
Abstract
Background
RNA-protein interactions (RPIs) play important roles in a wide variety of cellular processes, ranging from transcriptional and post-transcriptional regulation of gene expression to host defense against pathogens. High throughput experiments to identify RNA-protein interactions are beginning to provide valuable information about the complexity of RNA-protein interaction networks, but are expensive and time consuming. Hence, there is a need for reliable computational methods for predicting RNA-protein interactions.
Results
We propose RPISeq, a family of classifiers for predicting R NA-p rotein i nteractions using only seq uence information. Given the sequences of an RNA and a protein as input, RPIseq predicts whether or not the RNA-protein pair interact. The RNA sequence is encoded as a normalized vector of its ribonucleotide 4-mer composition, and the protein sequence is encoded as a normalized vector of its 3-mer composition, based on a 7-letter reduced alphabet representation. Two variants of RPISeq are presented: RPISeq-SVM, which uses a Support Vector Machine (SVM) classifier and RPISeq-RF, which uses a Random Forest classifier. On two non-redundant benchmark datasets extracted from the Protein-RNA Interface Database (PRIDB), RPISeq achieved an AUC (Area Under the Receiver Operating Characteristic (ROC) curve) of 0.96 and 0.92. On a third dataset containing only mRNA-protein interactions, the performance of RPISeq was competitive with that of a published method that requires information regarding many different features (e.g., mRNA half-life, GO annotations) of the putative RNA and protein partners. In addition, RPISeq classifiers trained using the PRIDB data correctly predicted the majority (57-99%) of non-coding RNA-protein interactions in NPInter-derived networks from E. coli, S. cerevisiae, D. melanogaster, M. musculus, and H. sapiens.
Conclusions
Our experiments with RPISeq demonstrate that RNA-protein interactions can be reliably predicted using only sequence-derived information. RPISeq offers an inexpensive method for computational construction of RNA-protein interaction networks, and should provide useful insights into the function of non-coding RNAs. RPISeq is freely available as a web-based server at http://pridb.gdcb.iastate.edu/RPISeq/.
Similar content being viewed by others

Background
Most of the essential molecular functions of cells are governed by interactions of proteins with other proteins, nucleic acids and small ligands. Computational studies of protein interaction data have helped identify protein-protein interaction PPI networks in various organisms [1, 2]. Similarly, studies on DNA-protein interactions have allowed construction of transcription factor-gene regulatory networks [3, 4]. In contrast, although several ribonucleoprotein (RNP) complexes have been extensively characterized (e.g., the ribosome, the spliceosome), post-transcriptional regulatory networks that are mediated by RNA-protein interactions (RPIs) are much less well studied [5–9]. In addition to their roles in controlling gene expression at the post-transcriptional level, RPIs regulate numerous fundamental biological processes, ranging from DNA replication and transcription, to pathogen resistance, to viral replication [10–13]. Recently, high-throughput experiments have provided evidence for large numbers of RNA binding proteins in cells, and are beginning to identify and characterize pairs of RNAs and proteins that participate in RPIs [14–19]. At present, however, our understanding of RNA binding proteins lags far behind our knowledge of regulatory DNA binding proteins, such as transcription factors and replication factors.
Computational studies of RNA-protein interactions have largely focused on the "interface prediction problem", i.e., the problem of identifying the amino acid residues in a protein that are likely to bind to an RNA [20–22]. Only a few studies to date have focused on the "partner prediction problem", i.e., identification of specific RNA interaction partner(s) for a known RNA binding protein, or protein binding partner(s) for non-coding RNAs (ncRNAs). Although large-scale experimental analyses of RPIs such as RNAcompete [23], RIP-Chip [24], HITS-CLIP [25], PAR-CLIP [8] are now providing valuable data about networks of RNA-protein interactions, these experiments are expensive and time-consuming. Thus, there is a compelling need for computational methods to accurately predict RPIs and to construct RNA-protein interaction networks. Given the limited number of structurally characterized RNA-protein complexes available in the PDB [26] at present (1,092 as of June 13, 2011) and the current availability of only one database of ncRNA-protein interactions (NPInter [27]), it would be especially valuable to develop sequence-based methods that can be used to identify potential RNA-protein partners in the absence of experimental structural information regarding either partner.
Machine learning offers one of the most cost-effective approaches to constructing predictive models in settings where experimentally validated training data are available. At present, however, it is unclear whether the available experimental data regarding RNA-protein interactions are sufficient for successfully training classifiers using machine learning algorithms. Against this background, this study explores machine learning approaches to train sequence-based classifiers for predicting RPIs.
Results
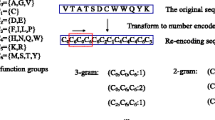
As a first step towards computational construction of RPI networks, we focused on the following question: Given the sequence of an RNA-binding protein, can we predict whether it interacts with a given RNA sequence? In developing sequence-based methods to answer this question, we considered several reduced and alternative alphabet representations of the input protein and RNA sequences. Shen et al. [28] used a Conjoint Triad Feature (CTF) representation to successfully predict protein-protein interactions. The CTF representation essentially encodes each protein sequence using the normalized 3-gram frequency distribution extracted from a 7-letter reduced alphabet representation of the protein sequence (See Methods for details). A recent study [29] demonstrated the utility of the CTF representation for predicting whether a given protein is an RNA binding protein. Inspired by these studies, we chose to encode each protein sequence using the normalized k-gram frequency distributions extracted from the 7-letter reduced alphabet representation of the sequence. The choice of k = 3 yielded the best results. We also explored several alternative representations of RNA sequences and settled on encoding each RNA sequence using normalized 4-gram frequencies extracted directly from the 4-letter ribonucleotide alphabet representation of the RNA sequence.
Our choice of Random Forest (RF) and Support Vector Machine (SVM) classifiers was motivated by several studies that have successfully used them on classification tasks that are closely related to the RPI prediction [30–33]. To rigorously evaluate the performance of these methods, we generated two non-redundant benchmark datasets, RPI2241 and RPI369, from PRIDB [34], a comprehensive database of RNA-protein complexes extracted from the PDB [26]. Most of the RNA-protein pairs in RPI2241 correspond to RPIs involving rRNAs or ribosomal proteins; the rest correspond to RPIs involving other ncRNAs or mRNAs. RPI369 corresponds to RPIs extracted from non-ribosomal complexes in RPI2241. "Negative" examples of non-interacting RNA-protein pairs were generated by randomly pairing proteins with RNAs and excluding the known interacting pairs (see Methods for details).
RPISeq classifiers can reliably predict RNA-protein interactions
We compared the performance of RPISeq-SVM and RPISeq-RF classifiers to predict RPIs, using the benchmark datasets described above. Table 1 summarizes the prediction results obtained in 10-fold cross-validation experiments. On the RPI2241 dataset, the prediction accuracy was 89.6% (RF) and 87.1% (SVM); precision and recall for both classifiers was greater than 87%. On the RPI369 dataset, performance of both classifiers was considerably lower with an average accuracy of only 76.2% (RF) and 72.8% (SVM). Notably, values of the F-measure (weighted average of precision and recall) were greater than 0.70 for both classifiers on both datasets. Thus, the performance of classifiers estimated using 10-fold cross-validation on the larger RPI2241 dataset, which includes ribosomal data, is considerably better than that estimated using the RPI369 dataset, from which ribosomal data have been excluded. We also performed leave-one-out cross validation for the RF classifier. The results were not significantly different from 10-fold cross-validation experiments.
The performance statistics reported in Table 1 were obtained using classifiers designed to provide high prediction accuracy. By varying the classification threshold value, the prediction specificity can be increased at the expense of a decrease in sensitivity. The corresponding trade-off between true positive rate and false positive rate can be seen from the receiver operating characteristic (ROC) curve shown in Figure 1. Consistent with the results in Table 1, ROC AUCs of 0.97 (RF) and 0.92 (SVM) were obtained for predictions on the RPI2241 dataset, with lower values of 0.85 (RF) and 0.81 (SVM) on the RPI369 dataset. For both classifiers, the AUC of ROC is significantly greater than 0.50 (random), indicating the feasibility of predicting RPIs using only sequence information from the RNA and protein as input.

Performance of RPISeq classifiers in predicting RPIs. Receiver operating characteristic (ROC) curves for RPI predictions, illustrating the trade-off between true positive rate and false positive rate for RPISeq-RF (random forest) and RPISeq-SVM (support vector machine) classifiers, using two datasets, RPI2241 and RPI369. The area under the curve (AUC) of each ROC is shown next to the curve. The AUC for a perfect classifier is 1, and for a random classifier = 0.5.
Comparison with other methods for predicting RNA-protein interactions
Bellucci et al. [35] used a variety of physicochemical properties (e.g., hydrogen-bonding propensities, secondary structure propensities) of proteins and RNAs to predict the interaction propensities for individual residues in the RNA and protein sequences of a potentially interacting pair. Because the catRAPID server [http://tartaglialab.crg.cat] does not directly report predictions as to whether or not a specific RNA-protein pair is expected to interact (the "partner prediction problem"), we were not able to directly compare our results with their method [35].
Pancaldi and Bähler et al. [36] also employed RF and SVM classifiers, but their method uses more than 100 different features of mRNA and proteins, extracted from the literature or computed from the protein and RNA sequences to make predictions. Examples of such features include mRNA half-life, predicted protein secondary structure, Gene Ontology annotation, relative abundance of each amino acid, codon bias. Using a dataset of 5,166 positive mRNA-protein RPI partners derived from Hogan et al. [10], and 5,166 randomly generated negative examples of mRNA-protein pairs, Pancaldi and Bähler reported an average accuracy of 70% in 2-fold cross-validation tests using an RF classifier based on 500 trees, and 68% using an SVM classifier using an RBF kernel with optimized parameters [36]. They also reported that 5-fold and leave-one-out experiments gave comparable results. We performed 10-fold cross-validation experiments on the same dataset using RPISeq-RF, which uses only sequence information. Our RF classifier achieved an accuracy of 68%, based on 500 trees, results comparable to the 70% reported for the RF classifier of Pancaldi and Bähler [36]. Our SVM classifier, using a normalized polykernel, gave less accurate predictions (61%) than the SVM of Pancaldi and Bähler (68%) [36].
In the Pancaldi and Bähler study, only 5,166 out of a total of 13,243 positive mRNA-protein pairs were actually used for prediction, because some of the features required by the classifiers were not available for the remaining 8,000 pairs [36]. When we tested our method using all 13,243 pairs for cross-validation, the prediction accuracies increased to 78% for the RF and 65% for SVM classifier. Taken together, our experiments indicate that the sequence-based method proposed here and the multiple feature-based method of Pancaldi and Bähler have comparable performance in predicting mRNA-protein interactions. Further, our results suggest that sequences of mRNAs and proteins carry sufficient information to allow reasonable predictions regarding whether or not a given mRNA and protein interact. Because feature information required by the method of Pancaldi and Bähler may not be available in many cases, our proposed method complements theirs, and may be more generally applicable for predicting ncRNA-protein partners, in addition to mRNA-protein partners.
Predicting ncRNA-protein interaction networks
An important potential application of RPISeq is computational construction of RNA-protein interaction networks. Recently, Nacher and Araki [37] used RPIs from the NPInter database [27], a database of non-coding RNA-protein interactions, to construct non-coding RNA-protein networks for several different model organisms. Their study revealed significant similarities between ncRNA-protein and transcription factor-gene regulatory networks. To explore whether RPISeq could be useful for constructing networks of ncRNA-protein interactions, we evaluated our method in predicting RPIs in networks derived from NPInter. Because the NPInter RPI pairs do not include any pairs derived from ribosomes, in this experiment, we also compared the performance of models trained on the RPI369 (which lacks ribosomal sequences) versus RPI2241, to evaluate the potential effect of strong ribosomal sequence bias on performance.
Tables 2 and 3 show the number of RPI pairs correctly predicted for each organism. When trained on the RPI2241 dataset (Table 2), the RF classifier correctly predicted ~ 80% (1,349 of 1,681 total interactions). The output probabilities of RPISeq are estimates of interaction propensities for a specific RNA-protein pair. In Tables 2 and 3, the probability threshold used for "positive" interactions was 0.50. Among the 1,349 interactions predicted by the RF classifier, only 119 were predicted with probabilities ≥ 0.80, and another 1,230 interactions were predicted with probabilities in the range 0.50-0.80. The SVM classifier generally had slightly lower performance, correctly predicting ~ 66% of the interactions.
In contrast, when trained on the RPI369 dataset, the SVM classifiers out-performed the RF classifiers (Table 3). Overall, the SVM classifier correctly predicted 1,402 (83%) and the RF classifier correctly predicted 1,115 (66%) of the interactions. Among the 1,402 interactions correctly predicted by SVM classifier, more than 850 interactions were predicted with probabilities ≥ 0.80, and another 525 interactions were predicted with probabilities in the range 0.50 to 0.80. For the RF classifier, only 50 interactions were predicted with probabilities ≥ 0.80.
With regard to the effects of ribosomal sequence bias, these results are somewhat difficult to interpret. The best "overall" prediction performance was obtained using the SVM classifier trained on the RPI369 dataset (which lacks ribosomal sequences), with 83.4% interactions correctly predicted; the RF classifier trained on the RPI2241 dataset (which includes ribosomal sequences) correctly predicted 80.2% of the total interactions. Differences in performance of classifiers trained on the two different datasets are significant when predictions for each model organism are considered individually. For example, for D. melanogaster, substantially better predictions were obtained with an RF classifier trained on the RPI2241 dataset (98.8%) versus an RF classifier trained on the RPI369 dataset (46.9%). In contrast, for predicting human and mouse RNA-protein interactions, SVM classifiers trained on the RPI369 dataset (which excludes the ribosomal sequences) provide the best prediction performance. For yeast RPIs, both the RF and SVM classifiers trained on RPI2241 generated excellent predictions, 98.0% and 99.2%, respectively, whereas classifiers trained on RPI369 made more errors, with correct predictions for 66.1% (RF) and 89.0% (SVM) of the cases.
Figure 2 shows the ncRNA-protein interaction network from S. cerevisiae, based on the data in NPInter. In Figure 2A, RPISeq predictions obtained using classifiers trained on the RPI2241 dataset are mapped onto the network. As described above, the SVM classifier (right) makes more correct predictions (green edges) and fewer incorrect predictions, i.e., false negatives, (red edges) than the RF classifier (left). In Figure 2B, RPISeq predictions made using classifiers trained on the RPI369 dataset, which results in more errors, are shown.

A. Predicted interactions using classifiers trained on the RPI2241 dataset. Among 254 known interactions, RPISeq-RF and RPISeq-SVM classifiers correctly predicted all except 5 and 2 edges, respectively. A protein hub, highlighted in yellow, shows interactions of a helicase (SEN1) with several snoRNAs. One of several RNA hubs, highlighted in purple, illustrates interactions of an snRNA (u4560) with various Sm-like proteins in the LSM complex.
B. Predicted interactions using classifiers trained on RPI369 dataset. Among 254 known interactions, RPISeq-RF classifier correctly predicted 168 (66%) and RPISeq-SVM correctly predicted 226 (89%). A protein hub highlighted in yellow, shows interactions of a helicase (SEN1) with 8 snoRNAs. One of several RNA hubs, highlighted in purple, illustrates interactions of an snRNA (u4560) with various Sm-like proteins in the LSM complex.
C. An enlarged view of the protein (SEN1) and RNA (snRNA) hubs described in B. above. Edges are labelled with the interaction probabilities predicted by RPISeq-RF (left) and RPISeq-SVM (right) classifiers, providing estimates of the relative pairwise interaction propensities.
One protein hub (highlighted in yellow), which appears as a green square node with connections to several RNA nodes (pink circles), is apparent in these views of the network. It corresponds to the yeast SEN-1 helicase, which is known to interact with several snoRNAs [38]. Several RNA hubs, represented by red circular nodes, each connected to several green protein nodes, are also apparent. One of these RNA hubs (highlighted in purple), corresponds to snRNA u4560, which interacts with various Sm-like proteins in the LSM complex [39].
Figure 2C shows an enlarged view of these hubs, extracted from Figure 2B. Edges are labelled with the interaction probabilities predicted by each classifier. Using classifiers trained on the RPI369 dataset, the RF classifier made more errors (i.e., predicted a known interaction with probability < 0.5) than the SVM classifier in both cases: for SEN-1 helicase, the RF classifier correctly identified only 4 out of 8 known snoRNA interactions, whereas the SVM classifier correctly identified 6 out of 8. Similarly, of 8 proteins known to interact with snRNA u4560 in yeast, the RF classifier identified 6, while the SVM classifier correctly identified all 8 interaction partners. Notably, as shown in Figure 2A, both RF and SVM classifiers trained on the RPI2241 dataset correctly identified all 8 RNA interaction partners of the SEN-1 helicase, and both classifiers missed only 1 of 8 protein interaction partners of the snRNA u4560.
Discussion
Regulation of gene expression at the post-transcriptional level is often mediated by interactions between RNA binding proteins and mRNAs or ncRNAs [5, 11, 40]. In this work, we present a new method, RPISeq, for predicting RNA-protein interaction partners, using only sequence information, with up to 90% average accuracy. We also demonstrate, that RPISeq can effectively predict RNA-protein interaction networks, based on evaluation using available data from five model organisms.
Sequence-based prediction of RNA-protein interactions
While several computational methods for predicting networks of protein-protein interactions have been developed [1, 2], very few studies have focused on computational analysis or prediction of RNA-protein interactions [3, 4]. One of the major challenges in solving the "partner prediction problem" for RNA-protein interactions is the limited amount of experimental data currently available. Unlike the "interface prediction problem," for which detailed structural information for more than 1,000 RNA-protein complexes is available in the PDB, mRNA partners for only a handful of RBPs are known [10]. Currently, two basic types of information regarding RNA-protein interaction partners are widely available: i) experimentally-determined structures of RNA-protein complexes, available in primary resources such as the PDB [26] and NDB [41], and secondary resources such as PRIDB [34] and BIPA [42]; and ii) experimental data from in vivo or in vitro cross-linking studies focused on individual proteins (e.g., SFRS1 [43], PUF [44]) or from high throughput RNA-binding microarrays [23], stored in repositories such as NPInter [27], CLIPZ [45] and RBPDB [46].
RPISeq requires only sequence information to generate predictions. In the current version of RPISeq, the classifiers were trained using only RPIs for which experimental structures are available. RPI2241 is a non-redundant training dataset consisting of 2241 interacting RNA-protein pairs, and includes a wide variety of different functional classes of proteins and RNA (e.g., rRNA, tRNA, miRNA, mRNA). rRNA-ribosomal protein pairs constitute ~ 40% of the total, reflecting the predominance of ribosomal structures in the current version of the PDB. To investigate the impact of this bias on machine learning methods for predicting RPIs, we also generated a smaller dataset of 369 RNA-protein partners (RPI369), from which all rRNA-containing complexes had been removed (see Methods for details).
We used RPI2241 and RPI369 as non-redundant benchmark datasets for developing and rigorously evaluating the performance of various machine learning classifiers. In cross-validation experiments, classifiers trained and tested on the larger dataset had superior prediction performance, indicating that the greater number and diversity of complexes in RPI2241, relative to RPI369, has a stronger positive effect on classification accuracy than the potentially negative effect of sequence bias in RPI2241. When we evaluated classifiers using independent datasets of RPIs from NPInter, however, classifiers trained on RPI369, in some cases, had better prediction performance. The basis for this observation is currently under investigation.
To identify sequence features of the proteins and RNA important in determining their specific interactions, we analyzed the features most frequently used by the Random Forest classifier to predict interacting partners (see Methods for details).
The four most often selected RNA tetrads were: AUUC, AGUG, UUUU and UCAA. Notably, these tetrads were found in the interfacial region in only 15% of the cases examined. The most frequently selected conjoint triad in protein sequences was {I, L, F, P}{A, G, V}{R, K}, which represents twenty-four possible amino acid triplets (e.g., IAR, IAK, IGR, IGK...). The complete list of important RNA and protein features is provided as Supplemental data S1 (Additional file 1). Although additional experiments and analyses of these features will be required to extract precise "rules" that specify a particular RNA-protein interaction, our current analysis indicates that at least 50 features (a combination of RNA and protein features) are required to accurately classify a given RNA-protein pair as interacting or not.
In this study, RPISeq accurately predicted RPIs in both cross-validation experiments using the benchmark datasets and in experiments on independent datasets. This suggests that normalized k-mer frequency distributions of RNA and protein sequences (specifically, reduced alphabet representations of protein sequences) in combination with appropriate machine learning methods, provide an effective approach to construct RPI predictors. Because the data used in this study represent only a small fraction of cellular RNA-protein complexes and interactions, we anticipate that more accurate predictions will be possible when larger and more diverse datasets of experimentally validated RPIs become available.
Comparison with other available methods
The method of Pancaldi and Bähler [36], which was developed to predict mRNA-protein interactions (rather than ncRNA-protein interactions), also uses RF and SVM classifiers, but requires a much more extensive set of features regarding the mRNAs and proteins. Input for the classifiers, which consists of a vector constructed by concatenating the features of potential RNA and protein partners (e.g., isoelectric point of protein, protein localization, mRNA half-life), cannot be extracted or calculated from sequence information alone. This requirement restricts the applicability of this method in practice: Pancaldi and Bähler were not able to extract the necessary features for a majority of interactions in their RPI dataset. The RPISeq methods do not suffer from this limitation because they require only sequence-derived features to make reliable predictions. In fact, the performance of RPISeq improved substantially (by 8% in accuracy) when evaluated on the entire dataset of Pancaldi and Bähler. Thus, for predicting mRNA-protein interactions, the sequence-based approach implemented in RPISeq provides performance comparable to that of classifiers that require a more extensive set of features, including those that cannot be extracted from RNA and protein sequences alone.
Application of RPISeq to constructing RNA-protein interaction networks
Encouraged by the success of RPISeq in predicting specific RPIs, we examined its effectiveness in constructing RNA-protein interaction networks in several model organisms, using only information derived from RNA and protein sequences. The networks were extracted from the "ncRNA binds protein" category of NPInter [27], currently the only available database of functional interactions of ncRNA with proteins. RPISeq was able to successfully predict the interactions of a single protein with multiple RNAs (protein hubs), as well as interactions of a single RNA with multiple proteins (RNA hubs).
In the case of the yeast, S. cerevisiae, RPISeq provided excellent predictions of RPIs: both the RF and SVM classifiers trained on the RPI2241 dataset correctly predicted > 98% of interactions in the NPInter database [27]. The RPISeq-RF classifier trained on the RPI2241 dataset also correctly identified a large majority of interactions in the D. melanogaster (99%) and E. coli (92%) networks. For human and mouse networks, however, classifiers trained on the RPI369 dataset gave better performance, with the RPISeq-SVM classifier correctly identifying 83% of the interactions in human and 93% in the mouse. It is important to note that these evaluations are based on predicting only known "positive" interactions currently available in NPInter [27]; "negative" data regarding non-interacting protein-RNA-protein pairs are not included in NPInter. Because the experimental data in NPInter are incomplete, it is problematic to assume that RNA-protein pairs not included in NPInter do not, in fact, interact. Also, some experimentally-determined RPIs included in NPInter could correspond to false positives.
Given the relatively small sizes of the RNA-protein networks analyzed in this study, differences in the results obtained using different classifiers to predict RPIs in different species must be interpreted with caution. It will be important to evaluate these methods on larger, more complete datasets of experimentally validated RNA-protein interactions as they become available. On the whole, our results suggest that RPISeq should be valuable for constructing and analyzing regulatory RNA-protein interaction networks.
Conclusion
In this work, we tested whether RPISeq, a family of purely sequence-based classifiers, can be used to predict whether a specific RNA-protein pair is likely to interact. Our results demonstrate that the corresponding RNA and protein sequences alone contain sufficient information to allow reliable prediction of RPIs. Such predictions can be used to: (i) identify putative RNA partners of a target protein, or protein partners of a target RNA; and (ii) computationally construct RNA-protein interaction networks. The datasets used in this study are relatively small compared with the large number of RNA-protein complexes and diverse interactions that occur in cells. The increasing availability of transcriptome-wide experimental data should lead to improvements in computational methods for predicting RNA-protein interactions and for modelling regulatory networks of RNA-protein interactions. RPISeq is freely available as a web-based server at http://pridb.gdcb.iastate.edu/RPISeq/.
Methods
RPI benchmark datasets derived from structure-based experimental data
For training and testing classifiers, two benchmark non-redundant datasets of RNA-protein interacting pairs were extracted from 943 protein-RNA complexes in PRIDB using an 8 Å distance cut-off [34]. PRIDB is a database of protein-RNA interfaces calculated from protein-RNA complexes in the PDB [26]. The original 943 complexes from PRIDB contained a total of 9,689 protein chains and 2,074 RNA chains; the final dataset RPI2241 (see below), which contains a total of 952 protein chains and 443 RNA chains, was derived from these complexes by applying the following criteria. Redundant protein sequences (i.e., with ≥ 30% sequence identity) interacting with similar RNA sequences (i.e., with ≥ 30% sequence identity) were discarded. Also, redundant RNA sequences (i.e., with ≥ 30% sequence identity) interacting with similar protein sequences (i.e., with ≥ 30% sequence identity) were discarded. Only proteins whose length is greater than 25 and RNAs at least 15 nucleotides long were retained. This resulted in a dataset of "positive" examples, RPI2241, consisting of 2241 experimentally validated RNA-protein pairs, and is provided as Supplemental data S2 (Additional file 2).
To generate a balanced dataset of "non-interacting RNA-protein pairs" (negative examples), we randomly paired the RNAs and proteins from the 943 protein-RNA complexes and removed similar interacting RNA-protein pairs (a randomly generated pair A-B was discarded if there exists a positive interaction pair C-B, and A and C share ≥ 30% sequence identity). Because ~40% of RNA-protein complexes in the PDB correspond to ribosomal structures, the RPI2241 dataset is also strongly biased towards ribosomal RPIs. Thus, we constructed a second dataset, RPI369, which is a subset of RPI2241 generated by removing all RPIs that contain ribosomal proteins or ribosomal RNAs and is provided as Supplemental data S3 (Additional file 3). RPI369 contains only non-ribosomal complexes (e.g., tRNA, mRNA, viral RNA, miRNA).
RPI datasets derived from non-structure-based experimental data
For evaluation of our method on independent RPI datasets, we used two datasets of RPIs obtained from RNA immunoaffinity purification and microarray experiments, published by Hogan et al[10]. One dataset comprises 5,166 mRNA-protein interactions; this dataset was also used in the study of Pancaldi and Bähler [36]. The second dataset is larger, consisting of 13,243 RPIs, and including all 5,166 interactions in the smaller dataset. Pancaldi and Bähler were not able to evaluate their method on this larger dataset because of missing feature information for RNAs and proteins involved in these interactions. Because RPISeq uses only sequence information, we were able to evaluate our method using all of the available data.
To test the ability of RPISeq to predict ncRNA-protein interaction networks, we used the NPInter database http://www.panrna.org/NPInter/, which includes eight different categories of functional interactions between non-coding RNAs, but excludes ribosomal RNAs and proteins. We extracted only those interactions for which there is experimental evidence for physical association of ncRNA with a protein, i.e. the 'ncRNA binds protein' category.
Alternative representations of protein and RNA sequences
Each RNA-protein pair is represented as a 599-feature vector, in which 343 features are used to encode the protein sequence and 256 features are used to encode the RNA sequence. Proteins are encoded using the conjoint triad feature (CTF) representation previously used by Shen et al[28]. In this method, the 20 amino acids are classified into 7 groups according to their dipole moments and the volume of their side chains: {A, G, V}, {I, L, F, P}, {Y, M, T, S}, {H, N, Q, W}, {R, K}, {D, E}, {C}. Each protein sequence is then encoded using the 7-letter reduced alphabet. Each protein feature represents the normalized frequency of the corresponding conjoint triad, i.e., 3-mer in the 7-letter reduced alphabet representation of the protein sequence. Thus, each protein sequence is represented by a 343 (7 × 7 × 7) dimensional vector, where each element of the vector corresponds to the normalized frequency of the corresponding 3-mer in the sequence (see [28] for details). Based on results of preliminary tests comparing the normalized k-mer frequency representation of RNA sequences for different values of k, we chose to encode RNA sequences using a 4 × 4 × 4 or 256-dimensional vector, in which each feature represents the normalized frequency of the corresponding 4-mer appearing in the RNA sequence (e.g., AAUG, CGAU, GGCC)
Machine Learning Algorithms
The SVM classifier [47] classifies input samples represented in the form of n-dimensional vectors into two classes using a hyperplane in a feature space. If the patterns are not separable in the original n-dimensional input space, a suitable non-linear kernel function is used to implicitly map the patterns in the n-dimensional input space into a typically higher (finite or even infinite) dimensional kernel-induced feature space in which the patterns become separable or nearly separable. Given a training set consisting labeled examples of the form (Xi, yi) where Xi is an n-dimensional input vector and yi = 0/1 is its label (i.e., the desired output of the SVM classifier for input Xi), the SVM learning algorithm effectively selects the hyperplane that maximizes the margin of separation between the training samples of the two classes from among all separating hyperplanes. If the examples are not perfectly separable in the kernel-induced feature space, a user-chosen parameter C is used to trade off training error (the number of misclassified training examples) against margin for the correctly classified training examples.
In our study, the input to the SVM classifier is a 599-dimensional vector that encodes features of a given pair of RNA and protein sequences as described above. The output of the SVM is a binary label indicating whether the given RNA-protein pair interact or not. We used the Sequential Minimal Optimization SMO implementation in Weka 3.7 [48] to train the SVM classifier. After some preliminary experiments which showed that the normalized polykernel performed better than RBF kernel on our data, we chose the normalized polykernel function of order 2 with ε = 1.0E-12. We set C = 1.0 and tolerance parameter T = 0.0010. We then used the option to fit a logistic model to the output of the resulting SVM classifier to obtain the posterior probability of class from the SVM output for any given input.
RandomForest (RF) [49] is an ensemble of many classification trees. Each tree in the ensemble is trained on a subset of training examples that are randomly sampled from the given training set. At each node the best split is chosen from a set of m variables selected at random from the set of input features. Given a query instance, the majority vote of all the classifiers is returned as the RF prediction. We used the Random Forest implementation in Weka 3.7. By default, Weka builds a RF classifier as an ensemble of 10 trees and sets the value of m = log_2 (number of features) + 1. For most of our experiments, we set the number of trees to 20 and 10 features were evaluated at each node. For comparison with the method of Pancaldi and Bähler [36], we set the number of trees to 500.
For performing feature selection, we used AttributeSelection class in Weka toolkit. We used wrapper subset evaluator in combination with Random Forest classifier and best first search method.
Performance Evaluation
Standard 10-fold cross-validation procedures were used to evaluate and compare classifier performance on the benchmark datasets. For the RF classifier, we also performed leave-one-out cross-validation; results were not significantly different from those obtained using 10-fold cross-validation (data not shown).
We computed the following statistics, as described in Baldi et al. [50], to measure the performance of the classifiers.
where TP is the number of true positives, FP is the number of false positives, TN is the number of true negatives, and FN is the number of false negatives.
The F-Measure is a composite indicator of performance that attempts to "balance" precision and recall. F-Measure values range from 0 to 1, with values close to 1 indicating better performance. The area under the curve (AUC) of the receiver operating characteristic curve (ROC) was also computed. AUC values also range from 0 to 1: the AUC = 1 for a perfect classifier and for a random classifier = 0.5.
References
Lees JG, Heriche JK, Morilla I, Ranea JA, Orengo CA: Systematic computational prediction of protein interaction networks. Phys Biol 2011, 8: 035008. 10.1088/1478-3975/8/3/035008
Wang T-Y, He F, Hu Q-W, Zhang Z: A predicted protein-protein interaction network of the filamentous fungus Neurospora crassa. Mol Biosyst 2011.
Lee TI: Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002, 298: 799–804. 10.1126/science.1075090
Martínez-antonio A: Escherichia coli transcriptional regulatory network. Netw Biol 2011, 1: 21–33.
Kishore S, Luber S, Zavolan M: Deciphering the role of RNA-binding proteins in the post-transcriptional control of gene expression. Brief Funct Genomics 2010, 9: 391–404. 10.1093/bfgp/elq028
Mittal N, Roy N, Babu MM, Janga SC: Dissecting the expression dynamics of RNA-binding proteins in posttranscriptional regulatory networks. Proc Natl Acad Sci USA 2009, 106: 20300–20305. 10.1073/pnas.0906940106
Tsvetanova NG, Klass DM, Salzman J, Brown PO: Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS One 2010, 5: e12671. 10.1371/journal.pone.0012671
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T: Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141: 129–141. 10.1016/j.cell.2010.03.009
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T: PAR-CliP--a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp 2010.
Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO: Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol 2008, 6: e255. 10.1371/journal.pbio.0060255
Licatalosi DD, Darnell RB: RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 2010, 11: 75–87.
Sola I, Mateos-Gomez PA, Almazan F, Zuñiga S, Enjuanes L: RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol 2011, 8: 237–248. 10.4161/rna.8.2.14991
Li Z, Nagy PD: Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol 2011, 8: 305–315. 10.4161/rna.8.2.15391
Baroni TE, Chittur SV, George AD, Tenenbaum SA: Advances in RIP-chip analysis: RNA-binding protein immunoprecipitation-microarray profiling. Methods Mol Biol 2008, 419: 93–108. 10.1007/978-1-59745-033-1_6
Barkan A: Genome-wide analysis of RNA-protein interactions in plants. Methods Mol Biol 2009, 553: 13–37. 10.1007/978-1-60327-563-7_2
Charon C, Moreno AB, Bardou F, Crespi M: Non-protein-coding RNAs and their interacting RNA-binding proteins in the plant cell nucleus. Mol Plant 2010, 3: 729–739. 10.1093/mp/ssq037
Kaymak E, Wee LM, Ryder SP: Structure and function of nematode RNA-binding proteins. Curr Opin Struct Biol 2010, 20: 305–312. 10.1016/j.sbi.2010.03.010
Kim MY, Hur J, Jeong S: Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep 2009, 42: 125–130. 10.5483/BMBRep.2009.42.3.125
Pacheco A, Martinez-Salas E: Insights into the biology of IRES elements through riboproteomic approaches. J Biomed Biotechnol 2010. doi:10.1155/2010/458927
Terribilini M, Lee J-H, Yan C, Jerniga RL, Honavar V, Dobbs D: Prediction of RNA binding sites in proteins from amino acid sequence. RNA 2006, 12: 1450–62. 10.1261/rna.2197306
Pérez-Cano L, Fernández-Recio J: Optimal protein-RNA area, OPRA: a propensity-based method to identify RNA-binding sites on proteins. Proteins 2010, 78: 25–35. 10.1002/prot.22527
Zhou P, Zou J, Tian F, Shang Z: Geometric similarity between protein-RNA interfaces. J Comput Chem 2009, 30: 2738–2751. 10.1002/jcc.21300
Ray D, Kazan H, Chan ET, Castillo LP, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR: Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nature Biotechnol 2009, 27: 667–70. 10.1038/nbt.1550
Keene JD, Komisarow JM, Friedersdorf MB: RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nature protoc 2006, 1: 302–7. 10.1038/nprot.2006.47
Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Blume JE, Wang X, Darnell JC, Darnell RB: HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456: 464–9. 10.1038/nature07488
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res 2000, 28: 235–42. 10.1093/nar/28.1.235
Wu T, Wang J, Liu C, Zhang Y, Shi B, Zhu X, Zhang Z, Skogerbø G, chen L, Lu H, Zhao Y, Chen R: NPInter: the noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res 2006, 34: D150–2. 10.1093/nar/gkj025
Shen J, Zhang J, Luo X, Zhu W, Yu K, Chen K, Li Y, Jiang H: Predicting protein-protein interactions based only on sequences information. Proc Natl Acad Sci USA 2007, 104: 4337–41. 10.1073/pnas.0607879104
Shao X, Tian Y, Wu L, Wang Y, Jing L, Deng N: Predicting DNA-and RNA-binding proteins from sequences with kernel methods. J Theor Biol 2009, 258: 289–293. 10.1016/j.jtbi.2009.01.024
Wang Y, Wang J, Yang Z, Deng N: Sequence-based protein-protein interaction prediction via support vector machine. J Syst Sci Complex 2010, 23: 1012–1023. 10.1007/s11424-010-0214-z
Hwang H, Vreven T, Whitfield TW, Wiehe K, Weng Z: A machine learning approach for the prediction of protein surface loop flexibility. Proteins: Struct Funct Bioinf 2011., 79: doi: 10.1002/prot.23070
Chen X-W, Liu M: Prediction of protein-protein interactions using random decision forest framework. Bioinformatics 2005, 21: 4394–400. 10.1093/bioinformatics/bti721
Liu Z-P, Wu L-Y, Wang Y, Zhang X-S, Chen L: Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 2010, 26: 1616–1622. 10.1093/bioinformatics/btq253
Lewis BA, Walia RR, Terribilini M, Feguson J, Zheng C, Honavar V, Dobbs D: PRIDB: a Protein-RNA Interface Database. Nucleic Acids Res 2011, 39: D277–82. 10.1093/nar/gkq1108
Bellucci M, Agostini F, Masin M, Tartaglia GG: Predicting protein associations with long noncoding RNAs. Nature Methods 2011, 8: 444–445. 10.1038/nmeth.1611
Pancaldi V, Bähler J: In silico characterization and prediction of global protein-mRNA interactions in yeast. Nucleic Acids Res 2011, 1–11.
Nacher JC, Araki N: Structural characterization and modeling of ncRNA-protein interactions. Biosystems 2010, 101: 10–9. 10.1016/j.biosystems.2010.02.005
Ursic D, Chinchilla KJSF, Culbertson MR: Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA. Nucleic Acids Res 2004, 32: 2441–2452. 10.1093/nar/gkh561
Vidal VP, Verdone L, Mayes AE, Beggs JD: Characterization of U6 snRNA-protein interactions. RNA 1999, 5: 1470–81. 10.1017/S1355838299991355
Blencowe B, Brenner S, Hughes T, Morris Q: Post-transcriptional gene regulation: RNA-protein interactions, RNA processing, mRNA stability and localization. Pac Symp Biocomput 2009, 545–548.
Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh S-H, Srinivasan AR, Schneider B: A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J 1992, 63: 751–759. 10.1016/S0006-3495(92)81649-1
Lee S, Blundell T: BIPA: a database for protein-nucleic acid interaction in 3D structures. Bioinformatics 2009, 25: 1559–1560. 10.1093/bioinformatics/btp243
Sanford JR, Wang X, Mort M, VanDyun N, Cooper DN, Mooney SD, Edenburg HJ, Liu Y: Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res 2009, 19: 381–94.
Gerber AP, Herschlag D, Brown PO: Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol 2004, 2: E79. 10.1371/journal.pbio.0020079
Khorshid M, Rodak C, Zavolan M: CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res 2010, 39: 245–252.
Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR: RBPDB: a database of RNA-binding specificities. Nucleic Acids Res 2010, 39: 301–308.
Vapnik V: The Nature of Statistical Learning Theory. New York: Springer; 1995.
Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten IH: The WEKA data mining software: An update. SIGKDD Explorations 2009, 11: 10–18. 10.1145/1656274.1656278
Breiman I: Random Forests. Mach Learn 2001, 45: 5–32. 10.1023/A:1010933404324
Baldi P, Brunak S, Chauvin Y, Andersen CAF, Nielsen H: Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16: 412–424. 10.1093/bioinformatics/16.5.412
Acknowledgements and Funding
We thank Benjamin Lewis, Pete Zaback and Rasna Walia for valuable suggestions and comments on the manuscript. We also thank Yasser EL-Manzalawy for critical reading of the manuscript and other members of the Honavar research group for interesting discussions. The work of Vasant Honavar while working at the National Science Foundation was supported by the National Science Foundation. Any opinion, finding, and conclusions contained in this article are those of the authors and do not necessarily reflect the views of the National Science Foundation. This work was partially supported by funding from National Institutes of Health (GM066387 to VGH and DD) and Iowa State University's Center for Integrated Animal Genomics (to UKM and DD). Partial funding for open access charges was provided by Iowa State University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
UKM conceived the study (with DD and VGH), carried out the experiments, implemented the RPISeq webserver and prepared the initial draft of the manuscript. DD and VGH contributed to the experimental design, supervised the work, and edited the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12859_2011_5035_MOESM1_ESM.TXT
Additional file 1:List of RNA and protein features important for distinguishing interacting and non-interacting RNA-protein pairs (S1). (TXT 859 bytes)
12859_2011_5035_MOESM2_ESM.TXT
Additional file 2:Positive RPIs in the RPI2241 dataset. This is a tab-delimited file with two columns. The first column is a list of proteins and the second column is a list of corresponding RNAs (S2). (TXT 31 KB)
12859_2011_5035_MOESM3_ESM.TXT
Additional file 3:Positive RPIs in RPI369 dataset. This is a tab-delimited file with two columns. The first column is a list of proteins and the second column is a list of corresponding RNAs (S3). (TXT 5 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Muppirala, U.K., Honavar, V.G. & Dobbs, D. Predicting RNA-Protein Interactions Using Only Sequence Information. BMC Bioinformatics 12, 489 (2011). https://doi.org/10.1186/1471-2105-12-489
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-12-489