME: Stepping-stone gene flow in the solitary coral Balanophyllia elegans: equilibrium and nonequilibrium at different spatial scales. Marine Biology. 1995, 123: 573-581. 10.1007/BF00349236.
Beagley CT, Okimoto R, Wolstenholme DR: The mitochondrial genome of the sea anemone Metridium senile (Cnidaria): introns, a paucity of tRNA genes, and a near- standard genetic code. Genetics. 1998, 148: 1091-1108.
Cairns SD: A generic revision and phylogenetic analysis of the Dendrophylliidae (Cnidaria: Scleractinia). Smithson Contrib Zool. 2001, 615: 75-
Martin AP, Naylor GJP, Palumbi SR: Rates of mitochondrial DNA evolution in sharks are slow compared with mammals. Nature. 1992, 357: 153-155. 10.1038/357153a0.
Brown WM, Pranger EG, Wand A, Wilson AC: Mitochondrial DNA sequences of primates: tempo and mode of evolution. J Mol Evol. 2982, 18: 225-239. 10.1007/BF01734101.
Moriyama EN, Powell JR: Synonymous substitution rates in Drosophila: mitochondrial versus nuclear genes. J Mol Evol. 1997, 45: 378-391. 10.1007/PL00006256.
Hellberg ME: Sympatric sea shells along the sea's shore: the geography of speciation in the marine gastropod Tegula. Evolution. 1998, 52: 1311-1324.
Sole-Cava AM, Thorpe JP, Kaye JG: Reproductive isolation with little genetic divergence between Urticina (= Tealia) felina and U. eques (Anthozoa: Actinaria). Mar Biol. 1985, 85: 279-284. 10.1007/BF00393248.
Mackenzie JB, Munday PL, Willis BL, Miller DJ, van Oppen MJH: Unexpected patterns of genetic structuring among locations but not colour morphs in Acropora nasuta (Cnidaria; Scleractinia). Mol Ecol. 2004, 13: 9-20. 10.1046/j.1365-294X.2003.02019.x.
Baums IB, Miller MW, Hellberg ME: Regionally isolated populations of an imperiled Caribbean coral, Acropora palmata. Mol Ecol. 2005, 14: 1377-1390. 10.1111/j.1365-294X.2005.02489.x.
Cairns S: A revision of the shallow-water zooxanthellate Scleractinia of the Western Atlantic. Stud Nat Hist Carib. 2000, 75: 1-240.
Hellberg ME, Balch DP, Roy K: Climate-driven range expansion and morphological evolution in a marine gastropod. Science. 2001, 292: 1707-1710. 10.1126/science.1060102.
Romano SL, Palumbi SR: Evolution of scleractinian corals inferred from molecular systematics. Science. 1996, 271: 640-642.
Govindarajan AF, Halanych KK, Cunningham CW: Mitochondrial evolution and phylogeography in the hydrozoan Obelia geniculata (Cnidaria). Marine Biology. 2005, 146: 213-222. 10.1007/s00227-004-1434-3.
Hebert PDN, Cywinska A, Ball SL, DeWaard JR: Biological identifications through DNA barcodes. Proc Roy Soc Lond B. 2003, 270: 313-321. 10.1098/rspb.2002.2218.
Hebert PDN, Ratnasingham S, deWaard JR: Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Roy Soc Lond B. 2003, 270 (Suppl): S96-S99.
Clark-Walker GD: Contrasting mutation rates in mitochondrial and nuclear genes of yeast versus mammals. Curr Genet. 1991, 20: 195-198. 10.1007/BF00326232.
Schröder HC, Efremova SM, Itskovich VB, Belikov S, Masuda Y, Krasko A, Müller IM, Müller WEG: Molecular phylogeny of the freshwater sponges in Lake Baikal. J Zool Syst Evol Res. 2003, 41: 80-86. 10.1046/j.1439-0469.2003.00199.x.
Cho Y, Mower JP, Qiu Y-L, Plamer JD: Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci USA. 2004, 101: 17741-17746. 10.1073/pnas.0408302101.
Palmer JD, Adams KL, Cho Y, Parkinson CL, Qiu Y-L, Song K: Dynamic evolution of plant mitochondrial genomes: mobile genes and introns and highly variable mutation rates. Proc Natl Acad Sci USA. 2000, 97: 6960-6966. 10.1073/pnas.97.13.6960.
Bridge D, Cunningham CW, Schierwater B, DeSalle R, Buss LW: Class-level relationships in the phylum Cnidaria: evidence from mitochondrial genome structure. Proc Natl Acad Sci USA. 1992, 89: 8750-8753.
Medina M, Collins AG, Silberman JD, Sogin ML: Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc Natl Acad Sci USA. 2001, 98: 9707-9712. 10.1073/pnas.171316998.
Cunningham CW, Buss LW: Molecular evidence for multiple episodes of paedomorphosis in the Family Hydractiniidae. Biochem Syst Ecol. 1993, 21: 57-69. 10.1016/0305-1978(93)90009-G.
Cunningham CW, Blackstone NW, Buss LW: Evolution of king crabs from hermit crab ancestors. Nature. 1992, 355: 539-542. 10.1038/355539a0.
Cunningham CW, Buss LW, Anderson C: Molecular and geological evidence of shared history between hermit crabs and the symbiotic genus Hydractinia. Evolution. 1991, 45: 1301-1316.
Dawson MN, Hamner WM: Rapid evolutionary radiation of marine zooplankton in peripheral environments. Proc Natl Acad Sci USA. 2005, 102: 9235-9240. 10.1073/pnas.0503635102.
Dawson MN, Jacobs DK: Molecular evidence for cryptic species of Aurelia aurita (Cnidaria, Scyphozoa). Biol Bull. 2001, 200: 92-96.
Schroth W, Jarms G, Streit B, Schierwater B: Speciation and phylogeography in the cosmopolitan marine moon jelly, Aurelia sp. BMC Evol Biol. 2002, 2: 1-10.1186/1471-2148-2-1.
Holland BS, Dawson MN, Crow GL, Hofmann DK: Global phylogeography of Cassiopea (Scyphozoa: Rhizostomeae): molecular evidence for cryptic species and multiple invasions of the Hawaiian Islands. Mar Biol. 2004, 145: 1119-1128. 10.1007/s00227-004-1409-4.
Blair D, Agatsuma T, Watanobe T, Okimoto M, Ito A: Geographical genetic structure within the human lung fluke, Paragonimus westermani, detected from DNA sequences. Parasitology. 1997, 115: 411-417. 10.1017/S0031182097001534.
Martin AP: Substitution rates of organelle and nuclear genes in sharks: implicating metabolic rate (again). Mol Biol Evol. 1999, 16: 996-1002.
Schön I, Martens K: No slave to sex. Proc Roy Soc Lond B. 2003, 270: 827-833.
van Oppen MJH, Catmull J, McDonald BJ, Hislop NR, Hagerman PJ, Miller DJ: The mitochondrial genome of Acropora tenuis (Cnidaria; Scleractinia) contains a large group I intron and a candidate control region. J Mol Evol. 2000, 55: 1-13. 10.1007/s00239-001-0075-0.
Lang B, Gray MW, Burger G: Mitochondrial genome evolution and the origin of eukaryotes. Ann Rev Genet. 1999, 33: 351-397. 10.1146/annurev.genet.33.1.351.
Eisen JA: A phylogenomic study of the MutS family of proteins. Nuc Acids Res. 1998, 26: 4291-4300. 10.1093/nar/26.18.4291.
Reenan RAG, Kolodner RD: Characterization of insertion mutations in the Saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics. 1992, 132: 975-985.
Pont-Kingdon GA, Okada NA, Macfarlane JL, Beagley CT, Wolstenholme DR, Cavalier-Smith T, Clark-Walker GD: A coral mitochondrial mutS gene. Nature. 1995, 375: 109-111. 10.1038/375109b0.
Eisen JA, Hanawalt PC: A phylogenomic study of DNA repair genes, proteins, and processes. Mut Res. 1999, 435: 171-213.
Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA: Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005, 309: 481-484. 10.1126/science.1112125.
Harman D: The aging process. Proc Natl Acad Sci USA. 1981, 78: 7124-7128.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG: Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004, 429: 417-423. 10.1038/nature02517.
Balaban RS, Nemoto S, Finkel T: Mitochondria, oxidants, and aging. Cell. 2005, 120: 483-495. 10.1016/j.cell.2005.02.001.
Fadlallah Y: Population dynamics and life history of a solitary coral, Balanophyllia elegans, from central California. Oecologia. 1983, 58: 200-207. 10.1007/BF00399217.
Gerrodette T: Dispersal of the solitary coral Balanophyllia elegans by demersal planular larvae. Ecology. 1981, 62: 611-619.
Hellberg ME, Taylor MS: Genetic analysis of sexual reproduction in the dendrophylliid coral Balanophyllia elegans. Marine Biology. 2002, 141: 629-637. 10.1007/s00227-002-0861-2.
Fenner D, Banks K: Orange cup coral Tubastrea coccinea invades Florida and the Flower Garden Banks, northwestern Gulf of Mexico. Coral Reefs. 2004, 23: 505-507.
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R: DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol. 1994, 3: 294-299.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetic Analysis and sequence alignment. Bioinformatics. 2004, 5: 150-163.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Hellberg ME, Vacquier VD: Rapid evolution of fertilization selectivity and lysin cDNA sequences in teguline gastropods. Mol Biol Evol. 1999, 16: 839-848.
Knowlton N, Weigt LA, Solórzano LA, Mills DK, Bermingham E: Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science. 1993, 260: 1629-1632.
McCartney MA, Keller G, Lessios HA: Dispersal barriers in tropical oceans and speciation in Atlantic and eastern Pacific sea urchins of the genus Echinometra. Mol Ecol. 2000, 9: 1391-1400. 10.1046/j.1365-294x.2000.01022.x.
Bermingham E, McCafferty SS, Martin AP: Fish biogeography and molecular clocks: perspectives from the Panamanian Isthmus. Molecular Systemtics of Fishes. Edited by: Kocher TD, Stepien CA. Academic, San Diego, 113-128.
McGovern TM, Hellberg ME: Cryptic species, cryptic endosymbionts, and geographic variation in chemical defenses in the bryozoan Bugula neritina. Mol Ecol. 2003, 12: 1207-1215. 10.1046/j.1365-294X.2003.01758.x.
Funk DJ: Molecular systematics of cytochrome oxidase I and 16S from Neochlamisus leaf beetles and the importance of sampling. Mol Biol Evol. 1999, 16: 67-82.
Tarjuelo I, Posada D, Crandall KA, Pascual M, Turon X: Cryptic species of Clavelina (Ascidiacea) in two different habitats: harbours and rocky littoral zones in the northwestern Mediterranean. Mar Biol. 2001, 139: 455-462. 10.1007/s002270100587.
Acknowledgements
Thanks to D. Ayre, J. Geller, E. Gleason, J. Larkin, J. Neigel, D. Pollock, and M. Taylor for discussions and comments, to M. Taylor and R. Toonen for collections, and to B. Bryan, N. Crochet and P. Arbour-Reily for technical help.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hellberg, M.E. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol 6, 24 (2006). https://doi.org/10.1186/1471-2148-6-24
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-6-24